

Abstract
Parkinson's disease (PD), dementia with Lewy bodies, and multiple system atrophy, collectively referred to as synucleinopathies, are associated with a diverse group of genetic and environmental susceptibilities. The best studied of these is PD. α–Synuclein (α–syn) plays a key role in the pathogenesis of both familial and sporadic PD, but evidence linking it to other predisposition factors is limited. Here we report a strong genetic interaction between α–syn and the yeast orthologue of the PD-linked gene PARK9 (ATP13A2). Dopaminergic neuron loss caused by α–syn overexpression in animal and neuronal PD models is rescued by co-expression of PARK9. Further, knockdown of the PARK9 orthologue in C. elegans enhances α–syn misfolding. These data provide a direct functional connection between α–syn and another PD susceptibility locus. Manganese exposure is an environmental risk factor linked to PD and PD-like syndromes. Remarkably, we discovered that yeast PARK9 helps to protect cells from manganese toxicity. Our studies reveal a striking connection between PD genetics (α–syn and PARK9) and an environmental risk factor (PARK9 and manganese). Finally, we show that additional genes from our yeast screen, with diverse functions, are potent modifiers of α–syn-induced neuron loss in animals, establishing a diverse, highly conserved interaction network for α–syn.
Introduction
Compelling evidence implicates α-syn in the pathogenesis of PD 1, including the identification of point mutations and locus duplication 2,3 and triplication 4 in familial forms, the abundance of α-syn in Lewy Bodies 5, and neurodegeneration resulting from increased expression of α-syn in multiple animal models 6-9. Likewise, expression of α-syn in yeast cells results in dosage-dependant toxicity 10. Several features of this toxicity, including production of reactive oxygen species (ROS), lipid droplet accumulation, and vesicle trafficking defects, are reminiscent of α–syn toxicity in mammalian neurons 10. Therefore, yeast cells afford the opportunity to rapidly screen for modifier genes with the hope that the identified genes will point to common cellular mechanisms of toxicity and suggest avenues for therapeutic intervention 11.
We recently reported the identification of a set of conserved genes functioning in vesicular trafficking between the endoplasmic reticulum (ER) and Golgi that were potent α-syn toxicity modifiers in yeast. One of these, Ypt1/Rab1, was tested in neuronal models of PD and was able to prevent dopaminergic neuron loss 12. We then expanded this screen to include 5,000 yeast genes. In addition to vesicular trafficking genes, we identified several other categories of α-syn toxicity modifiers, many with clear human orthologues, which reveal additional complexities to synuclein pathology and that complement numerous studies of human synucleinopathies 13.
A major question in the field is whether the genetic loci linked to PD interact with each other or whether multiple independent insults simply happen to result in a common phenotype (dopaminergic neuron loss and resulting parkinsonism). There is emerging evidence for a genetic interaction between PD-linked genes parkin and pink1 in Drosophila 14-16 as well as interactions between α–syn and DJ-1, another PD-linked protein 17-20. Recently, the molecular nature of PARK9, the gene responsible for early-onset parkinsonism with pyramidal degeneration and dementia (Kufor-Rakeb syndrome; OMIM 606693), was elucidated and found to encode ATP13A2, a predicted lysosomal P-type transmembrane cation transporting ATPase 21-23.
We have recently completed an unbiased screen to find modifiers of α–syn toxicity 12,13. Here we show that the yeast homologue of human PARK9/ATP13A2 can suppress α–syn toxicity in yeast. This genetic interaction between PARK9 and α–syn is conserved in neurons because PARK9 expression in animal models of PD is sufficient to rescue neurodegeneration. We also show that the yeast PARK9 gene can protect cells from manganese toxicity, suggesting an intimate connection between genetic and environmental causes of neurodegeneration. Finally, we find other diverse modifiers of α–syn toxicity from our screen, which are able to suppress α–syn induced neurodegeneration in animal and neuronal models of PD. These include an E3 ubiquitin ligase (HRD1/SYVN1), ubiquitin protease (UBP3/USP10), phosphodiesterase (PDE2/PDE9A), polo-like kinase (CDC5/PLK2), and a casein kinase (YCK3/CSNK1G3). Thus, our data establish α–syn as part of a highly conserved, multi-faceted pathway.
Results
Yeast PARK9 homologue suppresses α–syn toxicity
As part of our unbiased genetic screen for modifiers of α–syn toxicity 12, we discovered the yeast orthologue of human PARK9/ATP13A2, an uncharacterized yeast gene designated YOR291W, to be a suppressor of α-syn toxicity (Fig. 1a). We have therefore named this yeast gene YPK9 (for Yeast PARK9). This and other emerging work 13,17,19,24 suggest the possibility of many more connections between α–syn and other known causes of PD.
Figure 1.
