

Abstract
The presence of FoxP3+ regulatory T cells (Tregs) is necessary for control of deleterious immune responses in the steady-state; however, mechanisms for maintaining the frequency and quality of endogenous Tregs are not well-defined. In this study, we used in vivo modulators of the CD28 and CTLA4 pathways administered to intact mice to reveal mechanisms controlling the homeostasis and phenotype of endogenous Tregs. We demonstrate that expression of the negative costimulatory regulator CTLA4 on FoxP3+ Tregs in vivo is a direct consequence of their rapid, perpetual homeostasis. Upregulation of CTLA4 expression occurs only on FoxP3+ Tregs undergoing extensive proliferation and can be abrogated by inhibiting the CD28 pathway, coinciding with a reduction in FoxP3+ Treg proliferation and frequency. We further demonstrate that CTLA4 negatively regulates steady-state Treg homeostasis, as inhibiting CTLA4 signaling with an anti-CTLA4 blocking antibody greatly enhances Treg proliferation and overall Treg frequency. Our findings provide new insight into the origin and role of CTLA4 expression on natural FoxP3+ Tregs, and reveal that costimulation modulators can alter the steady-state level and quality of Tregs, with implications regarding their effects on endogenous Tregs in patients receiving immunotherapy.
Keywords: T cells, Costimulation, Tolerance/Suppression/Anergy, Immunotherapy
INTRODUCTION
Regulatory T cells (Tregs) constitute 6–12% of peripheral CD4 T cells and function to maintain self-tolerance and control antigen-specific responses (1). The FoxP3 transcription factor delineates Treg development and functional capacity (2–4), with genetic mutations in FoxP3 manifesting as a fatal autoimmune lymphoproliferative disease in both humans and mice (5, 6). Tregs have been shown to suppress immune responses to infectious pathogens (7), tumors (8, 9), transplants (10–12), and also ameliorate inflammatory bowel disease (IBD) (13–15) and autoimmune diabetes (16–18). Moreover, Tregs have an active role in maintaining immune homeostasis during healthy conditions in the absence of exogenous antigens, as in vivo ablation of FoxP3+ Tregs in immunocompetent adult mice results in a wasting, lymphoproliferative scurfy-like disease with multi-organ immune infiltration (19, 20). At present, it is not known how Tregs maintain control of deleterious immune responses in steady-state conditions, and what in turn, controls the maintenance and quality of Tregs during healthy conditions.
The identification of FoxP3+ Tregs as crucial cellular controllers of immunity suggests that direct manipulation of Tregs in vivo can result in the enhancement or suppression of specific immune responses. Two potential targets for Treg manipulation are the costimulatory molecules CD28 and CTLA4, which promote and inhibit naive T-cell activation, respectively (21), and play distinct roles in Treg generation, function and homeostasis (22, 23). CD28 is constitutively expressed on all T cells and binds the B7-1 (CD80) and B7-2 (CD86) ligands, providing the necessary “second signal” for TCR-mediated activation, differentiation, and survival of naive T cells (24) (25, 26). CD28 signaling in Tregs has been shown to promote FoxP3+ Treg generation from developing thymocytes (27) as well as peripheral Treg homeostasis and expansion (28, 29). However, other studies have shown that FoxP3+ Tregs can be generated in CD28−/− and B7.1/7.2−/− mice (28–30), suggesting the existence of CD28-independent pathways for Treg generation and/or homeostasis.
CTLA4 (CD152), a homolog of CD28, is upregulated on naive T cells after stimulation and acts to suppress T cell activation upon binding its B7 ligands (31). CTLA4 is also expressed constitutively on a subset of Tregs (32), although the origin of CTLA4 expression on Tregs is not known. CTLA4 expression on Tregs does not appear to be a direct consequence of activation by the FoxP3 transcription factor (33) (34), suggesting that additional factors drive the upregulation of CTLA4 expression on Tregs. Whether CTLA4 expression is required for Treg generation or intrinsic regulatory capacity likewise remains unresolved (23). CD4+FoxP3+ Tregs are generated in CTLA4-deficient mice, and these CTLA4-deficient Tregs can suppress T cell responses in vitro and in vivo (15, 35). However, CTLA4 upregulation is associated with enhanced Treg activity in IBD models (15, 32) and is required in antibody-mediated therapies for promoting allograft survival (36). In addition, the factors or pathways that induce or maintain CTLA4 expression on Tregs are not well defined.
Both the CD28 and CTLA4 pathways are targets for current immunomodulatory therapies: the B7-binding fusion protein CTLA4Ig (37) is an approved immunosuppressant for treatment of rheumatoid arthritis (38), and an anti-CTLA4 blocking antibody is in trials for immune enhancement in cancer (39). While these treatments have specific effects on antigen-driven activation of conventional naive and memory T cells (40–42), systemic administration has the potential to affect non-targeted T cell responses, such as those mediated by Tregs which are required to maintain immune homeostasis in healthy individuals.
In this study, we examined steady-state Treg turnover and used in vivo modulators of the CD28 and CTLA4 pathways administered to intact mice to reveal new mechanisms controlling CTLA4 expression and steady-state homeostasis of endogenous Tregs. We demonstrate that constitutive CTLA4 expression on peripheral FoxP3+ Tregs is a consequence of their rapid, steady-state homeostasis that is driven by CD28 signaling. Inhibiting CD28 costimulation with the B7-binding reagent CTLA4Ig blocked endogenous Treg proliferation and decreased CTLA4 expression on the remaining FoxP3+ Tregs. Conversely, inhibition of CTLA4 signaling with an anti-CTLA4 blocking antibody greatly enhanced Treg homeostasis and overall frequency, revealing a novel role for CTLA4 in the control of Treg turnover. Our findings provide new insight into the origin and role of CTLA4 expression on natural FoxP3+ Tregs, and reveal that costimulation modulators can alter the steady-state level and quality of Tregs, with implications regarding the role of endogenous Tregs in immunotherapies.
