

Abstract
Candidate HIV-1 vaccines currently being evaluated in clinical trials are designed to elicit HIV-1-specific cellular immunity. Intracellular cytokine staining (ICS) assays allow sensitive, quantitative ex vivo assessments of antigen-specific T cells including immunophenotyping of responding cells and measurement of multiple effector functions. Additionally, the use of banked cryopreserved PBMC samples makes this assay attractive in the setting of large efficacy trials where it is less feasible to perform immunoassays on freshly isolated samples. Here we describe extensive studies to optimize and quantitatively validate the 8-color ICS assay for use in clinical trials of candidate vaccines, which includes measurement of viable IFN-γ, IL-2, TNF-α and IL-4 secreting CD4+ and CD8+ T cells. We show that omission of viability dye staining results in an over-estimate of the true antigen-specific T cell response by up to two-fold. After optimization, the 8-color assay was validated for specificity, precision, linearity, limit of quantitation and robustness. The assay has a lower quantitation limit, generally below 0.04%, depending on the cytokine subset. Additionally, with appropriate gating, the 8-color assay gives comparable cytokine-positive responses to those observed with the conventional 4-color assay. In conclusion, we provide the first description of a quantitatively validated ICS assay, which permits quantitative and qualitative evaluation of vaccine-induced immunogenicity and analysis of immune correlates of protection.
Keywords: Flow cytometry, intracellular cytokine staining, vaccine, immunogenicity, assay validation
Introduction
The AIDS pandemic has created an urgent global need to produce an efficacious prophylactic HIV-1 vaccine. Unfortunately, sterilizing immunity will only be produced with broadly reactive HIV-specific neutralizing antibodies which, for numerous reasons, are difficult to induce by vaccination (discussed in (Burton et al., 2004)). Indeed, this was emphasized by the recent data from the Vaxgen, Inc. phase III trial showing no efficacy of this envelope-based vaccine (Cohen, 2003). Thus, most current clinical trials assessing potential HIV vaccines are designed to elicit T cell immunity against HIV-1. Recent studies in rhesus macaques have implicated vaccine-induced CD8+ T cell responses in increased survival following SIV infection (Letvin et al., 2006; Mattapallil et al., 2006). Studies in mouse models have demonstrated that maintenance of effective CD8+ CTL requires the presence of antigen-specific CD4+ T helper cells (Sun and Bevan, 2003; Sun et al., 2004). Thus, a successful vaccine may need to induce both T cell subset populations.
Measurement of antigen-specific T cell responses induced by vaccination has relied on the IFN-γ ELISpot assay, which has been extensively validated for use in clinical trials (Russell et al., 2003). However, this assay is limited in that it gives no information regarding the immunophenotype of the responding cells, and only measures production of a single effector molecule. The ICS assay provides sensitive and quantitative phenotypic and polyfunctional data on responding T cell populations, making the assay more valuable for functional profiling of low-level vaccine-induced T cell responses. Since the correlates of protection against HIV-1 infection/progression are not known, and since subtle differences in frequency and/or function of HIV-1-specific T cells may have profound effects on the capacity to protect and on subsequent HIV-1 disease course, this information is of consequence. Significantly, like the ELISpot assay, the ICS assay can be performed using cryopreserved PBMC, which enables retrospective analyses to measure quantitative and qualitative differences between different vaccine regimens and to determine whether cellular immune responses correlate with protection from infection or control of infection.
Two ICS assays were chosen for optimization and validation. Initial optimization experiments were restricted to 4-color staining because of limited access to flow cytometers capable of examining more than four colors. The 4-color panel consists of antibodies specific for CD3, CD4 and CD8 detected on separate colors, and IFN-γ and anti-IL-2 on the same color. In this way, the total cytokine response for the two cytokines is detected, although the co-expression of one or both cytokines cannot be separately revealed. We expanded our staining panel to eight colors when an instrument capable of examining more than four colors became available to our laboratory. In this panel, anti-IFN-γ and anti-IL-2 are included on separate colors to evaluate single and co-expression of these cytokines. In addition, antibodies specific for TNF-α and IL-4 are included. These two cytokines have been useful for identifying discrete functional profiles in previous studies (De Rosa et al., 2004; Betts et al., 2006). Finally, a viability marker is included to identify and exclude dead cells that can bind antibodies, which result in non antigen-specific cytokine responses.
Advancement of T cell vaccines to phase III efficacy trials requires validation of immunoassays that measure the frequency and function of the induced antigen-specific T cells. While assays for detecting cellular immune responses in humans have recently been included as primary endpoints for clinical trials, the validation of these assays has generally not been approached in a manner that satisfies the guidelines provided in the Industry for Guidance — Bioanalytical Method Validation (http://www.fda.gov/CDER/GUIDANCE/4252fnl.htm, 2001) or the ICH Q2A and Q2B documents (http://www.fda.gov/cder/guidance/index.htm, 1996). These documents provide detailed definitions of eight parameters that must be investigated experimentally in order to validate a bioanalytical assay: (1) Specificity/Selectivity, (2) Accuracy, (3) Precision, (Repeatability, Intermediate precision, reproducibility), (4) Detection Limit, (5) Quantification Limit, (6) Linearity, (7) Range and (8) Robustness. Here we provide extensive optimization and, to our knowledge, the first quantitative validation data for an 8-color ICS assay designed to allow qualitative and quantitative evaluation of vaccine-induced T cell responses.
Methods
Study Participants
Most validation experiments were performed on CMV-seropositive, HIV-seronegative individuals, chosen because they most closely represent the populations participating in HIV Vaccine Trials Network (HVTN) clinical trials. The majority of optimization experiments were performed on three CMV-seropositive individuals who underwent leukapheresis, providing sufficient cryopreserved PBMC from a single time point for all validation studies. Qualitative validation experiments also included individuals with chronic HIV-1 infection, some of whom were receiving anti-retroviral treatment. The subjects were recruited and enrolled at the Seattle HIV Vaccine Trials Unit. The Institutional Review Board approved the study, and prior to enrollment volunteers provided written consent after being informed of the nature and possible consequences of the study.
PBMC Sample Processing
PBMC were isolated and cryopreserved either from whole blood or from a leukapheresis product within 8 hours of venipuncture using standard procedures as previously described(Bach and Brashler, 1970; Boyum, 1976). PBMC were thawed and rested overnight at 37°C/5%CO2 in R10 [RPMI 1640 (GibcoBRL, NY, USA) containing 10% FCS (Gemini Bioproducts, CA), 2 mM L-glutamine (GibcoBRL), 100 U/ml penicillin G, 100 μg/ml streptomycin sulfate] prior to stimulation. A minimum cell viability of 66% measured after overnight resting on the day following thaw was required for use in ICS assays. This threshold is arbitrary and was chosen because this is the threshold currently used for PBMC samples processed within the HIV Vaccine Trials Network (HVTN).
