

Abstract
Background
Genome median and genome halving are combinatorial optimization problems that aim at reconstructing ancestral genomes as well as the evolutionary events leading from the ancestor to extant species. Exploring complexity issues is a first step towards devising efficient algorithms. The complexity of the median problem for unichromosomal genomes (permutations) has been settled for both the breakpoint distance and the reversal distance. Although the multichromosomal case has often been assumed to be a simple generalization of the unichromosomal case, it is also a relaxation so that complexity in this context does not follow from existing results, and is open for all distances.
Results
We settle here the complexity of several genome median and halving problems, including a surprising polynomial result for the breakpoint median and guided halving problems in genomes with circular and linear chromosomes, showing that the multichromosomal problem is actually easier than the unichromosomal problem. Still other variants of these problems are NP-complete, including the DCJ double distance problem, previously mentioned as an open question. We list the remaining open problems.
Conclusion
This theoretical study clears up a wide swathe of the algorithmical study of genome rearrangements with multiple multichromosomal genomes.
Background
The gene order or syntenic arrangement of ancestral genomes may be reconstructed based on comparative evidence from present-day genomes – the phylogenetic approach – or on internal evidence in the case of genomes descended from an ancestral polyploidisation event, or from a combination of the two. The computational problem at the heart of phylogenetic analysis is the median problem, while internal reconstruction inspires the halving problem, and the combined approach gives rise to guided halving. How these problems are formulated depends (1) on the karyotypic framework: the number of chromosomes in a genome and whether they are constrained to be linear, or if circular chromosomes are also permitted, and (2) on the objective function used to evaluate possible solutions. This function is based on some notion of genomic distance, either the number of adjacent elements on a chromosome in one genome that are disrupted in another – the breakpoint distance – or the number of evolutionary operations necessary to transform one genome to another.
While the karyotypes allowed in an ancestor vary only according to the dimensions of single versus multiple chromosome, and linear versus circular versus mixed, the genomic distances of interest have proliferated according to the kinds of evolutionary operations considered, from the classic, relatively constrained, reversals/translocations distance to the more inclusive Double Cut-and-Join (DCJ) measure, and many others [1].
The computational complexity of some of these problems has been settled for some specific distances and karyotypic contexts, and it is sometimes taken for granted that these results carry over to other combinations of context and distance. This is not necessarily the case. In this paper, we survey the known results and unsolved cases for three distance measures in three kinds of karyotype. We include several results presented here for the first time, as well as discussions on the definitions of the distances. The results contain both new polynomial-time algorithms and NP-hardness proofs. This paper is the full version of an extended abstract that has appeared in [2], which announced the results without giving all the proofs. In particular, a full discussion on the breakpoint distance definition, as well as the proofs of Theorem 2, Theorem 4, and Theorem 6 are added here, which makes this version a complete and definitive one.
Genomes, breakpoints and rearrangements
Multichromosomal genomes
We follow the general formulation of a genome in [3]. A gene A is an oriented sequence of DNA, identified by its tail At and its head Ah. Tails and heads are the extremities of the genes. An adjacency is an unordered pair of gene extremities. A genome Π is a set of adjacencies on a set of genes. Each adjacency in a genome means that two gene extremities are consecutive on the DNA molecule. In a genome, each gene extremity is adjacent to zero or one other extremity. An extremity x that is not adjacent to any other extremity is called a telomere, and can be written as an adjacency x∘ with a null symbol ∘. The adjacency x∘ is called a telomeric adjacency. For a genome Π on a set of genes 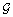 , consider the graph GΠ whose vertices are all the extremities of the genes, and the edges include all the non telomeric adjacencies in Π as well as an edge joining the head and the tail of each gene. This graph is a set of disjoint paths and cycles. Every connected component is called a chromosome of Π. A chromosome is linear if it is a path, and circular if it is a cycle. A genome with only linear, or only circular, chromosomes is called a linear or circular genome, respectively. An example of a graph GΠ is given in Figure 1.