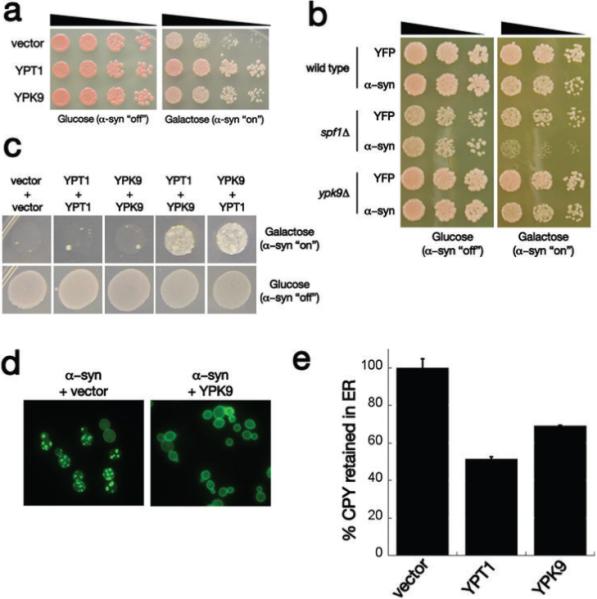
Interaction between 〈–syn and the yeast PARK9 homolog. a) Spotting assays with yeast 〈–syn toxicity modifier genes YPT1 and YPK9 showing their ability to suppress toxicity compared to empty vector control. Five-fold serial dilutions of yeast cells were spotted onto glucose (〈–syn expression repressed) or galactose (〈–syn expression induced). b) Deletion of Ypk9 has no effect on 〈–syn toxicity, however deleting the closely related ATPase Spf1 enhances 〈–syn toxicity. c) Synergistic genetic interaction between 〈–syn toxicity modifiers Ypt1 and Ypk9. In a high toxicity (HiTox) 2 copy 〈–syn yeast strain, expression of Ypt1 or Ypk9 alone is not sufficient to rescue toxicity. However, their co-expression restores growth to this strain. d) Ypk9 overexpression eliminates 〈–syn inclusions. 〈–Syn-YFP-expressing cells contain many vesicular inclusions when transformed with an empty vector and these are greatly diminished in cells transformed with a Ypk9 expression plasmid. e) The ability of Ypk9 to suppress the 〈–syn-induced block in ER-Golgi was measured by carboxypeptidase Y (CPY) maturation assay. Ypk9 significantly improved the trafficking of CPY from ER to Golgi.
Knockout of YPK9 did not enhance α–syn toxicity (Fig. 1b). Reasoning that this might be due to redundancy/overlap in function, we performed a BLAST search of the yeast genome. This identified the SPF1/COD1 gene as encoding a highly related P-type ATPase (30% identical, 49% similar, e-value=3×10−80). There are several P-type ATPases in yeast, but SPF1 and YPK9 are by far the most closely related to PARK9 (Supplemental Fig. S3). Overexpression of SPF1 did not suppress α-syn toxicity (data not shown) and deletion of SPF1 itself exerted a slight growth defect in yeast (Fig. 1b). Strikingly, however, the SPF1 deletion was nearly lethal in cells expressing a single copy of α–syn, which is by itself below the dosage threshold for toxicity in our yeast model (Fig. 1b); the combination of the SPF1 deletion and a single copy of α–syn resulted in a growth defect that was far greater than the sum of their individual toxicities. The double knockout of SPF1 and YPK9 did not exhibit a synthetic lethal phenotype (Supplemental Fig. S1), however overexpression of YPK9 was sufficient to rescue the increase in α–syn toxicity caused by deleting SPF1 (data not shown), clearly establishing a functional overlap between them. There are many possible explanations for the different effects of deletion and overexpression of these genes. For example, it might be that Spf1 expression or function is feedback regulated, preventing overexpression from being efficacious, but leaving cells still vulnerable to deletion. In any case, in yeast, the two genes most closely related to human PARK9 have diverged such that YPK9 suppresses α–syn toxicity when overexpressed while SPF1 enhances toxicity when deleted.
Next we investigated the relationship between Ypk9 and another strong α–syn toxicity suppressor from our screen, Ypt1 12. Ypt1 is the yeast homolog of human RAB1A, a guanosine triphosphatase (GTPase) that regulates the trafficking of vesicles between the endoplasmic reticulum (ER) and the Golgi. As previously reported, overexpression of this protein rescues α–syn toxicity in both yeast and neuronal cells 12. To determine if YPK9 and YPT1 suppress α–syn toxicity in a mechanistically similar manner, we utilized a yeast strain expressing higher levels of α–syn 25. This HiTox strain exhibits correspondingly higher toxicity and allows the detection of synergistic effects between different suppressors that would not be possible in the less toxic (IntTox) screening strain because YPT1 fully rescues in that strain 12,25.
In the HiTox strain neither Ypt1 nor Ypk9 could suppress the toxicity of α–syn. Together, strong, albeit not complete, suppression was observed (Fig. 1c). Importantly, this was not simply because the two proteins provided additional levels of a redundant function that had a strong threshold effect: co-transforming cells with two copies of either gene itself did not rescue toxicity at all (Fig. 1c). Thus, the genetic interaction between Ypt1 and Ypk9 is synergistic, indicating that Ypk9 and Ypt1 function in mechanistically distinct ways to suppress the toxic effects of α–syn accumulation.
In yeast and humans, the toxicity of α–syn is very dosage sensitive. One way that Ypk9 might affect α–syn toxicity, therefore, would be to alter its accumulation. However, both immunoblotting, and fluorescent quantification of the α–syn fusion protein established that overexpression of Ypk9 did not affect steady state levels of α–syn (Supplemental Fig. S2). Ypk9 did, however, dramatically alter the localization of α–syn, largely restoring plasma membrane localization and reducing intracellular inclusions (Fig. 1d). Previously, immuno-electron microscopy established that the intracellular inclusions formed by α–syn in yeast are associated with clusters of mislocalized transport vesicles from various steps of the endocytic and exocytic pathways 25,26. Thus, these inclusions are a readout of vesicle trafficking defects elicited by α–syn accumulation, and may well relate to early events in α–syn pathology seen in PD 25,26. Our data suggest that rescuing the vesicle trafficking block by overexpressing Ypt1 or Ypk9 results in a reduction in the number of intracellular inclusions.