In Vitro Stimulations
PBMC were assessed for ex vivo responses to (1) a pool of CMV 15-mer peptides overlapping by 11 amino acids spanning the entire pp65 protein (kindly provided by the Division of AIDS, NIH); and/or (2) pools of HIV-1 15-mer peptides overlapping by 11 amino acids spanning Gag (HIV-1 HxB2), Nef (HIV-1 BRU) or Pol (HIV-1 HxB2) or covering potential T cell epitopes (PTE) for Env, Gag, Pol and Nef (Li et al., 2006). Staphylococcal enterotoxin B (SEB) stimulation served as a positive control. PBMC with peptide diluent (1% DMSO) served as the negative control. During the six-hour stimulation, Brefeldin A (10 μg/ml, Sigma, St. Louis, MO) and the co-stimulatory antibodies CD28 and CD49d (each at 1 μg/ml, Becton Dickinson (BD) Biosciences, San Jose, CA) were included.
ICS Protocol
All antibodies, except the CD3 ECD, were purchased from BD Biosciences. The CD3 ECD (or PE-TR) was purchased from Beckman-Coulter (Marseille, France). LIVE/DEAD Fixable Violet Dead Cell Stain was purchased from Invitrogen/Molecular Probes (Eugene, OR). The concentration of all antibodies was titrated prior to use. Staining panels assayed during optimization and validation are detailed in Table 1. The Cytokine Flow Cytometry (CFC) protocol from BD was chosen for further optimization and validation since extensive documented standardization of this assay already exists. All samples were acquired on a FACS Calibur flow cytometer (BD) capable of measuring four colors or an LSRII flow cytometer capable of measuring 18 colors (BD), collecting 100-200,000 lymphocyte-gated events. Samples were collected from 96-well plates using High Throughput Sample (HTS, BD) device for analysis by the LSRII. All FACS analyses were performed using FlowJo® software (Treestar, Inc; OR).
Table 1.
Staining panels used for validation experiments
| ICS Panel | Marker | Dye | Purpose |
|---|---|---|---|
| Panel A 4-color |
CD3 | APC | Defines lineage |
| CD4 | FITC | ||
| CD8 | PerCP-Cy5.5 | ||
| IFN-γ/IL-21 combined or IFN-γ or IL-2 or TNF-α in separate panels2 |
PE | Cytokine response | |
| Panel B3 4-color |
CD3 | APC | Defines lineage |
| CD4 or CD84 | PerCP-Cy5.5 | ||
| IFN-γ | FITC | Cytokine response | |
| IL-2 | PE | ||
| Panel C 5-color5 |
Violet viability dye5 | PacBlu channel | Excludes dead cells |
| CD3 | APC | Defines lineage | |
| CD4 or CD8 | PerCP-Cy5.5 | ||
| IFN-γ | FITC | Cytokine response | |
| IL-2 | PE | ||
| Panel D 8-color |
Violet viability dye6 | PacBlu channel | Excludes dead cells |
| CD3 | PE-TR | Defines lineage | |
| CD4 | FITC | ||
| CD8 | PerCP-Cy5.5 | ||
| IFN-γ | PE-Cy7 | Cytokine response | |
| IL-2 | PE | ||
| TNF-α | Alx-700 | ||
| IL-4 | APC | ||
PE-conjugated antibodies to IFN-γ and IL-2 are both used together to stain cells.
One of these cytokines used individually in this panel (e.g., CD3, CD4, CD8, TNF-α). A separate panel needs to be used to evaluate each of these cytokines.
A variation of this panel included only one cytokine on PE and CD69 on FITC.
Two 4-color panels are required to examine both CD4+ and CD8+ T cell responses. One panel includes CD4, and the other panel includes CD8.
Referred to as the modified 4-color panel in Figure 4C.
LIVE/DEAD Fixable Violet Dead Cell Stain (see methods).
Statistical Analysis
Graphs were prepared using JMP software (SAS Institute, Cary, NC). A paired T test (JMP software) was used to compare across the conditions shown in Figure 1 and Figure 4 C.
Figure 1.

A. Culture of cells overnight after thaw increases cytokine responses. Cryopreserved PBMC from four healthy individuals were thawed, washed, resuspended at 5 × 106 cells/ml and added to the wells of two 96-well round-bottom plates. For one plate, the cells were stimulated immediately for six hours with DMSO, CMV or SEB, including co-stimulation and Brefeldin A. Cells were then processed by ICS. The other plate was placed into culture overnight (rested), and then stimulated for six hours followed by ICS. Three 4-color staining panels were used that included CD3, CD4, CD8 and the cytokine (IFN-γ, IL-2 or Panel A in Table 1). The upper panels show the CD4+ T cell responses, and the lower panels show the CD8+ T cell responses. The left panels show responses to co-stimulation only (negative control), the middle panels show responses to the CMVpp65 peptide pool, and the right panels show the response to SEB. Each color represents a different subject, and the lines join responses without or with overnight rest. All data for CMV and SEB are background subtracted. Paired T test p values are shown on the graphs.
B. Washing cells after overnight rest decreases background for IL-2 and TNF-α, but does not decrease CMV-specific responses. Three variables were examined in this experiment: resting concentration, incubation vessel for rest and stimulation, and washing vs. not washing cells in the morning after the overnight rest. For the first condition (left column in each graph), cells were rested at 5 × 106/ml in a 96-well round-bottom plate and were not washed before stimulation the next morning. The second condition (middle column in each graph) was the same except that the cells were washed in the morning before stimulation. For the third condition, cells were rested at 2 × 106/ml in 50 ml conical tubes and were washed in the morning before stimulation. CD4+ T cell responses to IL-2 (upper panels) and TNF-α (lower panels) are shown. The left panels show responses for the negative control, and the right panels show the responses to CMV. Six different PBMC samples were tested, and data for each sample are connected by lines. The 8-color ICS panel was used. Paired T test p values for comparisons between each of the three conditions are shown on the graphs.
C. Co-stimulation with α—CD28/CD49d increases background responses, but does not increase antigen-specific responses after background is subtracted. PBMC samples from five participants in HVTN protocol 041 (subunit protein vaccine including Gp120 and a Nef/Tat fusion protein) (Goepfert et al., 2005) were stimulated with a gp120 peptide pool (right panels), with the CMVpp65 peptide pool (middle panels), or with DMSO (negative control, left panels). The percentages of CD4+ (upper) and CD8+ (lower) T cells producing IFN-γ and IL-2 are shown. Within each graph, responses with or without co-stimulation are compared. Lines connect responses from the same PBMC donor. Gp120 and CMV results are shown with background subtracted. For this experiment, cells were rested overnight at 5 × 106/ml and were washed before stimulation. Paired T test p values are shown for the negative control. Comparisons for the Gp120 and CMV are not significant.