, consider the graph GΠ whose vertices are all the extremities of the genes, and the edges include all the non telomeric adjacencies in Π as well as an edge joining the head and the tail of each gene. This graph is a set of disjoint paths and cycles. Every connected component is called a chromosome of Π. A chromosome is linear if it is a path, and circular if it is a cycle. A genome with only linear, or only circular, chromosomes is called a linear or circular genome, respectively. An example of a graph GΠ is given in Figure 1.
Figure 1.

The graph GΠΠ of a genome Π. Π is a genome on the set of genes {1,...,14}, containing three chromosomes, two of them being linear and one circular. Its adjacencies are the union of C1 = {12h4h, 4t14t, 14h1t, 1h7h, 7t8t}, C2 = {3t11t, 11h10t, 10h6t, 6h13h, 13t3h} and C3 = {9h2t, 2h5h}. It has four telomeres.
A Genome can also be represented as a set of strings, by writing the genes for each chromosome in the order in which they appear in the paths and cycles of the graph GΠ, with a bar over the gene if the head of the gene appears before the tail (we say it has negative sign), and none if the tail appears before the head (it has positive sign). For each linear chromosome, there are two possible equivalent strings, according to the arbitrary chosen starting point. One is obtained from the other by reversing the order and switching the signs of all the genes. For circular chromosomes, there are also two possible circular string representations, according to the direction in which the cycle is traversed. For example, chromosome C1 of the genome Π of Figure 1 may be written (12 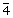 14 1
14 1 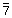 8) or (
8) or (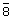 7
7 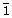
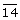 4
4 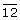 ).
).
A genome with only one chromosome is called unichromosomal. These correspond to signed permutations: the two string representations are (linear or circular) signed permutations.
Genomes with duplicates
A duplicated gene A is a couple of homologous oriented sequences of DNA, identified by two tails A1t and A2t, and two heads A1h and A2h. An all-duplicates genome Δ is a set of adjacencies on a set of duplicated genes.
For a genome Π on a gene set , a doubled genome Π ⊕ Π is an all-duplicates genome on the set of duplicated genes from such that if AxBy (x, y ∈ {t, h}) is an (possibly telomeric) adjacency of Π (Ax or By may be ∘), either A1xB1y and A2xB2y, or A2xB1y and A1xB2y, are adjacencies of Π ⊕ Π.
Note the difference between a general all-duplicates genome and the special case of a doubled genome: the former has two copies of each gene, while in the latter these copies are organised in such a way that there are two identical copies of each chromosome when we ignore the 1's and 2's in the A1x's and A2x's: it has two linear copies of each linear chromosome, and for each circular chromosome, either two circular copies or one circular chromosome containing the two successive copiesNote also that for a genome Π, there is an exponential number of possible doubled genomes Π ⊕ Π (exactly two to the power of the number of non-telomeric adjacencies in Π). These definitions correspond to duplicated and perfectly duplicated genomes found in [4], and slightly differs from the perfectly duplicated genome definition found in [5], as discussed in [4]. An example of an all-duplicates genome and a doubled genome is shown in Figure 2. Doubled genomes are the immediate result of an evolutionary event called Whole Genome Duplication (WGD), which is known to have occurred in many evolutionary lineages, from protists [6] to yeasts, to plants, to fish, to amphibians and even to mammals [7]. All-duplicates genomes derive from doubled genomes through a series of rearrangement events. Typically, all-duplicates genomes pertain to extant species, while doubled genomes are ancestral configurations inferred to exist immediately after the WGD, and that are to be reconstructed.
Figure 2.

The graphs and GΔ GΠ⊕Π of an all-duplicates genome Δ and a doubled genome Π ⊕ Π. (a) Δ has three chromosomes, while (b) Π ⊕ Π has four, and consist of two copies of two chromosomes, that have the same set of duplicated genes {1,...,7}.
In discussing all-duplicates genomes, we will sometimes contrast them with ordinary genomes which have a single copy of each gene.