We next asked whether the two proteins would have similar effects on the most immediate toxic defect that we have detected in cells expressing α–syn, a defect in ER to Golgi trafficking 12. We followed carboxypeptidase Y (CPY) as it trafficked through this pathway by performing a pulse-chase experiment. The subcellular location of CPY is easily determined by compartment-specific glycosylations and proteolytic cleavages that alter the molecular mass of the protein in a well-characterized manner. α–Syn inhibits ER-Golgi transport and prevents CPY from exiting the ER (Fig. 1e and ref. 12). Ypk9 overexpression significantly rescued the ability of proteins to leave the ER and traffic to the Golgi (less protein in the ER, Fig. 1e), although the effect was not as strong as that of Ypt1. Thus, Ypk9 (PARK9) and Ypt1 (RAB1A) have mechanistically distinct functions but both converge on vesicular transport to antagonize α–syn toxicity.
PARK9 rescues α–syn-induced dopaminergic neurodegeneration in C. elegans
To investigate the genetic relationship between α–syn and PARK9 in dopaminergic (DA) neurons, a cell type directly relevant to human PD, we turned first to the nematode model, Caenorhabditis elegans (C. elegans). Development in the nematode is highly stereotyped and wild-type animals invariably have exactly the same number of dopaminergic (DA) neurons. Expression of α–syn from the dopamine transporter (dat-1) gene promoter resulted in an age-dependent progressive loss of DA neurons 7, with approximately 85% of animals having reduced numbers of DA neurons at the 7-day stage (Fig. 2a,c). Expression of W08D2.5 (the C. elegans PARK9 orthologue) alone did not induce any change in the number of DA neurons (data not shown). Co-expression of W08D2.5 and α–syn, from the same promoter (dat-1), partially rescued this neurodegeneration in each of four independent transgenic lines (Fig. 2b,c).
Figure 2.
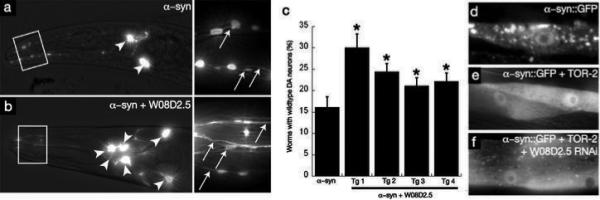
PARK9 antagonizes α–syn-mediated dopaminergic neuron degeneration in C. elegans. (a, b) Anterior DA neurons in worms expressing Pdat-1::GFP + Pdat-1::α–syn at the day 7 stage. Arrowheads and arrows depict cell bodies and neuronal processes, respectively. WT worms have 6 anterior DA neurons. a) α–Syn toxicity is depicted by the loss of anterior DA neurons. b) DA neurons are protected when Pdat-1::FLAG-W08D2.5 cDNA is co-expressed. c) Quantification of C. elegans PARK9 rescue of α–syn-induced neurodegeneration in 4 independent transgenic lines displaying all six anterior DA neuron. P < 0.05, Student's t test. d) Overexpression of α–syn in Punc-54::α–syn::GFP results in misfolding and aggregation of α–syn in body wall muscle cells at the young adult stage. e) Co-overexpression of TOR-2, a protein with chaperone activity, attenuates the misfolding of the α–syn::GFP protein. f) The misfolding of α–syn::GFP is enhanced following RNAi targeting W08D2.5.
We also used C. elegans to explore the consequences of PARK9 loss-of-function. Unfortunately, neuronal cells of this organism are refractory to RNAi-mediated inhibition of gene expression 27. However, our work with yeast and neuronal model systems establishes that α–syn toxicity is the result of general cellular defects, to which neuronal cells are simply more sensitive 25. We therefore took advantage of another cell type that has been extensively exploited for studies of protein homeostasis in this organism 28-31 and is readily affected by RNAi.
As previously described, body-wall muscle cells that express a human α–syn::GFP fusion exhibit age-dependent α–syn aggregation (Fig. 2d). Co-expressing tor-2, a chaperone protein that reduces α–syn aggregation, provides a sensitized genetic background within which an enhancement of α–syn misfolding is readily visualized (Fig. 2e and 28,32,33). In this sensitized background, we knocked down the expression the C. elegans PARK9 ortholog (W08D2.5) by RNAi. This profoundly enhanced the misfolding of human α–syn and did so in an age-dependent manner (Fig. 2f). Importantly, it did so without modifying the expression levels of α–syn or tor-2 (Fig. S4). These data provide further evidence for an intimate functional interaction between PARK9 and α-syn.
PARK9 rescues α–syn-induced dopaminergic neurodegeneration in rat primary midbrain neuron cultures
To validate our findings in mammalian dopaminergic neurons, we employed primary neuronal cultures prepared from the midbrain region of rat embryos at stage 17. Although this assay is far more laborious than those using stable tissue culture cell lines, the toxicity of α-syn in this setting is more robust and reproducible, likely because the cells retain apoptotic mechanisms that are lost in immortalized cell lines. Also, unlike the nematode model, these cultures provide an opportunity to assess toxicity to dopaminergic (DA) neurons relative to other neurons. Transduction of these cells with lentivirus encoding a PD linked mutant α–syn (A53T) causes a reduction in the total number of neurons (MAP2+ staining), including those utilizing γ-amino butyric acid (GABA+) as a neurotransmitter. But tyrosine hydroxylase-positive (TH) dopaminergic (DA) neurons were even more severely affected 12,34. Co-transduction with a lentivirus encoding ATP13A2 (human PARK9) was potently protective. This was apparent from the increased percentage of TH positive cells (Fig. 3a), from the dramatic restoration of neuronal processes in cells expressing TH, and from the restoration of more normal neuronal morphology throughout the culture (Fig. 3b). ATP13A2 also increased the ratio of TH-positive neurons compared to MAP2-positive neurons (Fig. 3a). Thus, the relationship between PARK9 function and α–syn pathobiology that we had discovered in yeast is conserved in mammals.
Figure 3.