Figure 4.

Dead cells can cause non-specific cytokine staining, especially for PE-conjugated reagents. A. Shown are the IL-2 (upper) and IFN-γ (lower) CD4+ T cell responses to CMV in one PBMC sample. The expression of CD8 is shown on the abscissa to display the different staining pattern of CD8 on the dim cytokine-staining vs. the bright cytokine-staining cells. The percent of CD4+ T cells in an open cytokine gate or a high gate is shown. Four-color staining panels including CD3, CD4, CD8 and the cytokine on PE were used (the first staining panel in table 1 with the PE IFN-g omitted, analyzed on the LSRII). B. Shown are the responses to CMV stimulation for the same PBMC sample as shown in A, determined by the 8-color ICS panel. The left panels show the expression of the cytokine vs. CD8. The middle panels show the expression of the cytokine vs. the viability marker. C. The percentage of CD4+ T cells expressing IL-2 is shown for seven PBMC samples stimulated with CMV (left) or with DMSO (negative control, right). Within each graph, the results for the 8-color panel using a high cytokine gate are shown. The other three columns show the results for staining with a modified 4-color panel that includes the violet viability marker (the third staining panel in table 1), and compares 1) the frequency of IL-2-producing CD4+ T cells when dead cells are not gated out and a standard open cytokine gate is used (left column); 2) when dead cells are gated out and the standard cytokine gate is used (next column); or 3) when dead cells are not gated out and the high cytokine gate is used (third column). Cells have been gated as CD3+, and CD4+CD8-.
Method for qualitative validation of responses from ICS assays
PBMC from 50 HIV-seronegative individuals and from between 10 and 20 HIV-seropositive individuals were examined. For each sample and for each HIV-1 peptide pool, the responses were categorized as either positive or negative. Positivity was based on comparisons of the percentage of T cells with positive cytokine staining between the experimental well and the negative control well. Positivity was determined for each cytokine subset; e.g. IFN-γ+IL-2-. If at least one cytokine subset was positive, the overall peptide pool was considered positive.
Two-by-two contingency tables were derived for each comparison between the stimulated and negative control data for each cytokine subset. The four entries in the table are the number of cells positive for the cytokine subset and the number of cells negative for the cytokine subset, for both the stimulated and the negative control data. A one-sided Fisher’s Exact Test was applied to each table, testing whether the number of cytokine-producing cells for the stimulated data was greater than that for the negative control data. Since many tests were conducted simultaneously for the multiple cytokine subsets, multiple T cell subsets and multiple peptide pools, a multiplicity adjustment was made using the discrete Bonferroni adjustment method (Westfall and Wolfinger, 1997; Westfall et al., 1999). The adjusted p-values were used to determine positivity, with values less than or equal to α = 10-5 indicating a positive response.
Method for quantitative validation of responses from ICS assays
We developed a plan to perform qualitative validation, and the experimental design is summarized in the results section. To assess the linearity of the assay, simple linear regression models were used to determine the relationship between assay read-out and sample concentration levels. Analyses of precision were done separately at each concentration level. Standard mixed effect models run in SAS Inc. were used to derive various precision estimates for inter-analyst, inter-day, inter-plate and within-plate variance components. These assessments were done for each analyst, and then combined. The range of reliable responses were defined as the range over which the precision estimates were within 30% at all concentration levels. Exploratory upper or lower limits of quantification that do not meet this criterion were excluded from the final analyses. Two outliers were excluded from the final analysis because of high cytokine responses for all cytokines, likely due to SEB contamination during the sample processing.
Results
Optimization of the ICS Assay
Many variables and conditions were evaluated during the optimization of the ICS assay (Table 2). For HIV Vaccine Trials Network (HVTN) clinical trials, PBMC are isolated and cryopreserved within eight hours of venipuncture to ensure optimal performance in immunoassay. Therefore, the ICS assay was optimized and validated using cryopreserved rather than freshly isolated PBMC although the results should be equally applicable for freshly isolated cells. The procedures used for freezing and thawing of PBMC samples affects the viabilities of the cells after thawing. Our previous studies have shown that lower viabilities are associated with a decrease in CMV-specific T cell responses as measured by the IFN-γ ELISpot assay (data not shown). In the studies reported here, the viabilities for the PBMC samples determined immediately after thaw or after overnight culture were on average above 90% and no samples had viabilities below 66%, the lower acceptable threshold used for the HVTN clinical trials.
Table 2.
Summary of variables and conditions tested during the optimization and validation of the ICS assay
| Variable | Conditions tested (Selected condition underlined) |
Reason chosen for assay | Figure/ Staining Panel1 |
|---|---|---|---|
| PBMC rest post-thaw |
Culture overnight (rest) No rest |
Increases Ag-specific cytokine responses, but not background |
1A Panel D |
| Resting condition |
5 × 106/ml in plates; no wash 5 × 106/ml in plates; wash 2 × 106/ml in tubes; wash |
Decreases background but not Ag- specific responses (mainly for IL-2 and TNF-α from CD4+ T cells); also allows for recounting after rest |
1B Panel D |
| Stimulation time |
4 hrs 6 hrs overnight |
Cytokine responses maximal (for IFN-γ, IL-2, TNF-α, IL-4); also for convenience |
N/A Panel D |
| Costimulation |
With αCD28/CD49d Without αCD28/CD49d |
May increases Ag-specific responses (but also increases background) |
1C Panel D |
| Peptide length | 15mers overlapping by 11 | Stimulates both CD4+ and CD8+ T cells (as reported previously) |
N/A Panel A |
| Peptide pool size |
50, 100, 150, 200 peptides | ≤100; moderate decrease in responses for >100 peptides per pool |
2 Panel A |
| CD4 and CD8 included in same 4-color panel |
CD3, CD4, cytokine (±CD69) CD3, CD8, cytokine (±CD69) CD3, CD4, CD8 cytokine |
Reduces non-specific staining because CD4/CD8 double positive cells excluded |
N/A Panel B |
| Cell viability marker |
Included Not included |
Decreases low-level non-specific staining, especially for IL-2 and IL-4 |
4 Panels C and D |
Staining panel as listed in Table 1
Processing of PBMC after thawing
Our previous experiments suggested that culturing thawed cells overnight at 37°C/5% CO2 prior to stimulation (denoted “resting cells”) results in increased detectable cytokine-producing cells by ICS. To confirm this, we compared CMV-specific and polyclonal (SEB) responses for PBMC stimulated immediately after thawing to those after thawing and overnight rest (Figure 1A). The percentage of both CD4+ and CD8+ T cells producing all three cytokines tested (IFN-γ, IL-2 and TNF-α) were increased by as much as three-fold after resting for SEB stimulation, supporting our earlier observations. Responses to CMV were not significantly increased, but there was a trend toward increased responses for some samples. Background levels of cytokine-producing cells, i.e., without antigen or SEB during the six-hour stimulation period, were not substantially affected whether cells had or had not been rested (Figure 1A).