The breakpoint distance
The breakpoint distance has been well-studied for permutations, i.e., unichromosomal genomes [8,9], but only a few published discussions have focused on how it should be defined for multichromosomal genomes (see [10] for one suggestion). The distance should depend not only on common adjacencies, or rather their absence, but also on common telomeres (or lack thereof) in two genomes. Here we propose a definition that we wish valid for all types of karyotypes, based on a most general approach integrating all possible informations from the two genomes. For two genomes Π and Γ on a set of n genes, suppose Π has NΠ chromosomes, and Γ has NΓ chromosomes. Let a(Π, Γ) be the number of common adjacencies, e(Π, Γ) be the number of common telomeres of Π and Γ. Then insofar as it should depend additively on these components, we may suppose the breakpoint distance has form
where β, θ and γ are positive parameters, while ψ may have either sign. Taking Π = Γ and imposing dBP (Π, Π) = 0 yields the relations β = 1 and 1 - 2θ + 2γ = 0, so θ = γ + 1/2, and the distance formula reduces to:
It is most plausible to count a total of 1 breakpoint for a fusion or fussion of linear chromosomes, which implies γ = ψ = 0, so the most natural choice of breakpoint distance between Π and Γ is
It might be argued that a fussion or fusion should count for as many as 2 breakpoints, or anything between 1 and 2, so that alternate values of γ and ψ might be entertained, provided γ ∈ [0, 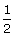 ], and ψ ∈ [0,1 - γ]. This may have an influence on how to calculate the number of breakages within a scenario, as discussed in [11]. For example, the parameters chosen in [10] are γ = and ψ =
], and ψ ∈ [0,1 - γ]. This may have an influence on how to calculate the number of breakages within a scenario, as discussed in [11]. For example, the parameters chosen in [10] are γ = and ψ = 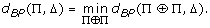 , giving rise to the disadvantage of there possibly being more breakpoints between two genomes than adjacencies in either one. For example, in comparing Π = (1 2 3 4 5) and Γ in which five linear chromosomes each contain one gene i ∈ {1,...,5}, the definition in [10] would count 9 breakpoints, which seems counterintuitive, while our definition counts 4, which seems more reasonable. Whether all the results presented in this paper also hold for the definition in [10] is open.
, giving rise to the disadvantage of there possibly being more breakpoints between two genomes than adjacencies in either one. For example, in comparing Π = (1 2 3 4 5) and Γ in which five linear chromosomes each contain one gene i ∈ {1,...,5}, the definition in [10] would count 9 breakpoints, which seems counterintuitive, while our definition counts 4, which seems more reasonable. Whether all the results presented in this paper also hold for the definition in [10] is open.
The definition of the breakpoint distance is easily transposable to the comparison of two all-duplicates genomes. For one all-duplicates genome Δ and one ordinary genome Π, the breakpoint distance between Π and Δ is the minimum breakpoint distance between Δ and a doubled genome Π ⊕ Π, that is,
The Double Cut-and-Join distance
Given a genome Π, a double-cut-and-join (DCJ) is an operation ρ acting on two adjacencies pq and rs (possibly some of p, q, r, s are ∘ symbols, so that telomeric adjacencies are considered; one adjacency can even be ∘∘). The DCJ operation replaces pq and rs either by pr and qs, or ps and qr. An example of DCJ operation on the genome Π of Figure 1 is drawn in Figure 3.
Figure 3.

A DCJ operation on the genome Π of Figure 1. Adjacencies 1h7h and 10t11h of the genome represented in (a) are cut and adjacencies 1h10t and 11h7h are joined to construct the genome represented in (b). This example shows how a DCJ operation can fuse two chromosomes into one.
A DCJ can reverse an interval of a genome, may cause the fussion of one chromosome into two, or the fusion of two chromosomes into a one, or a reciprocal translocation: the exchange of two telomere-containing segments between two chromosomes. Two consecutive DCJ operations, excising and circularising a chromosomal segment followed by a re-linearisation of the circular intermediate and reintegration on the same chromosome, using two new cut-points, results in a block interchange: two segments of the genome appear to simply exchange their positions. In the case these two segments are consecutive, the two DCJs result in a transposition, the apparent movement of a segment from one place on a chromosome to another. The DCJ operation is thus a very general framework, introduced by Yancopoulos et al. [12], as well as by Lin et al. in a special case [13], and since been adopted by Bergeron et al. [3,14] and many others, sometimes under other names such as spring [15] or "2-break rearrangement" [16].