PARK9 antagonizes α–syn-mediated dopaminergic neuron degeneration in rat primary midbrain neurons. a) Human PARK9 (ATP13A2) protects rat midbrain primary DA neurons from α–synA53T-induced toxicity. Primary rat embryonic midbrain cultures were either mock infected (control) or infected with lentivirus encoding LacZ, ATP13A2 alone, α–synA53T alone or α–synA53T and ATP13A2. Selective loss of DA neurons was determined immunocytochemically by comparing the percentage of MAP2-positive neurons that also stained positive for tyrosine hydroxylase (TH). N ≥ 3, # P < 0.05, ## P < 0.01, ### P < 0.001, one way analysis of variance with Newman-Keuls post-test (α–synA53T vs. control is also ###). b) ATP13A2/PARK9 rescues α–synA53T-induced DA neuron loss in rat primary midbrain cultures. Representative micrographs of cells stained for MAP2 (red) and TH (green). Arrows indicate TH+MAP2+ DA neurons. Scale bar = 20 μm.
Ypk9 localizes to the vacuole membrane and PARK9 patient-based mutations disrupt its localization and function
Homozygous mutations in ATP13A2/PARK9 have been identified as causing a hereditary form of parkinsonism with dementia 23. That both alleles must be mutant to cause disease suggests that a recessive loss of function is the root cause. However, PARK9 is expressed at a 10-fold higher level in the surviving neurons of the substantia nigra of patients with sporadic forms of PD 23. Therefore, it was also reasonable to suppose that the high expression of PARK9 in sporadic PD and the two mutant alleles in familial forms represent a proteotoxic gain of function with, in the latter case, two alleles required to cross a disease-threshold burden 23. The fact that in yeast overexpression of YPK9 suppressed α-syn toxicity supports the simpler view that it is a deficit of PARK9 function that leads to disease. To explore this further, we first determined the localization of the WT yeast and human proteins.
We used homologous recombination to chromosomally tag Ypk9 with the yellow fluorescent protein (YFP). YFP-Ypk9, expressed from its native promoter, localized strongly to the vacuole membrane (Fig. 4a), consistent with the localization of the human protein to the lysosome, the mammalian organellar equivalent of the yeast vacuole 23. We obtained similar results expressing GFP-Ypk9 fusion proteins from a low-copy (CEN) plasmid with a constitutive promoter (GPD) (Fig. 4a). Co-staining with a lipophilic dye, FM4−64, which concentrates at the vacuole membrane, confirmed this localization was vacuolar (data not shown).
Figure 4.

Ypk9 is localized to the vacuole in yeast and PARK9 patient-based mutations affect its ability to rescue α–syn toxicity. a) Fluorescence microscopy to visualize Ypk9 subcellular localization. A chromosomally tagged YFP fusion (YPK9-YFP) localizes to the vacuolar membrane, as does WT GFP-YPK9 expressed from the constitutive GPD promoter. PARK9 patient-based mutations 23 alter YPK9 localization but the ATPase-dead mutant (D781N) does not. GFP-tagged human ATP13A2 also localizes to the vacuole in yeast cells. b) Spotting assays with WT or 〈–syn-expressing cells. WT YPK9 overexpression suppresses 〈–syn toxicity but the two PARK9 patient-based mutant YPK9 proteins as well as the ATPase-dead mutant do not. Expressing mutant YPK9 in WT cells does not inhibit growth, supporting the idea that these are loss-of-function and not dominant negative mutations.
The human protein ATP13A2 also localized to the vacuole membrane in yeast cells (Fig. 4a). However, even with a high copy plasmid, it was expressed at lower levels than the yeast protein. This is common for multi-pass transmembrane proteins expressed across such large evolutionary distances (ATP13A2/PARK9 has 10 predicted transmembrane domains). Not unexpectedly, the human protein was unable to protect against α–syn toxicity. Therefore, to test the effect of the human mutations on PARK9 localization and function we took advantage of the homology between the proteins to introduce equivalent mutations into the yeast protein. Both patient-based mutant forms of yeast Ypk9 were aberrantly localized. Ypk9 (Δ833−1472) was expressed at lower levels than WT Ypk9 and was distributed throughout the cytosol, in a punctate pattern, whereas Ypk9 (Δ1329−1472) was retained in the ER (Fig. 4a).
Next we tested the ability of the PARK9 patient-based Ypk9 mutants to rescue α–syn toxicity in our yeast model. Overexpression of WT Ypk9 suppressed toxicity, but the two mutant Ypk9 proteins did not (Fig. 4b). Moreover, expressing the Ypk9 mutants in WT yeast cells (without α–syn expression) did not affect growth (Fig. 4b), further supporting the notion that these are loss-of-function and not dominant negative mutations. Human ATP13A2/PARK9 and yeast Ypk9 are predicted P-type ATPases (35 and Supplemental Fig. S3). We also mutated a conserved residue in Ypk9, predicted to abolish ATPase activity (D781N). This mutant localized properly to the vacuole (Fig. 4a) but was unable to rescue α–syn toxicity (Fig. 4b). Taken together, our data indicate that both vacuolar localization and ATPase activity are required for Ypk9 to antagonize α–syn toxicity.
ypk9Δ cells are hypersensitive to manganese
Little is known about the normal function of PARK9/ATP13A2 or how it might contribute to PD. To gain mechanistic insight into PARK9 function, we explored the function of the yeast homolog. Both the yeast and human proteins are predicted to be transmembrane cationic metal transporters, but their substrate specificity has remained elusive 23,35,36. We tested several metals to identify potential functions for Ypk9. We grew WT and ypk9Δ cells in media containing a wide range of metals and metal chelators at various concentrations, to determine the concentration that partially inhibited growth in our genetic background. This provided sensitized conditions to test the effects of Ypk9 (Fig. 5a and data not shown).
Figure 5.