For the overnight rest, cells are cultured at 2 × 106/ml in R10 in 50 ml conical tubes, washed the next morning, re-counted, and then distributed at 5 × 106/ml into the wells of a 96-well round-bottom plate (Costar, Corning NY) for the stimulation. To optimize this protocol, the effect of cell concentration, type of culture vessel and washing of the cells after overnight rest was evaluated. The type of culture vessel used for either the rest or the stimulation, whether tubes or 96-well round-bottom or v-bottom plates, had no effect on cytokine responses (data not shown). Incubating cells at concentrations of 2 or 5 × 106 cells/ml during the overnight rest did not result in different percentages of cytokine-secreting CD4+ or CD8+ T cells (Figure 1B). Washing cells after the overnight rest and prior to stimulation resulted in lower levels of background IL-2- and TNF-α-producing CD4+ T cells. The extent of the decrease varied between different PBMC samples; in some cases, the responses were decreased nearly two-fold after washing. Levels of background for IFN-γ-producing CD4+ T cells and CD8+ T cells producing all three cytokines were not substantially affected by washing (data not shown), nor did washing cells result in lower antigen-specific responses to CMV. Washing cells before stimulation provided the opportunity to recount the cells prior to stimulation and, therefore, to distribute the correct number of cells for stimulation.
Stimulation and Fixation/Permeabilization Protocols
The protocol chosen for the in vitro cellular stimulation is similar to protocols used by many laboratories performing ICS assays. Cells are cultured for six hours in the presence of a chemical that blocks golgi export (Brefeldin A), co-stimulatory antibodies (α-CD28/α-CD49d) and either pools of peptides covering the antigens of interest or a positive control stimulus (e.g., SEB). Brefeldin A was selected rather than monensin because the fluorescent intensity of the TNF-α-producing cells was greater when Brefeldin A was used ((O’Neil-Andersen and Lawrence, 2002), data not shown). A six hour stimulation was chosen because the intracellular cytokine production of the four cytokines (IFN-γ, IL-2, TNF-α and IL-4) peaked by six hours. The percentage of cells producing IFN-γ, IL-2 and TNF-α remained at the peak level for at least 18 hours, but those producing IL-4 began to decrease after six hours (data not shown).
The experiments described here use the Becton-Dickinson CFC protocol because BD had previously optimized this procedure. Several studies have been published describing optimization of the assay and describing use of the assay for various applications. Several of these studies describe the reproducibility and variability of the assay - in response to CMV lysate within and between assays (Nomura et al., 2000), in response to SEB in individuals over time (Dunn et al., 2002), and when performed across multiple laboratories (Maecker et al., 2005b). In addition, the assay has been used to evaluate responses using peptide mixes including peptides of different lengths (Maecker et al., 2001), to comparing responses when the assay is performed in tubes vs. plates (Suni et al., 2003), and when performed using fresh or frozen PBMC (Maecker et al., 2005a). The CFC protocol was also chosen because, after stimulation and fixation, the cells can be frozen before proceeding to permeabilization and staining. Because it allows all steps in the procedure to fit within 8-hour workdays, this technique is expedient for samples in large-scale trials.
Co-stimulation
We evaluated the effect of co-stimulatory antibodies on the level of detection of cytokine-producing antigen-specific T cells (Figure 1C). When cells were cultured for six hours with no antigen, the background control including co-stimulation increased the frequency of CD4+ T cells producing IFN-γ or IL-2 by as much as ten-fold. This “background” is generally greater for CD4+ T cells producing IL-2, and costimulation produced the largest proportional increase for this cytokine subset population. The detection of background cytokine-producing CD8+ T cells was also increased, but to a lesser extent. In general, co-stimulation did not increase antigen-specific responses to CMV or to Gp120 (in PBMC from individuals vaccinated with Gp120 protein) after background responses were subtracted. In only a few samples there were modest increases in these antigen-specific responses. CD4+ T cells producing TNF-α, which typically occur in higher frequencies without stimulation, were not tested in this experiment. Because co-stimulation is commonly used in many laboratories for these types of assays and because a few antigen-specific responses were increased, we include co-stimulation in our protocol despite the increase in background.
Peptide pool size
The peptides used for stimulation, were 15 amino acids in length, and overlapped by 11 amino acids. These 15-mer peptides used for stimulation were combined into pools. The effect of pool size, i.e., the number of peptides in the pool, was examined by determining the cytokine response to an individual peptide that was included in peptide pools of different sizes (Figure 2). The individual peptides were from the pp65 CMV protein (15-mers AGILARNLVPMVATV and TERKTPRVTGGGAMA), and the PBMC samples selected had previously observed responses to these CMV peptides. The other peptides included in the pools were derived from HIV proteins, and the PBMC tested were from HIV-uninfected subjects. The appropriate amount of DMSO was added for each stimulation condition to control for the larger amount of DMSO present for the largest pool size. Therefore, the response was specific only to the CMV peptide. In some instances, up to a two-fold decrease in the cytokine-producing CD8+ T cells was observed when the peptide was mixed with pools containing more than 100 peptides. In others, the larger pool sizes did not substantially decrease responses. Testing of a larger number of samples will be required to determine if our observations in these few samples are statistically significant. Nevertheless, in light of the anecdotal observations presented here and because we need the capability to detect low-level responses in our clinical trials, we include 100 peptides or fewer for the examination of our clinical trial samples, except when the number of peptides is too large for this to be feasible (e.g., for the PTE peptide pools used in the qualitative validation as discussed below).
Figure 2.

Cytokine-producing T cells recognizing an individual CMV pp65 15 amino acid peptide decrease as the total pool size increases. PBMC from two subjects with previously documented responses to individual peptides were examined [subject 1584 (red) responded to CMV peptide AGILARNLVPMVATV, and subject 2622 (blue) responded to CMV peptide TERKTPRVTGGGAMA]. Each of these peptides was mixed with increasing numbers of HIV-1 Gag and Pol peptides, as listed on the abscissa; and the IFN-γ, IL-2 and TNF-α CD8+ T cell responses were measured by 4-color ICS. The response to the peptide alone is shown as the first data point in each graph. One outlier for the IFN-γ response (10% response) to peptide TERKTPRVTGGGAMA in the 200 peptide pool size in subject 262 was removed, as indicated by the asterisk.