MATERIALS AND METHODS
Mice
BALB/c (Thy1.2+) mice were purchased from the National Cancer Institute (Frederick, MD), and BALB/c (Thy1.1+) congenic mice (University of North Carolina, Chapel Hill, NC) were bred and maintained under specific pathogen-free conditions and used at 6–10 weeks of age. All animal studies were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Maryland, Baltimore.
Antibodies
The following antibodies were purchased from Bio X Cell (West Lebanon, NH): anti-CD8 (TIB105), anti-CD4 (GK1.5), anti-I-Ad (212. A1), anti-Thy-1 (TIB238), murine IgG2a (C1.18.4), anti-FcγR (2.4G2), and anti-CTLA4 (UC10-4F10-11). FITC-conjugated anti-BrdU and anti-IgG1; PE-conjugated anti-CD25, anti-CTLA4, and anti-IgG1; PerCP-conjugated anti-CD4; and APC-conjugated anti-CD90.2 and anti-IgG2a antibodies were purchased from BD Pharmingen (San Diego, CA). PE-conjugated anti-FoxP3; PE-Cy-7-conjugated anti-CD4 and anti-CD90.2; APC-conjugated anti-FoxP3; APC-Cy-7-conjugated anti-CD25; and APC-Alexa Fluor 750-conjugated anti-CD25 antibodies were purchased from eBiosciences (San Diego, CA). Polyclonal hamster IgG antibody was purchased from Jackson Immunoresearch (West Grove, PA). Goat anti-rat IgG, anti-rat IgM, and anti-mouse IgG conjugated to magnetic beads were purchased from Polysciences, Inc. (Warrington, PA).
Treatment of mice with costimulation blockade and thymectomy
BALB/c mice were administered murine CTLA4Ig (250μg/dose, i.p., Bristol-Myers Squibb) or anti-CTLA4 (250μg/dose, i.p.) every other day for a total of 3 doses, with murine IgG2a, IgG, and PBS as controls. For CD28 inhibition, mice were administered a single-chain anti-CD28 blocking antibody (Wyeth Pharmaceuticals, Madison, NJ) in doses of 100μg twice daily for 7 days.
In some experiments, mice were thymectomized prior to treatment with CTLA4Ig. Briefly, mice were anesthetized i.p. with a 2% solution of 2,2,2-tribromoethanol (Sigma-Aldrich, St. Louis, MO) and the thymus removed with negative suction. Animals were allowed to recover for at least 7 days prior to further manipulation.
Isolation of Treg and non-Treg fractions
To isolate CD4+CD25+ Tregs, CD4 T cells were purified from the spleen by density centrifugation (Lymphocyte Separation Medium, Mediatech, Herndon, VA) and immunomagnetic depletion as described previously (43), stained with PE- or APC-Alexa Fluor 750-conjugated anti-CD25 antibody and anti-PE or anti-Cy-7 magnetic microbeads (Miltenyi Biotec, Auburn, CA), and positively selected by separation over a ferromagnetic column using AutoMACS (Miltenyi Biotec). Non-Tregs (CD4+CD25−) were isolated from splenic CD4 T cells after staining and conjugation to magnetic microbeads as above and depleted of CD4+CD25+ Tregs using AutoMACS. Isolated CD4+CD25+ fractions were >90% pure and CD4+CD25− were >95% pure.
In vivo BrdU labeling and flow cytometry
To assess the turnover of Tregs in comparison to non-Treg populations, CTLA4Ig-, IgG2a- and PBS-treated mice were injected with BrdU (1mg, i.p.) for 3 consecutive days prior to sacrifice. Splenic CD4 T cells were harvested at various timepoints (3–28 days), purified as above, and resuspended in stain buffer. Cells were surface stained with fluorescently-coupled antibodies specific for CD4, Thy1.2 and CD25, fixed and permeabilized (Cytofix/Cytoperm, Perm/Wash, BD Biosciences) for subsequent intracellular staining. For analysis of intracellular BrdU content, permeabilized cells were incubated with DNase (Sigma-Aldrich) for 60 minutes at 37°C, washed with Perm-Wash, incubated with Fc-block (Miltenyi) to block non-specific binding, followed by fluorescently-conjugated anti-BrdU, anti-FoxP3 and/or anti-CTLA4 antibody at 4°C. Cells were further washed and analyzed on LSRII using FACSDiva software (Becton Dickinson, San Jose, CA).
In vivo proliferation assays
To monitor the proliferation of T-cell subsets in vivo, CD4+CD25+ Tregs or CD4+CD25− non-Tregs were isolated from BALB/c (Thy1.2+) mice, labeled with 2.5μM CFSE (Invitrogen, Carlsbad, CA), and transferred via tail vein injection (1×106 cells/mouse) into congenic BALB/c (Thy1.1+) hosts undergoing treatment with CTLA4Ig, anti-CTLA4, anti-CD28, IgG2a, IgG or PBS. Splenic CD4 T cells were harvested and purified 6 days after adoptive transfer, stained for intracellular FoxP3 in accordance with the manufacturer’s protocol, and analyzed by flow cytometry. Treg proliferation was determined by gating on CD4+FoxP3+Thy1.2+ cells isolated from mice transferred with CD4+CD25+Thy1.2+ cells. Non-Treg proliferation was assessed by gating on CD4+FoxP3−Thy1.2+ cells from mice receiving CD4+CD25−Thy1.2+ cells.
Statistical Methods
Student’s two-tailed t-test was used to evaluate differences between treatment groups. A p-value<0.05 was considered significant.