PARK9 protects cells from elevated manganese levels. a) Examples of conditions used to identify the substrate specificity of YPK9. We identified ypk9Δ cells as being sensitive to manganese (Mn2+) relative to WT cells. b) The effect of various Mn2+ concentrations on ypk9Δ cells grown on rich (YPD) or synthetic (CSM) media. c) Expressing WT YPK9 in ypk9Δ cells is sufficient to rescue Mn2+ sensitivity but neither the PARK9 patient-based mutants nor the ATPase-dead mutant are able to rescue. Expressing YPK9 in WT yeast cells makes them more resistant to Mn2+ (compare top and bottom spottings). d) YFP- (top panel) and GFP-tagged (bottom panel) Ypk9 fusion proteins used for localization studies are functional because they are able to protect against Mn2+ sensitivity. e) ypk9Δ cells also exhibit sensitivity to Mn2+ when grown in liquid culture.
Strikingly, of all the conditions we tested, ypk9Δ cells were more sensitive to manganese (Mn2+) than were WT cells (Fig. 5a,b,e). This was detectable in rich media (Fig. 5a), even stronger in minimal media (Fig. 5b), and occurred in cells grown either on plates or in liquid (Fig. 5e). Interestingly, ypk9Δ cells were also slightly resistant to copper (Fig. 5a). Expressing WT Ypk9 from an extrachromosomal plasmid with a strong promoter was sufficient to rescue the Mn2+ sensitivity and indeed to make both ypk9Δ and WT cells more resistant to Mn2+ (Fig. 5c). We were unable to complement this phenotype with the human gene, which, as noted above, we attribute to our inability to express the human protein at sufficient levels in our yeast system. We therefore used the PARK9 patient-based mutations in the Ypk9 protein to test the effects of the human mutations on Ypk9's ability to protect against Mn2+ toxicity.
Whereas WT Ypk9 suppressed Mn2+ toxicity, expression of the disease-associated Ypk9 mutants did not (Fig. 5c). ATPase activity was also required to protect against Mn2+ toxicity because the ATPase-dead Ypk9D781N mutant failed to rescue the defect (Fig. 5c). Moreover, the GFP-tagged Ypk9 fusion proteins, which we used for the localization studies (Fig. 4a), were functional, because they were able to rescue Mn2+ sensitivity (Fig 5d). Thus, yeast Ypk9, and possibly human ATP13A2/PARK9, likely function as manganese transporters to protect cells from excess Mn2+ exposure.
Additional modifier genes from yeast screen protect against α–syn-induced neurodegeneration in animal PD models
The other α–syn toxicity modifier genes we discovered in our yeast screen 13 offer a multitude of promising possibilities for discovering new therapeutic strategies. But it is axiomatic that this approach will only work if hits from the yeast screen can be validated in neurons. As an initial step towards this goal, we chose a subset of genes from our screen for further analysis in neuronal PD models. Our sole criteria were to test representative genes 1) from diverse functional categories, 2) with different strengths of suppression, 3) with clear human orthologues (Table 1), and 4) with readily obtainable gene clones. We tested five suppressor genes, employing human expression clones because of their availability at the time the experiments were performed (yeast/human: Hrd1/SYVN1, Ubp3/USP10, Pde2/PDE9A, Cdc5/PLK2, Yck3/CSNK1G3), in the rat primary neuron lentiviral model and the C. elegans α–syn model. Remarkably, four out of five were efficacious. Two (PLK2 and PDE9A) even suppressed α–syn-induced DA neuron loss in the nematode (Supplemental Fig. S5). Because we used human expression clones for these studies it is not surprising that the suppressors were more effective in rat neurons than in nematode, which is separated from human by ∼800 million years. In any case, these studies establish a highly conserved genetic interaction network operating between α–syn and several genes of diverse function from yeast to mammals.
Table 1.
α-Syn toxicity modifiers tested in neuronal PD models
| Yeast Gene | Type | Predicted function | Human orthologue | Effect in Yeast | Effect in C. elegans | Effect in rat neurons |
|---|---|---|---|---|---|---|
| YPK9 | Suppressor | Lysosomal ATPase | ATP13A2 | suppress | suppress | suppress |
| HRD1 | Suppressor | E3 ubiquitin ligase | SYVN1 | suppress | no change | suppress |
| UBP3 | Suppressor | Ubiquitin protease | USP10 | suppress | no change | suppress |
| PDE2 | Suppressor | Phosphodiesterase | PDE9A | suppress | suppress | suppress |
| CDC5 | Suppressor | Polo-like kinase | PLK2 | suppress | suppress | suppress |
| YCK3 | Suppressor | Casein kinase | CSNK1G3 | suppress | no change | no change |
Discussion
There are no clear homologs of α–syn in either yeast or nematode. How relevant, then, is it to study this human PD gene in yeast? Very. Work from our laboratory 12,13,24,25 and others 26,37,38 indicates that α–syn, likely through its ability to bind lipids and associate with membranes, is involved in the control of vesicle trafficking, a core function conserved in all eukaryotes. We have discovered an α–syn interaction network in yeast consisting of proteins with very diverse functions (e.g. kinases, phosphatases, metal transporters, de-ubiquitinating enzymes) 12,13 and demonstrated its functional conservation in neurons of the rat and nematode, organisms separated from yeast by a billion years of evolution. This extraordinary and unexpected degree of conservation not only confirms a conserved and fundamentally important role for peripheral membrane proteins such as α–syn in normal vesicle trafficking, but also indicates that this function is deeply integrated with, and regulated by, other diverse, and conserved cellular functions.
α–Syn is a very small (14 kDa) protein that binds lipids and is peripherally associated with membranes. Although it folds when associated with membranes 39-41, it is otherwise natively unfolded, with a propensity to form toxic oligomeric species 42. We suggest that it is these very basic properties of α–syn that account for the conservation of its pathobiology from yeast to man. Yeast cells may have proteins with similar function. If so, they are likely constrained more by protein-lipid than protein-protein interactions and have simply diverged too greatly over the enormous evolutionary distances covered here to allow clear recognition of functional homologs by amino acid sequence.