Staining panels: 4- and 8 color; use of a viability dye
As noted in the introduction, initial optimization experiments were performed using 4-color staining panels. Later, an 8-color staining panel was developed and was used for all the qualitative and quantitative validation experiments described below. An example of the gating hierarchy and cytokine staining profiles of CD4+ and CD8+ T cells generated from the 8-color panel and the gating hierarchy is depicted in Figure 3.
Figure 3.

Staining profile for 8-color ICS. A. Shown is PBMC sample stimulated with SEB. The top row of plots shows sequentially, left to right, the gating hierarchy for PBMC, singlets (single cells), lymphocytes, live cells, CD3+ T cells, and CD4+ or CD8+ T cells. The next two rows show the gates used to identify cytokine-producing CD4+ (middle row) and CD8+ T cells (lower row). Each cytokine is gated individually. The co-expression of all the cytokines is determined in the software analysis by using Boolean gates based on these single-positive gates. B. Shown is the co-expression of IFN-γ and IL-2 for CD4+ and CD8+ T cells for the same PBMC sample shown in A after stimulation with SEB. C. Shown is the co-expression of IFN-γ and IL-2 for CD4+ and CD8+ T cells for a PBMC sample after stimulation with the CMV pp65 peptide pool. The percentage of either CD4+ or CD8+ T cells is indicated in each quadrant.
To determine whether the 4- and 8-color panels detect the same frequency of IFN-γ- and IL-2-producing T cells, we compared two 4-color panels (CD3, CD4, CD8, and either IFN-γ or IL-2) to the 8-color panel (Table 1). Since the 8-color panel included a viability dye, but the 4-color panels did not, this comparison also allowed us to determine whether dead cells contribute significantly to non-specific cytokine staining.
Examination of the staining profiles for the IL-2- and IFN-γ-producing cells using the 4-color panels revealed two levels of fluorescence intensity for these cells — cells with either bright or dull fluorescence (Figure 4A). We compared the percentage of cytokine-producing cells as determined by applying an open gate (including cells with either dim or bright cytokine staining) vs. a high gate that only included the bright cytokine staining cells. When an open gate was applied to the 4-color panel, the frequency of IFN-γ+ or IL-2+ cells was at least double the frequency seen vs. when a higher cytokine gate was applied (Figure 4A). The frequency of cytokine-positive cells determined with the higher cytokine gates was more comparable to the data obtained from the 8-color panel after excluding dead cells. When examined with the 8-color panel, the additional cytokine-dim cells that were included in the open gate were, in fact, the dead cells staining positive with the viability dye (Figure 4B). In this 8-color panel, the dim cytokine-staining dead cells were observed only for those producing intracellular IL-2, detected with an anti-IL-2 mAb conjugated with PE, but not those producing IFN-γ, using an anti-IFN-γ mAb conjugated with PE-Cy7. These results suggest that non-specific staining of dead cells may only occur with certain fluorochrome-antibody reagents.
This experiment demonstrates that when a standard open gate is applied, a viability marker is required to ensure that dead cells with low-level staining are eliminated (Figure 4C). Alternatively, use of a higher cytokine gate on a 4-color assay will also alleviate this problem. Use of a high cytokine gate on the 4-color assay provides frequencies of CMV-specific T cells similar to the 8-color assay with a viability marker. Use of the high gate also results in lower background, determined as the frequency of cytokine-producing CD4+ T cells when no antigen is included in the stimulation. For the 8-color assay, we use a high cytokine gate even though a viability marker is included in the assay. The use of a standard cytokine gate for the 8-color assay results in only a minimal increase in antigen-specific responses and in background, but also results in a higher false positive rate as determined in qualitative validation experiments (data not shown). This appears to be the result of low-level cytokine-staining cells that have been observed randomly without a recognized association with dead cells or other potential causes.
Qualitative and quantitative validation of the ICS assay
The optimization experiments described above allowed us to develop a Standard Operating Procedure (SOP) for the ICS assay, using either the 4-color or the 8-color panel, which is summarized in Figure 5. We next used the 8-color ICS SOP to perform qualitative and quantitative validation experiments. Some of the eight parameters required to validate bioanalytical assays cannot be assessed directly on the ICS assay. For example, accuracy determines whether the results of the assay agree with the true value. However, for the ICS assay, the true value of the cytokine response cannot be determined. As an alternative to comparing with a true value, the results of the assay can be compared with a “reference method”. In our laboratory, we plan to establish the current ICS assay as the reference method; and any future modifications to the assay will be compared to the results from the current assay.
Figure 5.

Summary of the ICS protocol is shown. This diagram outlines the major steps in the ICS protocol used for analysis of trial samples. The protocol requires three days. Plates frozen on day 2 can be kept at −80deg for at least four weeks before proceeding to the next steps as shown on day 3.
Qualitative Validation of the 8-color Assay
As a measure of specificity, we have performed experiments to determine the frequency of detection of HIV-specific responses in previously cryopreserved PBMC from 50 HIV-seronegative individuals. This determines the false positive rate for the assay for the particular HIV antigen tested. PBMC samples from HIV-seropositive individuals were also included to measure sensitivity of the assay. These experiments provide qualitative measurements only, determining whether a response is detected or is not detected.
We have used two methods to determine positivity. One is empirical based on our experience using the assay in the research laboratory. For this method, a sample is designated positive if the antigen-specific response is at least three-fold greater than the negative control (the “background”) and at least 0.05% above the background. For this method, a threshold value is necessary in order to reduce the false-positive rate. Additionally, we developed a statistical method based on the Fisher’s exact test. This method also includes a discrete Bonferroni correction for multiple testing (see methods). A sample is positive if the multiplicity adjusted p-value associated with the test is less than a specified alpha level (e.g. α = 10-5).
Using PBMC from 50 HIV-seronegative and 11 HIV-seropositive individuals, we assessed their recognition of HIV epitopes using peptide pools spanning Env, Gag, Pol and Nef proteins. The peptide sequences for these peptides were determined based on the Potential T Cell Epitope (PTE) method for global HIV sequences (Li et al., 2006). PBMC from all 11 HIV-1-seropositive individuals scored positive against at least one peptide pool. The false positive rate was 0% for all the peptide pools except for Pol2, Pol3 and Nef, where each had one false-positive (2%; Table 3). Thus the specificity of the assay is 94% when examining these nine peptide pools.
Table 3.
Qualitative validation of the PTE peptide pools1
| # positive in each pool2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV status |
# of subjects |
Env | Gag | Pol | Nef | Any | |||||
| 1 | 2 | 3 | 1 | 2 | 1 | 2 | 3 | ||||
| HIV- | 50 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 3 |
| HIV+ | 11 | 5 | 5 | 4 | 11 | 11 | 9 | 10 | 9 | 10 | 11 |
Sequences of peptides determined using the global potential T cell epitope (PTE) method.