RESULTS
Rapid steady-state turnover of Tregs in vivo is associated with upregulated CTLA4 expression
We initially analyzed the steady-state turnover of endogenous FoxP3+ Tregs compared to the FoxP3− non-Treg population using short-term BrdU incorporation. We injected intact, healthy BALB/c mice with BrdU for 3 days prior to harvest and analyzed BrdU uptake as a function of FoxP3 expression on splenic CD4 T cells. We found that a higher proportion of FoxP3+ Tregs incorporated BrdU compared to FoxP3− non-Tregs. Of the FoxP3+ CD4 T cells, 19% incorporated BrdU compared to only 6% of FoxP3− cells (Fig. 1A, right). Overall, FoxP3+ Tregs exhibited three-fold greater proliferation compared to FoxP3− non-Tregs, based on the extent of BrdU incorporation (Fig. 1B). These results demonstrate rapid and enhanced turnover of FoxP3+ CD4 T cells (Tregs) relative to their FoxP3− (non-Treg) counterparts.
FIGURE 1. Enhanced steady-state turnover of FoxP3+ Tregs in healthy mice.
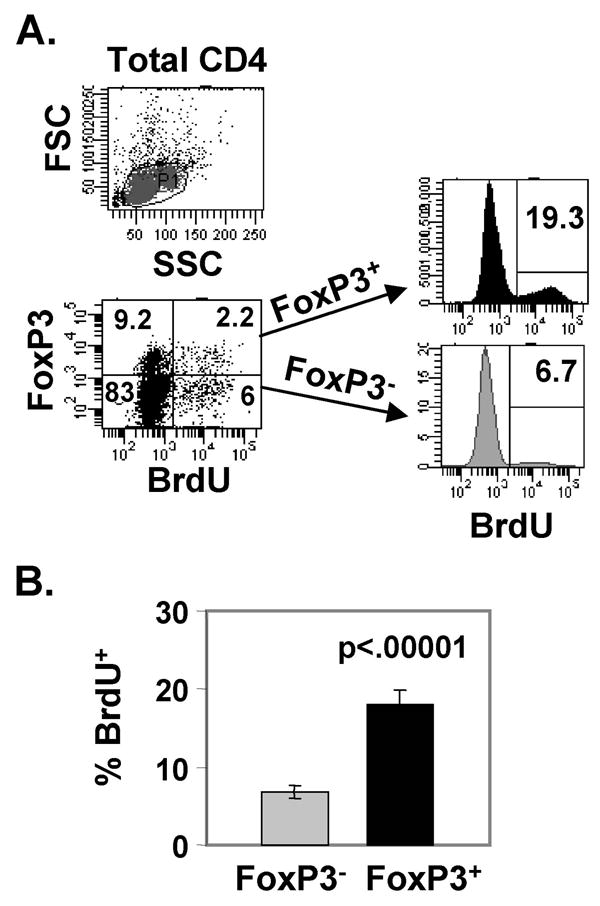
BALB/c mice were administered BrdU (1mg/dose, i.p.) for 3 consecutive days and splenic CD4 T cells were harvested the following day, stained intracellularly for FoxP3 and BrdU, and analyzed by flow cytometry. (A) Top: Forward (FSC) and side scatter (SSC) plots of total CD4 T cells with gating indicated. Bottom: FoxP3 versus BrdU staining of total live CD4 T cells, with percentages indicated in each quadrant. To the right of the dot plot are histograms showing BrdU incorporation gated on FoxP3+ and FoxP3− CD4 T cells. (B) Mean BrdU incorporation ± SD by FoxP3+ and FoxP3− CD4 T cells from 5 mice per group (representative of 3 experiments).
To more closely analyze the dynamics of steady-state Treg homeostasis, we examined the ability of Treg and non-Treg populations to proliferate in vivo when transferred into intact hosts. For these experiments, we isolated Treg and non-Treg populations from intact BALB/c (Thy1.2+) mice by sorting on CD25 expression, yielding CD4+CD25+ and CD4+CD25− populations, respectively. These subsets were CFSE-labeled, transferred into intact BALB/c (Thy1.1+) congenic hosts, and harvested after 6 days in vivo to assess spontaneous turnover of the transferred populations. In our analysis of the transferred populations (Fig. 2A), we further gated on FoxP3+ cells within the CD25+ transferred population, and FoxP3− cells within the CD25− transferred population to ensure that true Treg and non-Treg populations were being evaluated. We found extensive in vivo proliferation of transferred FoxP3+ Tregs (53% dividing) compared to non-Tregs (6%) after only 6 days in vivo in intact mice (Fig. 2A). These results demonstrate that the natural population of polyclonal FoxP3+ Tregs contains a subset capable of rapid and extensive proliferation in steady state conditions.
FIGURE 2. Rapid steady-state homeostasis of FoxP3+ Tregs is associated with increased CTLA4 expression.

CFSE-labeled CD4+CD25+ (Tregs) and CD4+CD25− (non-Tregs) from BALB/c (Thy1.2+) mice were transferred into BALB/c (Thy1.1+) congenic hosts (1×106 cells/mouse), harvested after 6 days, stained intracellularly for FoxP3 and additional Treg markers, and analyzed by flow cytometry. (A) CFSE dilution of transferred Thy1.2+ populations, gating on FoxP3+ cells for analysis of Tregs within the CD4+CD25+ transferred population, and gating on FoxP3− cells for analysis of non-Tregs within the CD4+CD25− transferred population. Mean percentage ± SD of divided (CFSElo) Tregs (CD4+FoxP3+Thy1.2+) and non-Tregs (CD4+FoxP3−Thy1.2+) are 53±1% and 6±1%, respectively (n=10 mice per group). (B) Expression of FoxP3, CD25, CTLA4, and GITR on maximally divided (CFSElo) and undivided (CFSEhi) FoxP3+ cells with mean fluorescence intensity (MFI) of each marker indicated. Bar graphs show mean MFI ± SD for each marker expressed by maximally-divided (black) and undivided (white) Tregs from 4 mice per group. Box highlights histogram plots of CTLA4 expression, with the most striking differences in expression between dividing and non-dividing Tregs.