The discoveries of single-gene mutations in familial forms of PD over the last ten years provides an extraordinary opportunity for further elucidating the fundamental mechanisms of PD 43. Our approach has revealed genetic interactions between human disease genes, encoding α–syn and PARK9, for which there was previously no known relationship. The fact that 5 of the 6 genes that we discovered in yeast also affect the toxicity of α–syn in neurons suggests that other modifiers recovered in our screen will also be relevant 13. It may, therefore, be useful to test polymorphisms in these genes for association in synucleinopathies. An additional challenge is to explore the complexities of gene-gene, and gene-environment interactions in PD. Our identification of a connection between α–syn, PARK9 and manganese also provides a toehold for these investigations.
A unifying theme emerging from our work, as well as that of several other laboratories, is that α–syn sits at a nodal point, integrating a multitude of seemingly diverse genetic and environmental lesions 13,44,45. We hope the ability to dissect the nature of these interactions, and how they contribute to disease in a variety of model systems, will forge new avenues for understanding disease mechanisms and suggest therapeutic approaches aimed at the fundamental biological lesions in the complex disorders associated with α–syn that intersect with diverse aspects of pathology.
Materials and Methods
Yeast Strains and Media
The α-synuclein expressing yeast strain we used in the modifier screen was IntTox: α–syn-WT, MATa can1−100 his3−11,15 leu2−3,112 trp1−1 ura3−1 ade2−1 pRS303Gal-α–synWT-YFP pRS304Gal-αSynWT-YFP. The α-synuclein expressing yeast strain we used for combinatorial gene analysis was HiTox: α–syn-WT, MATa can1−100 his3−11,15 leu2−3,112 trp1−1 ura3−1 ade2−1 pRS304Gal-α–synWT-GFP pRS306Gal-α–synWT-GFP. The Gal promoter reporter strain used to determine the effect of modifier genes on expression from galactose-regulated promoter was Gal-YFP, MATa can1−100 his3−11,15 leu2−3,112 trp1−1 ura3−1 ade2−1 pRS303Gal-YFP. For Ypk9 localization studies, a cassette containing a LoxP-flanked KanMX cassette followed by YFP (pDH22, a gift from Yeast Resource Center, University of Washington, Seattle, WA) was inserted in frame at the N-terminus of Ypk9 by homologous recombination in the BY4741 strain background. Correct insertion was checked by PCR and the KanMX gene was subsequently removed by transformation with a plasmid containing a GAL-inducible Cre recombinase (pSH47, a gift from Yeast Resource Center, University of Washington, Seattle, WA). The ypk9Δ strain was obtained by replacing the YPK9 coding region with the HIS3 gene in the BY4741 strain background. Colony PCR was used to verify correct gene disruption. Strains were manipulated and media prepared using standard techniques.
Plasmids
For Ypk9 localization studies, pAG416GPD-EGFP-Ypk9 was constructed by Gateway cloning using the Ypk9 entry clone (pDONR221-YPK9) and pAG416GPD-EGFP-ccdb destination vector 46 in an LR reaction. PARK9 patient-based mutations were introduced into pDONR221-YPK9 using the QuikChange Site Directed Mutagenesis Kit (Stratagene) and sequence verified. Del833−1472 corresponds to human mutation 1632_1653dup22 and Del1329−1472 corresponds to human mutation 3075delC. The ATPase-dead mutation is D781N. Using Gateway cloning, we subcloned WT and mutant Ypk9 into pAG416GPD-EGFP-ccdB (for localization and Mn2+ rescue), pAG416GPD (Mn2+ rescue), or pBY011 (α–synuclein rescue). Primer sequences are available upon request. For studies with human PARK9, pcDNA3.1V5-His-Topo-ATP13A2 was a generous gift from Christian Kubisch. The ATP13A2 coding region was PCR amplified and subcloned in pDONR221 to generate the entry clone pDONR221-ATP13A2. Subsequent LR Gateway reactions generated pBY011-ATP13A2, pAG416GPD-ATP13A2 and pAG416GPD-EGFP-ATP13A2.
Metals
Serial dilutions of WT (BY4741) or ypk9Δ cells were spotted onto YPD or CSM agar plates supplemented with excess concentrations of metals (Ca2+, Fe3+, Mn2+, Zn2+, Co2+, Cu2+) or metal chelators (10 mM EGTA, 0.75 mM EDTA) and growth was assessed after 2 to 3 days at 30 °C. For the Mn2+ toxicity rescue experiments, WT and ypk9Δ strains were transformed with the indicated plasmids and transformants spotted onto SD-URA plates containing different concentrations of MnCl2 (8, 10, 12, 14 mM). To assess ypk9Δ Mn2+ sensitivity in liquid culture, we used the Bioscreen (www.bioscreen.fi) to monitor growth. Yeast cells were pre-grown in YPD to mid-log phase, diluted to OD600 = 0.1, and dispensed to individual wells, in the presence of the indicated concentrations of MnCl2. OD600 measurements were taken every thirty minutes, and the plates were shaken every thirty seconds to aerate the cells. At least three independent runs were conducted for each growth condition, and each condition was tested in triplicate.
Phylogenetic tree
Protein sequences for all yeast and human P-type ATPases were retrieved from the UniProtKB/Swiss-Prot family/domain classification database (cation transport ATPase (P-type) family). A multiple sequence alignment was obtained using the ClustalW algorithm with default parameters. The phylogenetic tree was obtained using the PROML program (maximum likelihood algorithm with Jones-Taylor-Thornton probability model, constant rate of change among sites) in the PHYLIP package (v3.67).