If Π and Γ are two genomes on a set of n genes, the minimum number of DCJ operations needed to transform Π into Γ is called the DCJ distance and noted dDCJ(Π, Γ).
This DCJ distance is easily defined also for two all-duplicates genomes. For one all-duplicates genome Δ and one ordinary genome Π, the DCJ distance between Π and Δ is dDCJ(Π, Δ) = minΠ⊕Π dDCJ(Π ⊕ Π, Δ).
The reversal/translocation distance
The reversal/translocation distance was introduced by Hannenhalli and Pevzner [17], and is equivalent to the DCJ distance constrained to linear genomes.
If Π is a linear genome, a linear DCJ operation is a DCJ operation on Π that results in a linear genome. This allows reversals, chromosome fusions, fussions, and reciprocal translocations. DCJs that create circular intermediates, temporary circular chromosomes, and thereby mimic block interchanges and transpositions, are not allowed. Chromosome fusions and fussions are particular cases of translocations in this framework, justifying the appellation RT-distance. If Π and Γ are linear genomes, the RT distance between Π and Γ is the minimum number of linear DCJ operations that transform Π into Γ, and is noted dRT (Π, Γ).
Computational problems
The classical literature on genome rearrangements aims at reconstructing the evolutionary events and ancestral configurations that explain the differences between the organization of extant genomes. The focus has been on the genomic distance, median and halving problems. More recently the doubled distance and guided halving problems have also emerged as important. In each of the ensuing sections of this paper, these five problems are examined for a specific combination of distance d (breakpoint, DCJ or RT) and kind of multichromosomal karyotype (linear, circular, mixed).
1. Distance. Given two genomes Π, Γ, compute d(Π, Γ). Once the distance is calculated, an additional problem in the cases of DCJ and RT is to reconstruct the rearrangement scenario of length d(Π, Γ), i.e. the putative events that differentiate the genomes.
2. Double distance. Given an all-duplicates genome Δ and an ordinary genome Π, compute d(Δ, Π). This computation evaluates the evolutionary distance posterior to a WGD of the given genome Π, leading to an all-duplicates genome Δ, and locates the genes of the all-duplicates genome on chromosomes in one of the two ancestral copies of the ordinary genome. Because the assignment of labels "1" or "2" to the two identical (for our purposes) copies of a duplicated gene in Δ is arbitrary, the double distance problem is equivalent to finding such an assignment that minimises the distance between Δ and a genome Π ⊕ Π considered as ordinary genomes, where all the genes on any one chromosome in Π ⊕ Π are uniformly labeled "1" or "2" [16,18]. The double distance function is not symmetric because Δ is an all-duplicates genome and Π is an ordinary one, thus capturing the presumed asymmetric temporal and evolutionary relationship between the ancestor Π and the present-day genome Δ.
3. Median. Given three genomes Π1, Π2, Π3, find a genome M which minimises d(Π1, M) + d(Π2, M) + d(Π3, M). The median problem estimates the common ancestor of two genomes, given a third one as an outgroup. This is meaningful even in the "unrooted" case, where it is not specified which of the three genomes is the outgroup, because of the symmetry of the sum to be minimised.
4. Halving. Given an all-duplicates genome Δ, find an ordinary genome Π which minimises d(Δ, Π), the double distance mentioned above. The goal of a halving analysis is to reconstruct the ancestor of an all-duplicates genome at the time of a WGD event.