Given this heterogeneity in the ability of FoxP3+ Tregs to undergo rapid homeostasis, we asked whether FoxP3+ Tregs that underwent maximal division (CFSElo) differed in the expression of Treg functional markers compared to the undivided (CFSEhi) population. Indeed, we found that FoxP3+ CFSElo Tregs exhibiting extensive division expressed higher levels of FoxP3, CTLA4 and GITR, and lower levels of CD25 compared to non-proliferating Tregs (Fig. 2B). The most striking phenotypic difference between proliferating and non-proliferating Tregs was in CTLA4 expression, with maximally-dividing Tregs expressing high levels of CTLA4, and undivided Tregs exhibiting low CTLA4 expression (Fig. 2B, boxed). These results demonstrate a correlation between the level of CTLA4 expression on FoxP3+ Tregs and the extent of peripheral homeostasis, and also show that FoxP3 and GITR expression varies between actively dividing and quiescent Tregs.
Costimulatory inhibition with CTLA4Ig decreases Treg turnover, frequency, and CTLA4 expression in vivo
The association of CTLA4 expression with rapidly dividing Tregs suggests that CTLA4 upregulation could either occur as a direct result of in vivo turnover, or that CTLA4 directly signals for rapid turnover. To address the first possibility, we investigated whether inhibiting steady-state homeostasis would reduce CTLA4 expression on endogenous Tregs. As the CD28 pathway is important for peripheral turnover of CD4+CD25+ Tregs in vivo (28, 29), we used the B7-binding reagent CTLA4Ig (37) to downmodulate Treg homeostasis and determine potential effects on CTLA4 expression. We first examined the extent of CTLA4Ig-mediated inhibition of steady-state FoxP3+ Treg homeostasis in intact BALB/c hosts, as earlier studies had not addressed direct effects on in vivo proliferation of transferred or endogenous FoxP3+ Tregs (29). To analyze effects of CTLA4Ig treatment on transferred populations of Tregs and non-Tregs, we treated BALB/c (Thy1.1+) mice with murine CTLA4Ig or IgG2a isotype control antibody according to the schematic in Fig. 3A, transferred CFSE-labeled Thy1.2+ CD4+CD25+ and CD4+CD25− T cells into treated mice on day 1 (Fig. 3A), and assessed spontaneous in vivo turnover of the transferred populations after 6 days. While FoxP3+ CD4 T cells proliferated extensively when transferred into control-treated hosts as in Fig. 2A, CTLA4Ig dramatically inhibited this steady-state homeostasis by 75% (Fig. 3B). These results demonstrate a near-complete inhibition of FoxP3+ Treg proliferation in vivo by CTLA4Ig.
FIGURE 3. Steady-state homeostasis of FoxP3+ Tregs is blocked by CTLA4Ig-mediated inhibition of CD28 costimulation.

(A) Experimental scheme for measuring effects of CTLA4Ig on steady-state homeostasis of transferred and endogenous Treg populations. BALB/c (Thy1.1+) congenic hosts were treated with CTLA4Ig or IgG2a on days 0, 2 and 4. For Treg transfer experiments in (B), CFSE-labeled CD4+CD25+ or CD4+CD25− T cells from untreated BALB/c (Thy1.2+) mice were transferred on day 1 into Thy1.1+ mice undergoing treatment, harvested at day 7, and stained intracellularly for FoxP3 expression. To examine effects on endogenous FoxP3+ and FoxP3− populations, BrdU was administered to treated mice on days 4–6 and mice were harvested at day 7 in (C) and (D). (B) Representative CFSE histograms show percentage of divided Thy1.1+FoxP3+ Tregs and Thy1.1+FoxP3− non-Tregs gated as in Fig. 2A. Mean percentage ± SD of divided Thy1.2+FoxP3+ and Thy1.2+FoxP3− CD4 T cells are as follows: IgG2a 45±4% and 5±1%, and CTLA4Ig 10±2% and 2±1%, respectively (n=3–7 mice per group). (C) CTLA4Ig inhibits endogenous Treg homeostasis. Left: Representative histograms show BrdU incorporation by FoxP3+ and FoxP3− CD4 T cells in IgG2a and CTLA4Ig-treated mice, with the percentage of BrdU+ cells indicated. Right: Graph shows the mean proportion ± SD of FoxP3+ and FoxP3− CD4 T cells incorporating BrdU from 5 mice per group. (D) Reduced frequency of CD4+FoxP3+ T cells in CTLA4Ig-treated mice. Left: Representative histograms indicate the percentage of FoxP3+ Tregs among total CD4 T cells. Right: Graph showing the mean percentage ± SD of FoxP3+ Tregs of total CD4 T cells from 5 mice per group. Results are representative of 4 experiments.
To assess whether CTLA4Ig similarly inhibited endogenous FoxP3+ Treg turnover, we pulsed IgG2a- and CTLA4Ig-treated BALB/c mice with BrdU for 3 days prior to harvest, and analyzed FoxP3 expression in conjunction with BrdU incorporation by splenic CD4 T cells. In control-treated mice, as in intact mice, FoxP3+ Tregs divided more extensively than FoxP3− non-Tregs (16% versus 6% incorporating BrdU, respectively). However, in CTLA4Ig-treated mice, BrdU incorporation by FoxP3+ Tregs was almost completely inhibited, compared to the slight inhibition of BrdU incorporation by FoxP3− non-Tregs (Fig. 3C). Interestingly, CTLA4Ig treatment dramatically altered the ratio of Treg:non-Treg proliferation, from a 3-fold higher proliferation by FoxP3+ compared to FoxP3− cells in control-treated mice, to equivalent low-level proliferation of both FoxP3+ and FoxP3− cells in CTLA4Ig-treated hosts (Fig. 3C). This decrease in endogenous Treg turnover resulted in a net 50% decrease in overall FoxP3+ Treg frequency compared to control mice (Fig. 3D). These results indicate that the rapid steady-state proliferation of FoxP3+ Tregs is driven by CD28 engagement whereas the low-level turnover of the non-Treg population appears to be independent of CD28 costimulation.