α–Syn toxicity modifier screen
We performed the high-throughput yeast transformation protocol as described previously for a smaller library of genes 12 and as detailed in 13.
Combinatorial analysis
For the combinatorial analysis pAG413GPD, pAG413GPD-YPK9 or pAG413GPD-Ypt1 was co-transformed along with pAG415GPD, pAG415GPD-YPK9 or pAG415GPD-Ypt1 into the HiTox α–syn yeast strain using the standard lithium acetate technique. The transformants were plated onto SD-His/Leu agar plates and grown for 2 days. Cells were then normalized and spotted onto SD-His/Leu and SGal-His/Leu plates. Suppressors of α–syn-induced toxicity were identified on the galactose plates after 3 days of growth at 30°C.
ER-Golgi trafficking assay
We performed the carboxypeptidase Y (CPY) maturation assay as described previously 12.
C. elegans Experiments
Nematodes were maintained following the standard procedures 47. RNAi and fluorescent microscopy were performed as described 48 by feeding UA50 [baInl3; Punc-54::a-syn::gfp, Punc-54::tor-2, rol-6 (su1006)] worms with the RNAi clones (Geneservice, Cambridge, UK) corresponding to the worm orthologs of YPK9 and its interactors. RNA isolation, cDNA preparation, and semi-quantitative RT-PCR were conducted as described 51 with the following modification. Total RNAs from 50 young-adult control [RNAi bacteria HT115(DE3) with empty vector] and RNAi-treated worms were isolated to generate cDNAs. PCR was then performed using primers specific for amplifying cdk-5 as loading control, α–syn, and tor-2. For DA neurodegeneration analysis, strains UA51 [baEx42; Pdat-1::a-syn, Pdat-1::gfp, Pdat-1::FLAG-W08D2.5, rol-6 (su1006)] and UA108 [baEx83; Pdat-1::gfp, Pdat-1::FLAG-W08D2.5, Punc-54::mCherry] were generated by injecting 50 μg/ml of each expression plasmid into integrated Pdat-1::α–syn, Pdat-1::GFP as well as Pdat-1::GFP worms, respectively. The stable lines were analyzed for neurodegeneration as described previously 7,12,25.
Rat primary midbrain neuron culture experiments
Primary midbrain cultures were prepared, transduced with lentivirus, and analyzed immunocytochemically as described previously 12. All of the methods involving animal handling were reviewed and approved by the Purdue Animal Care and Use Committee. Relative dopaminergic cell viability was determined by counting MAP2- and TH-immunoreactive neurons in randomly chosen observation fields. The data were expressed as the percentage of MAP2+ neurons that were also TH+ (this ratiometric approach was used to correct for variations in cell density). Typically, 300−1500 MAP2+ cells were counted per experiment for each condition. In the control conditions, and in conditions where suppressors are efficacious, we typically count 500−1500 MAP2-positive neurons, a range that corresponds to 20−60 TH-positive neurons. It is more difficult to obtain such high cell counts from cultures expressing A53T α-synuclein alone because the cell viability is markedly reduced. In these cases we typically count 300−500 MAP2-positive neurons.
Preparation of primary mesencephalic cultures
Whole brains were dissected from day 17 embryos obtained from pregnant Sprague-Dawley rats (Harlan, Indianapolis). The mesencephalic region containing the substantia nigra and ventral tegmental area was isolated stereoscopically, and the cells were dissociated with trypsin (final concentration, 26 μg/mL in 0.9% [w/v] NaCl). The cells were plated on coverslips pretreated with poly-L-lysine (5 μg/mL) in media comprised of DMEM, 10% (v/v) fetal bovine serum, 10% (v/v) horse serum, penicillin (100 U/ml), and streptomycin (100 μg/ml). After a 4-day incubation, the cells were treated for 48 h with cytosine arabinoside (AraC) (20 μM) to suppress the growth of glial cells. Methods involving animal handling were approved by the Purdue Animal Care and Use Committee.
Preparation of lentiviral constructs
The ViraPower Lentivirus Expression System (Invitrogen) was used to generate lentiviruses encoding human α–syn (A53T), ATP13A2, CSNK1G3, USP10, PDE9A, and PLK2 as described previously 12. The insert from a pENTR-based entry construct was transferred into the pLENTI6/V5 DEST lentiviral expression vector (Invitrogen) via recombination. The lentiviral construct was sequenced using an Applied Biosystems DNA sequencer and packaged into virus via transient transfection of the 293FT packaging cell line. We showed in a previous study that lentiviruses prepared using this method have similar transduction efficiencies for MAP2- and TH-positive neurons (i.e. approximately 90% and 80%, respectively) 17.
Supplementary Material
Acknowledgments
We are grateful to Christian Kubisch for providing the human ATP13A2 cDNA and to the Yeast Resource Center for plasmids. A.D.G. was a Lilly Fellow of the Life Sciences Research Foundation and is currently a Pew Scholar in the Biomedical Sciences. A.D.G. is also supported by the NIH Director's New Innovator Award Program, part of the NIH Roadmap for Medical Research, through grant number 1-DP2-OD004417−01. A.C. is supported by a postdoctoral fellowship from the Parkinson's Disease Foundation. S.L. acknowledges support from the MGH/MIT Morris Udall Center of Excellence in Parkinson Disease Research, NS038372. M.L.G. was supported by a grant from the National Parkinson Foundation. C. elegans studies in the Caldwell lab were supported in part by grants from the Michael J. Fox Foundation, American Parkinson's Disease Foundation, and Bachmann-Strauss Dystonia and Parkinson Foundation. Research in the Rochet lab was supported by National Institutes of Health Grant NS049221 and a grant from the American Parkinson's Disease Association. S.L. is a founder of, a former member of the Board of Directors, and has received consulting fees from FoldRx Pharmaceuticals, a company that investigates drugs to treat protein-folding diseases. A.D.G., A.A.C, and S.L. are inventors on patents and patent applications that have been licensed to FoldRx. S.L. is also a member of the Board of Directors of Johnson & Johnson. A.A.C. and J.-C. R. have received consulting fees from FoldRx Pharmaceuticals and J.-C. R. has received payment from FoldRx for testing drugs in his laboratory.