5. Guided halving. Given an all-duplicates genome Δ and an ordinary genome Π, find an ordinary genome M which minimises d(Δ, M) + d(M, Π). The guided halving problem is similar to the genome halving problem for Δ, but it takes into account the ordinary genome Π of an organism presumed to share a common ancestor with M, the reconstructed undoubled ancestor of Δ. A variant of the guided halving problem introduced in [19] is to find an ordinary genome M that is a solution to genome halving, that is, minimises d(Δ, M), and which in addition minimises d(M, Π). This helps choosing, among the numerous solutions to the genome halving problem, the one that is closest to the outgroup. We do not study this variant here, and it is open for all genomic distances.
We will survey these five computational problems for the three distances that we have introduced, in the cases of multichromosomal genomes containing all linear chromosomes, all circular chromosomes, or permitting both. The latter are refered as mixed genomes.
While many problems are open for multichromosomal genomes, there is a huge amount of research on these problems for unichromosomal genomes, whether circular or linear (the two cases are often equivalent up to some transformations [1]). They are not systematically particular cases of the multichromosomal problems, as the constraint of keeping only one chromosome along a rearrangement scenario can result in more difficult problems. More precisely, unichromosomal DCJ problems reduce to RT multichromosomal ones. Indeed, the RT operations always transform a unichromosomal genome into a unichromosomal one. As this paper contains very few results on the RT distance, practically the unichromosomal cases are often independent and not generalized here. Results on unichromosomal genomes are summarised in Table 1, together with the results for the multichromosomal case we review or present here. A complete survey on these problems can be found in [1].
Table 1.
Results summary.
| problem context: distance, #chr, linear, circular or mixed |
distance | halving | double distance | median | guided halving |
| breakpoint unichr, circular or linear | P | open | open | NP [20,21] | open |
| breakpoint multichr, circular and mixed | P new | P new | P new | P new | P new |
| breakpoint multichr, linear | P new | open P? | P new | NP new | NP [27] |
| DCJ unichr, circular or linear | P [3,12] | P [16] | open | NP [22] | open |
| DCJ multichr, circular and mixed | P [3,12] | P [4,5] | NP new | NP new | NP new |
| DCJ multichr, linear | P [12] | open | open | open NP? | open NP? |
| RT unichr | P [39] | open | open | NP [22] | open |
| RT multichr | P [17,33-35] | P [32] | open NP? | open NP? | open NP? |
Status of complexity questions for five problems related to ancestral genome reconstruction, for eight genomic distances in the unichromosomal and multichromosomal contexts. Note that unichromosomal problems require that both input and output genomes be unichromosomal, so all problems involving doubled genomes are computationally defined in the circular case, when the doubled genome consists in a single circular chromosome composed of two successive occurences of the ordinary genome. Other versions of the halving problem are less restrictive [5,16,32]. P and NP stand for polynomial and NP-hard, respectively, and when followed by ?, represent our conjectures.
Results
Breakpoint distance, circular and mixed genomes
In this section, d = dBP, and genomes are considered in their most general definition, that is, multichromosomal with both circular and linear chromosomes allowed. All the results also stand for circular genomes, but not always for linear genomes, which will be considered in a following section. As the nuclear genome of a eukaryotic species, a mixed karyotype is rarely observed, so probably unstable. Nevertheless this case is of great theoretical interest, as it is the only combination of distance and karyotype where all five problems mentioned in the previous section prove to be polynomially solvable, including the median problem which is hard for almost every other variant. Furthermore, the solutions in this context may suggest approaches for other variants of the problems, as well as providing a rapid bound for other distances, through the Watterson et al. bound [8].
Distance and double distance
The distance computation follows directly from the definition, and is easily achievable in linear time. The double distance computation is also easy: let Π be a genome and Δ be an all-duplicates genome. Let a(Π, Δ) be the sum, for every adjacency xy in Π, of the number of adjacencies among x1y1, x1y2, x2y1, x2y2 in Δ. Let e(Π, Δ) be the sum, for every telomere x in Π, of the number of telomeres among x1 and x2 in Δ.