We asked whether the expression of FoxP3 or CTLA4 was altered within the endogenous Treg population in CTLA4Ig-treated mice, given their reduced frequency and in vivo turnover. We found that endogenous FoxP3+ Tregs from CTLA4Ig-treated mice had equivalent levels of FoxP3 expression as Tregs from IgG2a-treated mice (Fig. 4A). However, FoxP3+ Tregs from CTLA4Ig-treated mice exhibit greatly reduced CTLA4 expression compared to the high levels of CTLA4 expression in Tregs from IgG2a-treated hosts (Fig. 4B). These results indicate that CD28 engagement is required for CTLA4 upregulation on FoxP3+ Tregs in steady-state conditions.
FIGURE 4. Inhibition of steady-state homeostasis selectively reduces CTLA4 expression on Tregs while FoxP3 expression is maintained.

BALB/c mice were treated with IgG2a or CTLA4Ig as in Fig. 3A, administered BrdU on days 4–6, and splenic CD4 T cells were harvested at day 7, and analyzed for FoxP3 and CTLA4 expression. (A) Comparable level of FoxP3 expression in IgG2a- and CTLA4Ig-treated mice. Top: Histograms show the level of FoxP3 expression of CD4+CD25+ T cells from IgG2a- and CTLA4Ig-treated mice, with MFI of FoxP3 indicated in each histogram. Bottom: Graph shows the average MFI of FoxP3 expression from 5 mice per group. Results are representative of 4 experiments. (B) CTLA4Ig treatment reduces CTLA4 expression on Tregs. Top: Histograms show CTLA4 expression gated on FoxP3+ and FoxP3− CD4 T cells from IgG2a- and CTLA4Ig-treated mice with the percentage CTLA4+ shown based on isotype control staining indicated by marker. Bottom: Graph shows average MFI of CTLA4 expression by FoxP3+ Tregs from IgG2a- and CTLA4Ig-treated mice (n=5 mice per group). Results are representative of 6 experiments. (C) Steady-state homeostasis of CTLA4+ and CTLA4− CD4 T cells in IgG2a- and CTLA4Ig-treated mice. Histograms show percent BrdU incorporation by CD4+CTLA4+ and CD4+CTLA4− T cells from total CD4 T cells in IgG2a- and CTLA4Ig-treated mice (n=5 mice per group).
To determine whether CTLA4 expression directly correlated with in vivo homeostasis, we performed a similar BrdU incorporation analysis as in Fig. 1A, examining BrdU uptake in CD4 T cells as a function of CTLA4 expression. Thus, we injected intact BALB/c mice with BrdU for three consecutive days, harvested and analyzed splenic CD4 T cells as in Fig. 1. This analysis revealed that CTLA4 expression marks the population of cells undergoing rapid endogenous turnover in intact mice (Fig. 4C, top row), and that homeostasis of CTLA4+ T cells in CTLA4Ig-treated mice is inhibited by CTLA4Ig (Fig. 4C, bottom row). These results suggest that homeostasis drives CTLA4 expression, rather than CTLA4 signaling directing homeostasis.
The reduced proportion of CTLA4+FoxP3+ Tregs seen after CTLA4Ig treatment was also observed in thymectomized mice (Fig. 5A), indicating that CTLA4Ig-mediated effects on Treg homeostasis acted on peripheral Tregs. Furthermore, the decrease in FoxP3+ and CTLA4+FoxP3+ cells in CTLA4Ig-treated mice as described in Figs. 2 and 3, persisted between 14–28 days post-treatment (Fig. 5B). These results indicate that CD28-driven peripheral homeostasis is required for CTLA4 expression on Tregs and the accumulation of CTLA4+ Tregs in the periphery, although maintenance of FoxP3 expression is CD28-independent. We also found that CTLA4Ig-treated Tregs maintained in vitro suppressive capacity (data not shown), indicating that CTLA4− Tregs maintain suppressive function, presumably due to the maintenance of FoxP3 levels.
FIGURE 5. Abrogation of CTLA4 expression on Tregs by CTLA4Ig treatment is due to decreased peripheral homeostasis.

(A) Decreased Treg frequency and CTLA4 expression in thymectomized mice treated with CTLA4Ig. BALB/c mice were thymectomized, treated with CTLA4Ig or IgG2a as in Fig. 3A, and splenic CD4 T cells harvested 3 days post-treatment (day 7). Graphs show percentage of FoxP3+ (left) and CTLA4+FoxP3+ CD4 T cells (right) among total CD4 T cells from thymectomized IgG2a- and CTLA4Ig-treated mice. Results are an average of 4 mice per group and are representative of 2 experiments. The proportion of Tregs in IgG2a- and CTLAIg-treated sham-thymectomized controls were indistinguishable from intact mice similarly treated (data not shown). (B) Long-term reduction of FoxP3+ frequencies and CTLA4 expression on FoxP3+ Tregs following CTLA4Ig treatment. BALB/c mice were treated with IgG2a or CTLA4Ig as in Fig. 3A and the proportions of FoxP3+ (left) and CTLA4+FoxP3+ CD4 T cells (right) of total CD4 T cells were determined 3–28 days post-treatment. Results are expressed as mean percentage ± SD from 4–5 mice per group.
CTLA4 expression on Tregs controls their steady-state homeostasis
The above results strongly suggest that the lack of CTLA4 expression on Tregs is due to the lack of CD28-driven Treg homeostasis; however, it is important to consider that CTLA4Ig binds the B7 ligands, potentially inhibiting signaling through both CD28 and CTLA4. To test whether the association of CTLA4 expression with homeostasis was affected by CTLA4 engagement by Tregs, we treated mice with an anti-CTLA4 blocking antibody and assessed its effects on steady-state proliferation of adoptively transferred and endogenous Tregs. Remarkably, spontaneous turnover of adoptively transferred FoxP3+ Tregs was substantially increased in anti-CTLA4-treated mice with 80–90% of Tregs undergoing maximal division after only 6 days in vivo (Fig. 6A). In addition, steady-state homeostasis of FoxP3− non-Tregs was also increased, although remained significantly lower than that of Tregs (Fig. 6A). Consistent with the increased level of spontaneous Treg turnover in vivo, there was a substantial 2–3-fold increase in the proportion of endogenous FoxP3+ cells in anti-CTLA4-treated mice (24–28%) compared to controls (10–13%) (Fig. 6B). These results demonstrate that engagement of the CTLA4 pathway on Tregs negatively regulates steady-state homeostasis and limits Treg frequency in vivo, in contrast to the CD28 pathway which positively regulates Treg homeostasis and frequency. These results further establish that homeostasis drives CTLA4 expression and not the converse mechanism.