Positivity determined by the statistical method developed by SCHARP (see methods). The results are similar when positivity is determined by the empirical method used in our laboratory. The differences are: for the HIV-seronegative group, Gag1 had one response and Nef had no responses; for the HIV-seropositive group, Env1 had 7 responses, Gag2 had 10, and Pol3 had 8.
Quantitative Validation of the 8-color Assay
We performed quantitative validation to assess precision, linearity, limit of quantitation and robustness of the ICS assay together in the same experiments. PBMC from three HIV-1 seronegative subjects were chosen known to have high, intermediate and low frequency CMV-specific T cells in previous studies. For linearity, CMV-stimulated PBMC were diluted into unstimulated PBMC from the same donor using eight 2-fold dilutions (Figure 6A). Samples were examined in triplicate at each dilution level to determine intra-sample variability. Three technicians performed the experiments with the same samples on the same day to determine inter-operator variability. In addition, one technician examined responses of the same donor PBMC on three days to determine inter-day variability. Finally, three frozen aliquots of the same PBMC sample were examined by three technicians on the same day to determine inter-sample variability.
Figure 6.

A. Quantitative validation of the 8-color ICS assay demonstrates that the assay is linear in the detection of IFN-γ alone and in combination with IL-2 for CD4 and CD8+ T cells. Shown are data from one representative experiment where CMV-stimulated PBMC are diluted into un-stimulated autologous PBMC. Three donors were used for these experiments, chosen because they had three different levels of total cytokine response to CMV. The donor with the medium response is shown here. At each cell dilution, the assay was performed in triplicate. Cells producing both IF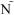 -γ and IL-2 (left panels) or only IFN-γ (right panels) for either CD4+ T cells (upper) or CD8+ T cells (lower) are shown. The Y-axis shows the percentage of CD4+ or CD8+ T cells producing the indicated cytokine(s). The cell dilution is shown on the X-axis as the percentage of undiluted CMV-stimulated cells in the dilution. The intra-sample variability in this one experiment is visualized by the range of responses at each dilution level. The standard least squares correlation is listed.
-γ and IL-2 (left panels) or only IFN-γ (right panels) for either CD4+ T cells (upper) or CD8+ T cells (lower) are shown. The Y-axis shows the percentage of CD4+ or CD8+ T cells producing the indicated cytokine(s). The cell dilution is shown on the X-axis as the percentage of undiluted CMV-stimulated cells in the dilution. The intra-sample variability in this one experiment is visualized by the range of responses at each dilution level. The standard least squares correlation is listed.
B. The CV for measurement of cytokine responses increases as the level of the cytokine response decreases and the limit of quantitation is determined as the lowest cytokine response for which the CV remains ≤30%. Shown is one of the experiments used to determine limit of quantitation. These are the same experiments used to determine linearity, and the four cytokine subsets are shown as in Figure 6A. Data shown here are from the PBMC donor with the high CMV response. The triplicates at each dilution level are averaged (shown in green), and the scale for this mean response is shown on the right vertical axis. The red line denotes the intra-sample CV at each dilution level for each cytokine subset. The scale for the CV is shown on the left vertical axis. The dashed red line indicates the acceptable upper limit of CV, 30%. The arrow points to the lowest mean cytokine response for which the CV is below 30%.
C. The limit of quantitation is calculated for each type of variation for each PBMC sample and for each cytokine subset. Cells producing both IF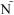 -γ and IL-2 (left panels) or only IFN-γ (right panels) for either CD4+ T cells (upper) or CD8+ T cells (lower) are shown. The type of variation is shown on the X-axis grouped by the three PBMC donors with low, medium or high CMV responses. For the medium responder, an additional type of variation, inter-sample, was evaluated. The limit of quantitation is shown on the Y-axis as the percent of CD4+ or CD8+ T cells. This was determined as in Figure 6B, i.e., the lowest cytokine response for which the CV remains ≤30%. One high outlier (0.6%, marked as *) was removed for the intra-sample analysis of the medium donor for the CD8+ IFN-γ+IL-2- T cell response.
-γ and IL-2 (left panels) or only IFN-γ (right panels) for either CD4+ T cells (upper) or CD8+ T cells (lower) are shown. The type of variation is shown on the X-axis grouped by the three PBMC donors with low, medium or high CMV responses. For the medium responder, an additional type of variation, inter-sample, was evaluated. The limit of quantitation is shown on the Y-axis as the percent of CD4+ or CD8+ T cells. This was determined as in Figure 6B, i.e., the lowest cytokine response for which the CV remains ≤30%. One high outlier (0.6%, marked as *) was removed for the intra-sample analysis of the medium donor for the CD8+ IFN-γ+IL-2- T cell response.
Although the 8-color assay includes four cytokines, only IFN-γ and IL-2 responses were evaluated in terms of the validation parameters listed above. We found that very few CD4+ or CD8+ T cells produced IL-2 alone in response to stimulation with the CMV pp65 peptide pool (See Figure 3C). Therefore, we could only evaluate four cytokine subsets: CD4+ and CD8+ T cells producing IFN-γ alone, or IFN-γ in combination with IL-2.
Linearity was assessed for each dilution experiment, for each PBMC sample tested and for each of the four cytokine subsets. For the majority of these analyses, linearity of the assay is determined by a hypothesis test using the good of fit statistic R2 >0.90. For the few analyses with R2 <0.9, the examination of linearity is inappropriate; for these analyses, the responses at most of the eight dilution levels were very low (well below 0.05%), and therefore in the range below the limit of quantitation (see below).
Robustness refers to the ability of the assay to generate reproducible results as operational parameters change. Many of these parameters cannot easily be categorized or defined, but are parameters that can be expected to change as the assay is repeated on different days and by different operators and in different labs. The quantitative experiments described here can be expected to incorporate variation due to these undefined parameters, and will therefore address the robustness of the assay. These experiments will not address differences between laboratories; therefore, results here only apply to the assay as performed in one laboratory.
Precision was examined by determining the coefficient of variation (CV) at each dilution level for each PBMC sample for each type of precision, as referred to above (intra- and inter-sample, inter-day, inter-operator). The CV increased as the level of response decreased (Figure 6B). We used a threshold of 30% as the acceptable upper limit for the CV. The limit of quantitation (LOQ) was determined as the lowest frequency of antigen-specific T cell response for which the CV was ≤30%. This was determined separately for each of the four cytokine subsets, for each type of variation and for each of the three PBMC samples with the three levels of CMV responses. As shown in Figure 6C, the LOQs range from near zero to 0.08%, and the distribution of LOQs is similar for all four cytokine subsets. The median for the distribution of all LOQs is less than 0.01%, and 75% of the LOQs are below 0.03% (data not shown). Overall, these validation experiments demonstrate that the 8-color ICS assay is linear, has a low LOQ, and detects few false-positive responses specific for HIV-1 Env, Gag, Pol and Nef using the PTE peptide pools.