Then we obtain
Indeed, it is a lower bound on the distance, because a(Π, Δ) and e(Π, Δ) are upper bounds on the number of common adjacencies and common telomeres, respectively, between Δ and any Π ⊕ Π. This lower bound is attained by constructing Π ⊕ Π in the following way: let xy be a possibly telomeric adjacency in Π (either x or y may be ∘ symbols); if x1y1 or x2y2 is an adjacency in Δ, choose x1y1 and x2y2 as adjacencies in Π ⊕ Π; If x1y2 or x2y1 is an adjacency in Δ, choose x1y2 and x2y1 as adjacencies in Π ⊕ Π; the two cases are either mutually exclusive if xy is not telomeric, or identical if xy is telomeric, so the assignment is made without ambiguity. For all adjacencies that have not been assigned, assign them arbitrarily.
Median
The following result contrasts with the NP-completeness proofs of almost all median problems in the literature [20-22] (see [23,24] for tractability results on some variants). The problem is NP-complete for unichromosomal genomes, that is, when the median genome M is required to be unichromosomal, whether the genomes are linear or circular [20,21], but the multichromosomal case happens to be easier.
Theorem 1. There is a polynomial time algorithm for the breakpoint median problem for multichromosomal genomes.
Proof. Let Π1, Π2, Π3 be three genomes on a gene set of size n. For any genome M on , let s(M) = d(Π1, M) + d(Π2, M) + d(Π3, M) be the median score of M.
Draw a graph G on the vertex set containing (1) all extremities of genes in , and (2) one supplementary vertex tx for every gene extremity x. For any pair of gene extremities x, y, draw an edge xy weighted by the number of genomes, among Π1, Π2, Π3, for which xy is an adjacency. Then there is an edge between each pair of gene extremities, weighted by 0, 1, 2, or 3. Now for any vertex x, draw an edge xtx weighted by half the number of genomes, among Π1, Π2, Π3, having x as a telomere. Each edge xtx is then weighted by 0, , 1, or 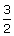 . Finally, put an edge of weight 0 between tx and ty for all pairs of gene extremities x, y. Let M be a perfect matching in G. Clearly, the edges joining gene extremities in M define the adjacencies of a genome, which we also call M. The relation between the weight of the perfect matching M and the median score of the genome M is easy to state:
. Finally, put an edge of weight 0 between tx and ty for all pairs of gene extremities x, y. Let M be a perfect matching in G. Clearly, the edges joining gene extremities in M define the adjacencies of a genome, which we also call M. The relation between the weight of the perfect matching M and the median score of the genome M is easy to state:
Claim 1. The weight w(M) of the perfect matching M in G is 3n - s(M).
Indeed, for any genome Πi, 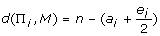 , where ai = a(Πi, M) is the number of common adjacencies between M and Πi, and ei = e(Πi, M) is the number of common telomeres between M and Πi. If M and Πi have a common adjacency or a common telomere, this accounts for 1 or , respectively, in the weight of the perfect matching M. So the weight of the matching M is
, where ai = a(Πi, M) is the number of common adjacencies between M and Πi, and ei = e(Πi, M) is the number of common telomeres between M and Πi. If M and Πi have a common adjacency or a common telomere, this accounts for 1 or , respectively, in the weight of the perfect matching M. So the weight of the matching M is  , which yields d(Π1, M) + d(Π2, M) + d(Π3, M) = 3n - w(M).
, which yields d(Π1, M) + d(Π2, M) + d(Π3, M) = 3n - w(M).
Conversely, any genome M can be extended to a perfect matching M in G such that s(M) = 3n - w(M): construct the matching M by including the edges xy and txty for each adjacency xy and an edge xtx for each telomere x.
Claim 1 implies that a maximum weight perfect matching M is a minimum score median genome. As the maximum weight perfect matching problem is polynomial [25], so is the breakpoint median problem. □
If the three genomes in the instance are circular, then it is possible to constrain the result to also be circular by restricting the graph G to the extremities of the genes. Then, in the same way, a perfect matching gives a circular solution to the median problem. This is not the case for linear genomes, since there is no way to guaranty that no chromosome in an instance is circular.