FIGURE 6. CTLA4 negatively regulates Treg homeostasis.

(A) Effect of anti-CTLA4 treatment on in vivo proliferation of Tregs. CFSE-labeled CD4+CD25+ or CD4+CD25− T cells from BALB/c (Thy1.2+) mice were transferred (1×106 cells/mouse) on day 1 into BALB/c (Thy1.1+) congenic mice undergoing treatment with anti-CTLA4 or IgG control using a similar treatment schedule as with CTLAIg (days 0, 2 and 4; Fig. 3A), and harvested after 6 days in vivo (day 7). Left: Representative CFSE histograms showing the division of transferred FoxP3+ Tregs and FoxP3− non-Tregs gated as in Fig. 2A. Right: Graph shows mean percentage ± SD of divided CD4+FoxP3+Thy1.2+ Tregs in IgG- and anti-CTLA4-treated mice (n=6–7 mice per group). (B) FoxP3 expression of endogenous CD4 T cells isolated from IgG- and anti-CTLA4-treated mice. Top two plots: Representative histograms showing FoxP3+ expression of total CD4 T cells from IgG- and Anti-CTLA4-treated BALB/c mice, with the percentage of CD4+FoxP3+ cells indicated. Bottom plot: Graph showing mean percentage ± SD of CD4+FoxP3+ T cells of 10 mice per group, compiled from three independent experiments (C) FoxP3 and CTLA4 expression of endogenous CD4+FoxP3+ cells from mice treated with anti-CD28, anti-CTLA4 or IgG2a depicted as representative histograms and graphs showing mean MFI ± SD of n=3–4 mice per group. Results are representative of 3 experiments.
To directly compare the effect of the CTLA4 versus CD28 pathway on CTLA4 expression by Tregs, we examined CTLA4 and FoxP3 expression by endogenous Tregs isolated from intact mice treated with anti-CTLA4 or anti-CD28 blocking antibodies. Anti-CD28 treatment resulted in significant downregulation of CTLA4 expression on Tregs and comparable levels of FoxP3 compared to control mice (Fig. 6C), similar to effects of CTLA4Ig on these markers (Figs. 4A and B). Additionally, anti-CD28 treatment resulted in a reduced frequency of endogenous CD4+FoxP3+ T cells, but not to the degree observed with CTLA4Ig due to the limited stability and partial receptor coverage of the single chain anti-CD28 antibody (data not shown). By contrast, anti-CTLA4 treatment resulted in higher levels of CTLA4 expression and slight increases in FoxP3 expression on endogenous Tregs (Fig. 6C). These results demonstrate opposing effects of the CD28 and CTLA4 pathways on the steady-state homeostasis, frequency, and expression of CTLA4 on FoxP3+ Tregs.
DISCUSSION
We demonstrate here that CD4+FoxP3+ T cells (Tregs) undergo rapid and dynamic proliferation during steady-state conditions which can be reciprocally modulated by targeting costimulatory pathways. We show a striking bias in the steady-state proliferation of FoxP3+ CD4 T cells and an association between CTLA4 expression and the subset of FoxP3+ Tregs undergoing rapid turnover. In addition, both Treg steady-state homeostasis and CTLA4 upregulation were dramatically inhibited by blocking the CD28 pathway. Conversely, Treg homeostasis and endogenous frequency were significantly enhanced when the CTLA4 pathway was blocked by in vivo administration of anti-CTLA4. Our findings demonstrate that Treg frequency and CTLA4 expression are determined by the extent of homeostasis during steady-state conditions and can be altered by immunotherapies in the absence of an antigenic stimulus. These findings have implications for the effects of costimulation-based immunotherapies on the endogenous host regulatory environment.
Our findings of biased incorporation of BrdU by FoxP3+ CD4 T cells in vivo over several days are consistent with reports of homeostatic turnover of CD4+CD25+ populations in lymphopenic or intact hosts (44, 45). However, we reveal here that steady-state turnover of endogenous FoxP3+ Tregs in intact hosts is rapid and continuous. We also demonstrate a direct and novel association between CTLA4 expression and the peripheral homeostasis of FoxP3+ Tregs, with the expression of FoxP3 and GITR also varying between actively dividing and quiescent Tregs. While CD28 signaling has been recognized as promoting Treg homeostasis (22, 29), our results indicate that CD28 signaling is required for both rapid steady-state Treg homeostasis and CTLA4 upregulation. Conventional (FoxP3−) T cells likewise require CD28 signaling for optimal CTLA4 upregulation during antigenic stimulation (46); however, homeostasis of FoxP3− cells during steady-state conditions was only slightly dependent on CD28 signaling and CTLA4 upregulation did not occur in the dividing FoxP3− population. These results indicate that homeostasis and CTLA4 expression on FoxP3+ and FoxP3− T cells is controlled by distinct mechanisms during steady-state conditions.
One mechanism to account for the difference in steady-state behavior of Tregs and conventional T cells is the TCR specificity. It has been shown that self-reactive TCRs are disproportionately represented within the FoxP3+ Treg population (47). Therefore, reaction with self antigens and CD28 may account for the rapidly dividing Treg population in healthy, normal conditions, as was also suggested by a study in which peripheral turnover of Tregs expressing a fixed, transgene-encoded TCR required endogenous expression of the peptide antigen (48). Upregulation of CTLA4 expression on conventional FoxP3− T cells requires both TCR triggering and CD28 costimulation (46), suggesting that a similar mechanism of CD28 triggering and TCR-ligation by endogenous antigens functions to upregulate CTLA4 expression by Tregs.