Discussion
Here we describe, to our knowledge, the first quantitative validation of the ICS assay. Validated assays are essential requirements by regulatory agencies for clinical trial assessments of vaccine-induced immunity in order for the product to ultimately meet licensure. Although alone they are unlikely to prevent HIV infection, HIV-specific CD8+ T cells may have an important impact in rapidly controlling viral replication following infection. Recent studies in rhesus macaques have shown the importance of controlling acute infection and maintaining memory CD4+ T cell responses (Letvin et al., 2006; Mattapallil et al., 2006), and have implicated the role of vaccine-induced CD8+ T cells in such control. The magnitude and functional phenotype of the vaccine-induced T cell responses are likely to be critical in defining responses that can control pathogenic challenge. Indeed, a certain threshold frequency of HIV-specific T cells may be required before control is observed. Such quantitative assessments of cellular immunity require quantitatively validated immunoassays. Alternatively, antigen-specific T cells may not represent the true mechanism of vaccine-induced protection but could serve as a surrogate for protective responses. Cellular assays have not generally been validated to the same rigorous extent as antibody assays presumably because these assays tend to have more biological variability making them harder to standardize.
Cytokine secretion profiles likely differ between CD4+ and CD8+ T cell populations, and between different memory subsets within either CD4+ or CD8+ T cell populations (Wherry and Ahmed, 2004). Moreover, little is known about whether cytokine secretion profiles are static over time and how these may be influenced by persistence of antigen. In the context of replication-competent vector constructs that may be desirable for inducing long-lived memory T cells, this is important and may also alter functionality of the induced responses due to antigen persistence. This may be less of an issue with the majority of candidate vaccines in current clinical trials, which are replication incompetent. It is likely that the functional profiles and frequencies of antigen-specific T cells induced by these vaccines will be different due to lack of persistent antigen.
Our studies suggest that the limit of quantitation of cytokine-producing T cells is extremely low (a median of less than 0.01% for the analyses performed here). However, these low cytokine responses may not be biologically relevant, and therefore, it is important to distinguish between the LOQ of the assay and the lower limit considered to be biologically relevant of the immune function of interest. For the measurement of vaccine-induced T cell responses, the lower limit can be empirically determined by comparing cytokine responses before and after vaccination and by comparing vaccine-induced responses with responses detected in control groups (i.e., placebo recipients). This will allow determination of the range of cytokine responses for which a true cytokine response in the absence of vaccination should not be present. Since validation of an assay is an ongoing process, parameters need to continue to be evaluated as the assay is used over time and for different applications.
Many investigators do not have access to sophisticated flow cytometers capable of 8 and more-color analysis. We have shown that, provided appropriate high cytokine gates are used in 4-color analysis to exclude low level staining from dead cells, the conventional 4-color ICS assay can give equivalent data to this 8-color assay. However, most 4-color assays are limited to using a combination of IFN-γ and IL-2 on the same fluorophore (usually PE); and, therefore, determining whether T cells are secreting either cytokine alone or in combination is impossible unless separate 4-color panels are used for examining CD4+ and CD8+ T cell responses. This differentiation is important since recent reports suggest that different functional profiles of CD4+ T cells are associated with either resolution or persistence of antigen (Harari et al., 2005).
One of the variables we evaluated is the use of co-stimulation with antibodies to CD28 and CD49d during the in vitro stimulation. Our results are inconclusive in terms of the benefit of including co-stimulation. With co-stimulation, the number of responding cells in the negative control (i.e., the background) was increased, especially for IL-2-producing CD4+ T cells. The number of antigen-specific responding cells in general increased to the same extent so that after background subtraction the responses were equivalent with or without co-stimulation. However, there were a few samples where the antigen-specific responses were increased to a greater extent than the background, and for this reason we have included co-stimulation in our validated panel. In addition, previous published data demonstrated enhanced responses with co-stimulation (Waldrop et al., 1997). In this prior publication, whole CMV antigen was used for stimulation as opposed to peptides, and our contrasting results may indicate that co-stimulation is not required when stimulating with peptides. Another potential concern related to our finding of increased background due to co-stimulation, is the possibility that co-stimulation may decrease the sensitivity of the assay. In order to determine this, more testing with a larger number of samples with low-level responses is required.
In conclusion, we have optimized, and qualitatively and quantitatively validated an 8-color ICS assay. This assay has a very low false positive rate, a very low limit of detection, and high sensitivity, reproducibility and linearity, making it suitable for qualitative and quantitative analysis of cellular immune responses in clinical trials of candidate HIV-1 vaccines. The quantitative and qualitative data produced from this assay should enable more in-depth characterization of vaccine-induced T cell responses, and aid in determining if cellular immunity contributes to vaccine efficacy.
Acknowledgements
We would like to acknowledge John Hural, Patricia D’Souza, Isaac Rodriguez and Jean Novak for helpful discussions concerning ICS validation. We thank Phyllis Stegall for help with editing. We also thank the clinicians at the Seattle HIV Vaccine Trials Unit. Special appreciation goes to the study volunteers for donating their time and blood as part of the global effort to develop an HIV vaccine.