Note that a generalisation of this algorithm remains valid if the median of more than three genomes is to be computed. The phylogeny problems, both "big" and "small" versions, which also generalise the median problem for three genomes, remain open. The big problem is the search for a Steiner tree in the space of genomes, minimising the sum of the distances on its branches, while in the small problem, presumably easier, the graph-theoretical structure of the tree, namely its vertex set and edge or branch set, are given, and only the genomes corresponding to the extra vertices (not corresponding to the given genomes) need to be reconstructed.
Halving
To our knowledge, the genome halving with breakpoint distance has not yet been studied. In this framework, it has an easy solution, using a combination of elements from the maximum weight perfect matching technique in the solution of the median problem presented above, and the double distance computation. Let Δ be an all-duplicates genome on a gene set , and G be the graph on the vertex set containing (1) all the extremities of the genes in , and (2) one supplementary vertex tx for every gene extremity x. For any pair of gene extremities x, y, draw an edge in G weighted by zero, one or two according to the number of adjacencies in Δ among x1y1, x1y2, x2y1, and x2y2. Now for any vertex x, draw an edge xtx weighted by half the number of telomeres among x1 and x2 in Δ. Finally, put an edge of weight 0 between txty for all pairs of gene extremities x, y.
For a genome M on , define a perfect matching, also called M, by including edges xy and txty for each adjacency xy, and an edge xtx for each telomere x. Let w(M) be the weight of the matching M.
Claim 2. For a genome M on , the perfect matching M thus constructed satisfies w(M) = 2n - d(Δ, M).
Indeed, the score of the perfect matching M is 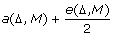 , that is, 2n - d(Δ,M), according to the double distance formula (see above in this section).
, that is, 2n - d(Δ,M), according to the double distance formula (see above in this section).
Conversely, it is easy to see that any perfect matching on G defines a genome M such that w(M) = 2n - d(Δ, M). This implies that the maximum weight perfect matching solves the genome halving problem in the breakpoint distance context.
Again, it is possible to solve the problem on only circular genomes by restricting the graph G to the gene extremities, dropping the tx supplementary vertices.
Guided Halving
As is the case for the median problem, this context provides the only polynomial result for the guided genome halving problem up to our knowledge. The solution combines elements of the three previous results, on the double distance, median and halving problems.
Let Δ be an all-duplicates genome on a gene set , and Π be an ordinary genome on . Let G be the graph on the vertex set containing (1) all the extremities of the genes in , and (2) one supplementary vertex tx for every gene extremity x.
For any pair of gene extremities x, y, there is an edge in G weighted by the number of adjacencies among x1y1, x1y2, x2y1, x2y2 in Δ, and xy in Π. Now there is an edge xtx for any gene extremity x weighted by half the number of telomeres among x1, x2 in Δ and x in Π. So each edge between gene extremities has an integer weight in {0, 1, 2, 3}, and xtx edges may have weight 0, , 1, or . Add 0-weight edges txty for all pairs x, y of gene extremities.
For any genome M, let s(M) = d(Δ, M) + d(M, Π). It is possible to construct a perfect matching M in G from genome M by choosing edges xy and txty for every adjacency xy in M. Its weight is denoted w(M).
Claim 3. For a genome M, the perfect matching thus constructed satisfies w(M) = 3n - s(M).
Indeed, the weight of the perfect matching M is 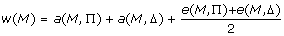 . According to the double distance formula (see above in this section), this yields w(M) = 3n - s(M).
. According to the double distance formula (see above in this section), this yields w(M) = 3n - s(M).
Conversely, if M is a perfect matching in G, its edges between gene extremities define the adjacencies of a genome M which satisfies s(M) = 3n - w(M). This implies that the maximum weight perfect matching solves the guided genome halving problem in the breakpoint distance context.
As is the case for the median problem, it is possible to generalise this statement for an arbitrary number of ordinary outgroup genomes. The phylogenetic problems are open.
Again, we can solve the problem on circular genomes by dropping the tx supplementary vertices in the graph G.