The role of CTLA4 expression of Tregs has been actively investigated. While CTLA4-deficient mice possess FoxP3+ Tregs (35) and CTLA4-deficient T cells exhibit regulatory activity in vivo (15), blockade of CTLA4 can abrogate Treg function (13, 15). Our finding that Treg homeostasis is enhanced following anti-CTLA4 blockade suggests that CTLA4 signaling downmodulates steady-state Treg turnover. A role for CTLA4 in controlling peripheral Treg frequency is also implied by a previous report showing an association between a single nucleotide polymorphism in CTLA4 and an increase in Treg frequency (49). Our results suggest that impaired CTLA4 signaling may lead to an increase in Treg proliferation and an augmented Treg frequency, with potential consequences on immune regulation.
We demonstrate here that the a subset of FoxP3+ Tregs exhibits rapid steady-state proliferation that greatly exceeds that of FoxP3− non-Tregs and propose that this rapid and extensive turnover comprises the homeostatic “noise” associated with a healthy, regulated state (Fig. 7). We show here that immunotherapies targeting costimulation can affect the homeostatic turnover, frequency, and quality of FoxP3+ Tregs, raising the question whether their effect on steady-state Treg homeostasis can predict their effect on immune responses. Inhibiting the CD28 pathway with CTLA4Ig reduces steady-state proliferation of FoxP3+ Tregs more extensively than FoxP3− T cells, resulting in an equal frequency of proliferating cells in both FoxP3+ and FoxP3− populations (Fig. 7). However, these treated mice do not develop autoimmunity up to two months post-treatment (data not shown), and clinically, CTLA4Ig treatment results in dampening of autoimmune-induced inflammation in rheumatoid arthritis (38) and in suppressing allogeneic responses in transplantation (50, 51), indicating that the net effect of CTLA4Ig treatment is immunosuppressive. Conversely, while anti-CTLA4 promotes Treg homeostasis and increases Treg frequency, it also increases non-Treg turnover under healthy conditions (Fig. 7). However, in tumor models, the net result of anti-CTLA4 treatment is an increase in tumor-specific immune responses (52, 53), although an increase in both effector and Treg numbers has been observed (54). Thus, CTLA4Ig decreases and anti-CTLA4 increases Treg frequency and homeostasis, which appear to be in opposition to their therapeutic outcomes in disease models.
FIGURE 7. Treg and non-Treg turnover during steady-state conditions and during modulation of the CD28 or CTLA4 costimulation pathways.

Regulatory T cells (Tregs) and conventional T cells (non-Tregs) undergo steady-state homeostasis, with the proliferation of Tregs exceeding that of non-Tregs in healthy conditions. CD28 (triangle) is expressed by both subsets and CTLA4 (rectangle) is only expressed by proliferating Tregs in the steady state. Treatment with CD28 pathway inhibitors reduces Treg proliferation to the low level seen with non-Tregs, thereby inhibiting CTLA4 upregulation. Conversely, inhibition of the CTLA4 pathway increases both Treg and non-Treg homeostasis, with an increased proportion of CTLA4+FoxP3+ T cells.
We propose that the predominant outcome of immunomodulation of the CD28/CTLA4 costimulatory pathways in a disease state with an active antigen response is determined primarily by the effects on the non-Treg population. Moreover, our results suggest that the immune system demonstrates plasticity with regard to the frequency and steady-state levels of Tregs. However, long-term use of these costimulation modulators in the clinic, particularly once the disease state is ameliorated, may result in their effects on Tregs dominating over those on the non-Treg population. Further studies are warranted to establish whether spontaneous Treg proliferation serves as a mechanism for maintaining proper immune homeostasis in the steady-state, or is a by-product of the CD28 and/or TCR signals encountered in the periphery.
Acknowledgments
The authors wish to thank Drs. Stephen T. Bartlett and David Scott for critical reading of this manuscript, Emily Welty for anti-CD28 coverage studies, and Wendy Lai for mouse colony maintenance.
Footnotes
supported by a grant from Bristol-Myers Squibb and NIH AI050632 awarded to D.L.F.