Abbreviations
- (ICS)
Intracellular Cytokine Staining
- (PBMC)
peripheral blood mononuclear cells
- (HVTN)
HIV Vaccine Trials Network
- (CMV)
Cytomegalovirus
- (HIV-1)
Human Immunodeficiency Virus
- (SEB)
Staphylococcal enterotoxin B
- (PTE)
potential T cell epitope
- (IFN-γ)
interferon-γ
- (IL-2)
interleukin-2
- (TNF-α)
tumor necrosis factor-α
- (IL-4)
interleukin-4
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bach MK, Brashler JR. Isolation of subpopulations of lymphocytic cells by the use of isotonically balanced solutions of Ficoll. I. Development of methods and demonstration of the existence of a large but finite number of subpopulations. Exp Cell Res. 1970;61:387–96. doi: 10.1016/0014-4827(70)90462-3. [DOI] [PubMed] [Google Scholar]
- Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyum A. Isolation of lymphocytes, granulocytes and macrophages. Scand J Immunol Suppl. 1976;5:9–15. [PubMed] [Google Scholar]
- Burton DR, Desrosiers RC, Doms RW, Koff WC, Kwong PD, Moore JP, Nabel GJ, Sodroski J, Wilson IA, Wyatt RT. HIV vaccine design and the neutralizing antibody problem. Nat Immunol. 2004;5:233–6. doi: 10.1038/ni0304-233. [DOI] [PubMed] [Google Scholar]
- Cohen J. Public health. AIDS vaccine trial produces disappointment and confusion. Science. 2003;299:1290–1. doi: 10.1126/science.299.5611.1290. [DOI] [PubMed] [Google Scholar]
- De Rosa SC, Lu FX, Yu J, Perfetto SP, Falloon J, Moser S, Evans TG, Koup R, Miller CJ, Roederer M. Vaccination in humans generates broad T cell cytokine responses. J Immunol. 2004;173:5372–80. doi: 10.4049/jimmunol.173.9.5372. [DOI] [PubMed] [Google Scholar]
- Dunn HS, Haney DJ, Ghanekar SA, Stepick-Biek P, Lewis DB, Maecker HT. Dynamics of CD4 and CD8 T cell responses to cytomegalovirus in healthy human donors. J Infect Dis. 2002;186:15–22. doi: 10.1086/341079. [DOI] [PubMed] [Google Scholar]
- Goepfert PA, Horton H, McElrath MJ, Gurunathan S, Ferrari G, Tomaras GD, Montefiori DC, Allen M, Chiu YL, Spearman P, Fuchs JD, Koblin BA, Blattner WA, Frey S, Keefer MC, Baden LR, Corey L. High-dose recombinant Canarypox vaccine expressing HIV-1 protein, in seronegative human subjects. J Infect Dis. 2005;192:1249–59. doi: 10.1086/432915. [DOI] [PubMed] [Google Scholar]
- Harari A, Vallelian F, Meylan PR, Pantaleo G. Functional heterogeneity of memory CD4 T cell responses in different conditions of antigen exposure and persistence. J Immunol. 2005;174:1037–45. doi: 10.4049/jimmunol.174.2.1037. [DOI] [PubMed] [Google Scholar]
- Guidance for Industry: Bioanalytical Method Validation. 2001 http://www.fda.gov/CDER/GUIDANCE/4252fnl.htm.
- International Conference on Harmonisation Q2A Text on Validation of Analytical Procedures and Q2B Validation of Analytical Procedures: Methodology. 1996 http://www.fda.gov/cder/guidance/index.htm.
- Letvin NL, Mascola JR, Sun Y, Gorgone DA, Buzby AP, Xu L, Yang ZY, Chakrabarti B, Rao SS, Schmitz JE, Montefiori DC, Barker BR, Bookstein FL, Nabel GJ. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science. 2006;312:1530–3. doi: 10.1126/science.1124226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Malhotra M, Gilbert P, Hawkins N, Duerr A, McElrath M, Corey L, Self S. Peptide selection for human immunodeficiency virus type 1 CTL-based vaccine evaluation. Vaccine. 2006 doi: 10.1016/j.vaccine.2006.06.009. in press. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Dunn HS, Suni MA, Khatamzas E, Pitcher CJ, Bunde T, Persaud N, Trigona W, Fu TM, Sinclair E, Bredt BM, McCune JM, Maino VC, Kern F, Picker LJ. Use of overlapping peptide mixtures as antigens for cytokine flow cytometry. J Immunol Methods. 2001;255:27–40. doi: 10.1016/s0022-1759(01)00416-1. [DOI] [PubMed] [Google Scholar]
- Maecker HT, Moon J, Bhatia S, Ghanekar SA, Maino VC, Payne JK, Kuus-Reichel K, Chang JC, Summers A, Clay TM, Morse MA, Lyerly HK, DeLaRosa C, Ankerst DP, Disis ML. Impact of cryopreservation on tetramer, cytokine flow cytometry, and ELISPOT. BMC Immunol. 2005a;6:17. doi: 10.1186/1471-2172-6-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maecker HT, Rinfret A, D’Souza P, Darden J, Roig E, Landry C, Hayes P, Birungi J, Anzala O, Garcia M, Harari A, Frank I, Baydo R, Baker M, Holbrook J, Ottinger J, Lamoreaux L, Epling CL, Sinclair E, Suni MA, Punt K, Calarota S, El-Bahi S, Alter G, Maila H, Kuta E, Cox J, Gray C, Altfeld M, Nougarede N, Boyer J, Tussey L, Tobery T, Bredt B, Roederer M, Koup R, Maino VC, Weinhold K, Pantaleo G, Gilmour J, Horton H, Sekaly RP. Standardization of cytokine flow cytometry assays. BMC Immunol. 2005b;6:13. doi: 10.1186/1471-2172-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil JJ, Douek DC, Buckler-White A, Montefiori D, Letvin NL, Nabel GJ, Roederer M. Vaccination preserves CD4 memory T cells during acute simian immunodeficiency virus challenge. J Exp Med. 2006;203:1533–41. doi: 10.1084/jem.20060657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura LE, Walker JM, Maecker HT. Optimization of whole blood antigen-specific cytokine assays for CD4(+) T cells. Cytometry. 2000;40:60–8. doi: 10.1002/(sici)1097-0320(20000501)40:1<60::aid-cyto8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- O’Neil-Andersen NJ, Lawrence DA. Differential modulation of surface and intracellular protein expression by T cells after stimulation in the presence of monensin or brefeldin A. Clin Diagn Lab Immunol. 2002;9:243–50. doi: 10.1128/CDLI.9.2.243-250.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell ND, Hudgens MG, Ha R, Havenar-Daughton C, McElrath MJ. Moving to human immunodeficiency virus type 1 vaccine efficacy trials: defining T cell responses as potential correlates of immunity. J Infect Dis. 2003;187:226–42. doi: 10.1086/367702. [DOI] [PubMed] [Google Scholar]
- Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–42. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection. Nat Immunol. 2004;5:927–33. doi: 10.1038/ni1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suni MA, Dunn HS, Orr PL, de Laat R, Sinclair E, Ghanekar SA, Bredt BM, Dunne JF, Maino VC, Maecker HT. Performance of plate-based cytokine flow cytometry with automated data analysis. BMC Immunol. 2003;4:9. doi: 10.1186/1471-2172-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldrop SL, Pitcher CJ, Peterson DM, Maino VC, Picker LJ. Determination of antigen-specific memory/effector CD4+ T cell frequencies by flow cytometry: evidence for a novel, antigen-specific homeostatic mechanism in HIV-associated immunodeficiency. J Clin Invest. 1997;99:1739–50. doi: 10.1172/JCI119338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall P, Tobias R, Ram D, Wolfinger R, Hochbert Y. Multiple comparisons and multiple tests using the SAS system. SAS Institute; Cary, NC: 1999. [Google Scholar]
- Westfall P, Wolfinger R. Multiple tests with discrete distributions. The American Statistician. 1997;51:3–8. [Google Scholar]
- Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–45. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]