References
- 1.Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–8. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 3.Ibanez P, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–71. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 4.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 6.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–8. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 7.Cao S, Gelwix CC, Caldwell KA, Caldwell GA. Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci. 2005;25:3801–12. doi: 10.1523/JNEUROSCI.5157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masliah E, et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–9. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 9.Lo Bianco C, Ridet JL, Schneider BL, Deglon N, Aebischer P. alpha - Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson's disease. Proc Natl Acad Sci U S A. 2002;99:10813–8. doi: 10.1073/pnas.152339799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–5. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gitler AD. Beer and Bread to Brains and Beyond: Can Yeast Cells Teach Us about Neurodegenerative Disease? Neurosignals. 2008;16:52–62. doi: 10.1159/000109759. [DOI] [PubMed] [Google Scholar]
- 12.Cooper AA, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeger-Lotem E, et al. 2008. Manuscript submitted.
- 14.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 15.Park J, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 16.Yang Y, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–8. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu F, Nguyen JL, Hulleman JD, Li L, Rochet JC. Mechanisms of DJ-1 neuroprotection in a cellular model of Parkinson's disease. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05333.x. [DOI] [PubMed] [Google Scholar]
- 18.Meulener MC, et al. DJ-1 is present in a large molecular complex in human brain tissue and interacts with alpha-synuclein. J Neurochem. 2005;93:1524–32. doi: 10.1111/j.1471-4159.2005.03145.x. [DOI] [PubMed] [Google Scholar]
- 19.Batelli S, et al. DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson's disease and involvement of HSP70. PLoS ONE. 2008;3:e1884. doi: 10.1371/journal.pone.0001884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–8. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 21.Di Fonzo A, et al. ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology. 2007;68:1557–62. doi: 10.1212/01.wnl.0000260963.08711.08. [DOI] [PubMed] [Google Scholar]
- 22.Lees AJ, Singleton AB. Clinical heterogeneity of ATP13A2 linked disease (Kufor-Rakeb) justifies a PARK designation. Neurology. 2007;68:1553–4. doi: 10.1212/01.wnl.0000265228.66664.f4. [DOI] [PubMed] [Google Scholar]
- 23.Ramirez A, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- 24.Su LJ, et al. Chemical Suppressors of alpha-Synuclein Toxicity Link Defects in ER-Golgi Trafficking and Mitochondria. Submitted. 2008 [Google Scholar]
- 25.Gitler AD, et al. The Parkinson's disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc Natl Acad Sci U S A. 2008;105:145–50. doi: 10.1073/pnas.0710685105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soper JH, et al. {alpha}-Synuclein Induced Aggregation of Cytoplasmic Vesicles in Saccharomyces cerevisiae. Mol Biol Cell. 2008 doi: 10.1091/mbc.E07-08-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kennedy S, Wang D, Ruvkun G. A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature. 2004;427:645–9. doi: 10.1038/nature02302. [DOI] [PubMed] [Google Scholar]
- 28.Caldwell GA, et al. Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum Mol Genet. 2003;12:307–19. doi: 10.1093/hmg/ddg027. [DOI] [PubMed] [Google Scholar]
- 29.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 30.Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–72. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satyal SH, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLean PJ, et al. TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem. 2002;83:846–54. doi: 10.1046/j.1471-4159.2002.01190.x. [DOI] [PubMed] [Google Scholar]
- 33.Sharma N, et al. A close association of torsinA and alpha-synuclein in Lewy bodies: a fluorescence resonance energy transfer study. Am J Pathol. 2001;159:339–44. doi: 10.1016/s0002-9440(10)61700-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Outeiro TF, et al. Sirtuin 2 Inhibitors Rescue {alpha}-Synuclein-Mediated Toxicity in Models of Parkinson's Disease. Science. 2007 doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 35.Kuhlbrandt W. Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol. 2004;5:282–95. doi: 10.1038/nrm1354. [DOI] [PubMed] [Google Scholar]
- 36.Axelsen KB, Palmgren MG. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 37.Gosavi N, Lee HJ, Lee JS, Patel S, Lee SJ. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J Biol Chem. 2002;277:48984–92. doi: 10.1074/jbc.M208194200. [DOI] [PubMed] [Google Scholar]
- 38.Larsen KE, et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–22. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubo S, et al. A combinatorial code for the interaction of alpha-synuclein with membranes. J Biol Chem. 2005;280:31664–72. doi: 10.1074/jbc.M504894200. [DOI] [PubMed] [Google Scholar]
- 40.Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT., Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–15. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- 41.Eliezer D, Kutluay E, Bussell R, Jr., Browne G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol. 2001;307:1061–73. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 42.Volles MJ, Lansbury PT., Jr. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–8. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- 43.Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med. 2004;10:1055–63. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 44.Norris EH, et al. Pesticide exposure exacerbates alpha-synucleinopathy in an A53T transgenic mouse model. Am J Pathol. 2007;170:658–66. doi: 10.2353/ajpath.2007.060359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dauer W, et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc Natl Acad Sci U S A. 2002;99:14524–9. doi: 10.1073/pnas.172514599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alberti S, Gitler AD, Lindquist S. A suite of Gateway((R)) cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast. 2007;24:913–9. doi: 10.1002/yea.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–21. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.