References
- 1.Sakaguchi S, Sakaguchi N, Shimizu J, Yamazaki S, Sakihama T, Itoh M, Kuniyasu Y, Nomura T, Toda M, Takahashi T. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 2.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 3.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 4.Ziegler SF. FOXP3: of mice and men. Annu Rev Immunol. 2006;24:209–226. doi: 10.1146/annurev.immunol.24.021605.090547. [DOI] [PubMed] [Google Scholar]
- 5.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 6.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 7.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–360. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 8.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 9.Zhou G, Levitsky HI. Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol. 2007;178:2155–2162. doi: 10.4049/jimmunol.178.4.2155. [DOI] [PubMed] [Google Scholar]
- 10.Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003;9:1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 11.Graca L, Thompson S, Lin CY, Adams E, Cobbold SP, Waldmann H. Both CD4+CD25+ and CD4+CD25− regulatory cells mediate dominant transplantation tolerance. J Immunol. 2002;168:5558–5565. doi: 10.4049/jimmunol.168.11.5558. [DOI] [PubMed] [Google Scholar]
- 12.Ochando JC, Homma C, Yang Y, Hidalgo A, Garin A, Tacke F, Angeli V, Li Y, Boros P, Ding Y, Jessberger R, Trinchieri G, Lira SA, Randolph GJ, Bromberg JS. Alloantigen-presenting plasmacytoid dendritic cells mediate tolerance to vascularized grafts. Nat Immunol. 2006;7:652–662. doi: 10.1038/ni1333. [DOI] [PubMed] [Google Scholar]
- 13.Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh B, Read S, Asseman C, Malmstrom V, Mottet C, Stephens LA, Stepankova R, Tlaskalova H, Powrie F. Control of intestinal inflammation by regulatory T cells. Immunol Rev. 2001;182:190–200. doi: 10.1034/j.1600-065x.2001.1820115.x. [DOI] [PubMed] [Google Scholar]
- 15.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. 2006;177:4376–4383. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, von Herrath M. Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J Clin Invest. 2006;116:1371–1381. doi: 10.1172/JCI27191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- 19.Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, Sparwasser T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J Exp Med. 2007;204:57–63. doi: 10.1084/jem.20061852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J, Rasmussen J, Rudensky A. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–7. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 21.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 22.Boden E, Tang Q, Bour-Jordan H, Bluestone JA. The role of CD28 and CTLA4 in the function and homeostasis of CD4+CD25+ regulatory T cells. Novartis Found Symp. 2003;252:55–63. doi: 10.1002/0470871628.ch5. discussion 63–56, 106–114. [DOI] [PubMed] [Google Scholar]
- 23.Sansom DM, Walker LS. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol Rev. 2006;212:131–148. doi: 10.1111/j.0105-2896.2006.00419.x. [DOI] [PubMed] [Google Scholar]
- 24.Jenkins MK, Taylor PS, Norton SD, Urdahl KB. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J Immunol. 1991;147:2461–2466. [PubMed] [Google Scholar]
- 25.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 26.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 27.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6:152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 28.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 29.Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171:3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- 30.Lohr J, Knoechel B, Jiang S, Sharpe AH, Abbas AK. The inhibitory function of B7 costimulators in T cell responses to foreign and self-antigens. Nat Immunol. 2003;4:664–669. doi: 10.1038/ni939. [DOI] [PubMed] [Google Scholar]
- 31.Carreno BM, Collins M. The B7 family of ligands and its receptors: new pathways for costimulation and inhibition of immune responses. Annu Rev Immunol. 2002;20:29–53. doi: 10.1146/annurev.immunol.20.091101.091806. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 34.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 35.Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 36.Ariyan C, Salvalaggio P, Fecteau S, Deng S, Rogozinski L, Mandelbrot D, Sharpe A, Sayegh MH, Basadonna GP, Rothstein DM. Cutting edge: transplantation tolerance through enhanced CTLA-4 expression. J Immunol. 2003;171:5673–5677. doi: 10.4049/jimmunol.171.11.5673. [DOI] [PubMed] [Google Scholar]
- 37.Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, Singh C, Tepper MA. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257:792–795. doi: 10.1126/science.1496399. [DOI] [PubMed] [Google Scholar]
- 38.Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–1915. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- 39.Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, Comin-Anduix B, Reuben JM, Seja E, Parker CA, Sharma A, Glaspy JA, Gomez-Navarro J. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol. 2005;23:8968–8977. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- 40.Bluestone JA, St Clair EW, Turka LA. CTLA4Ig: Bridging the Basic Immunology with Clinical Application. Immunity. 2006;24:233–238. doi: 10.1016/j.immuni.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 41.Ndejembi MP, Teijaro JR, Patke DS, Bingaman AW, Chandok MR, Azimzadeh A, Nadler SG, Farber DL. Control of Memory CD4 T Cell Recall by the CD28/B7 Costimulatory Pathway. J Immunol. 2006;177:7698–7706. doi: 10.4049/jimmunol.177.11.7698. [DOI] [PubMed] [Google Scholar]
- 42.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr Opin Immunol. 2006;18:206–213. doi: 10.1016/j.coi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 43.Ahmadzadeh M, Hussain SF, Farber DL. Effector CD4 T cells are biochemically distinct from the memory subset: evidence for long-term persistence of effectors in vivo. J Immunol. 1999;163:3053–3063. [PubMed] [Google Scholar]
- 44.Fisson S, Darrasse-Jeze G, Litvinova E, Septier F, Klatzmann D, Liblau R, Salomon BL. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med. 2003;198:737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gavin MA, Clarke SR, Negrou E, Gallegos A, Rudensky A. Homeostasis and anergy of CD4(+)CD25(+) suppressor T cells in vivo. Nat Immunol. 2002;3:33–41. doi: 10.1038/ni743. [DOI] [PubMed] [Google Scholar]
- 46.Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157:4762–4770. [PubMed] [Google Scholar]
- 47.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 48.Cozzo C, Larkin J, 3rd, Caton AJ. Cutting edge: self-peptides drive the peripheral expansion of CD4+CD25+ regulatory T cells. J Immunol. 2003;171:5678–5682. doi: 10.4049/jimmunol.171.11.5678. [DOI] [PubMed] [Google Scholar]
- 49.Atabani SF, Thio CL, Divanovic S, Trompette A, Belkaid Y, Thomas DL, Karp CL. Association of CTLA4 polymorphism with regulatory T cell frequency. Eur J Immunol. 2005;35:2157–2162. doi: 10.1002/eji.200526168. [DOI] [PubMed] [Google Scholar]
- 50.Baliga P, Chavin KD, Qin L, Woodward J, Lin J, Linsley PS, Bromberg JS. CTLA4Ig prolongs allograft survival while suppressing cell-mediated immunity. Transplantation. 1994;58:1082–1090. [PubMed] [Google Scholar]
- 51.Levisetti MG, Padrid PA, Szot GL, Mittal N, Meehan SM, Wardrip CL, Gray GS, Bruce DS, Thistlethwaite JR, Jr, Bluestone JA. Immunosuppressive effects of human CTLA4Ig in a non-human primate model of allogeneic pancreatic islet transplantation. J Immunol. 1997;159:5187–5191. [PubMed] [Google Scholar]
- 52.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 53.Sotomayor EM, Borrello I, Tubb E, Allison JP, Levitsky HI. In vivo blockade of CTLA-4 enhances the priming of responsive T cells but fails to prevent the induction of tumor antigen-specific tolerance. Proc Natl Acad Sci U S A. 1999;96:11476–11481. doi: 10.1073/pnas.96.20.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116:1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]