Breakpoint distance, linear case
In this section, d = dBP and all genomes must be linear, as is most appropriate for modeling for the eukaryotic nuclear genome. In contrast to the model of the previous section, all the problems concerning at least three genomes are NP-complete.
Distance and double distance
The solutions to these problems are the same as in the previous section, where circularity was allowed. In the double distance computation, it is guaranteed that Π ⊕ Π is linear if Π is linear, because if x is a telomere in Π, then both x1 and x2 are telomeres in Π ⊕ Π.
Median
Whereas the median is polynomial in the circular and mixed cases, it changes complexity as soon as median genomes are required to be linear. This does not prevent the use of the polynomial algorithm described above as a lower bound, but all biologically relevant median problems seem in fact to be NP-complete.
Theorem 2. The breakpoint median problem for multichromosomal linear genomes is NP-hard.
Proof. We use a reduction from the 2-chromosome breakpoint median, for which NP-hardness is proved in Lemma 2.
The 2-chromosome breakpoint median problem takes as input three unichromosomal linear genomes Π1, Π2, and Π3 on a set of genes, all having the same pair of telomeres. It asks for a linear genome M on with at most two chromosomes, which minimises its median score s(M) = d(Π1, M) + d(Π2, M) + d(Π3, M). The following lemma states the difficulty of the breakpoint median problem compared to the 2-chromosome breakpoint median problem, and thus, together with Lemma 2, proves Theorem 2.
Lemma 1. Let Π1, Π2, Π3, be three unichromosomal linear genomes Π1, Π2, and Π3 all having the same pair of telomeres on the gene set = {1,...,n}, and k be a positive integer. There exists a genome M on with at most two linear chromosomes such that s(M) ≤ k if and only if there exists a multichromosomal linear genome M' on with s(M') ≤ k.
(⇒): This direction is trivial: simply take M' = M.
(⇐): Let M' be a linear multichromosomal genome satisfying s(M') ≤ k, that has as few chromosomes as possible. We will prove that M' has at most two chromosomes. Suppose M' has at least three chromosomes. Then it has at least six telomeres v1,...,v6. Among them, it is possible to identify two telomeres (say without loss of generality v1 and v2), that belong to different chromosomes and are not telomeres in Π1, Π2, or Π3, because by hypothesis, they all have the same two telomeres. Then the genome constructed from M' by adding the adjacency v1v2 has at most the same median score as M' and fewer chromosomes, contradicting the hypothesis on M'. So choosing M = M' gives a genome with at most two chromosomes such that s(M) ≤ k. □
We now need to prove the NP-hardness of the 2-chromosome breakpoint median problem. We use a reduction from the hamiltonian cycle problem for directed graphs with vertex degree at most three, similar to the proof of Bryant [21] for the breakpoint median problem for unichromosomal circular genomes.
Lemma 2. The 2-chromosome breakpoint median problem is NP-hard.
Proof. Given a directed graph with maximum degree 3, deciding if it has a hamiltonian directed cycle is an NP-complete problem [26]. Let thus G0 be such a digraph. We will construct an instance of the 2-chromosome breakpoint median problem from G0.
First, let G be the directed graph with vertex set V (G) = V (G0) ∪ {xe : e ∈ E(G0)} ∪ {p, q}, and arc set
Note that G0 has a hamiltonian cycle if and only if there is a cycle in G covering all vertices but p and q. Given a subset X ⊆ E(G) of the arcs of a graph G, let GX denote the graph with vertex set V(G) and arc set X.
Construct three subsets A, B, C of arcs of G such that every arc of G belongs to exactly one of A, B, C, and A, B, C are either hamiltonian cycles of G or sets of vertex disjoint paths in G. The procedure is straightforward: for all vertex v of G which is a vertex of G0, put all incoming arcs in different subsets, and all outgoing arcs in different subsets. It can be done independently for every vertex because from the construction of G, no two vertices of G0 are neighbors. Eventually put arc pq in any subset.
Now perform a series of modifications of G to obtain a graph with three hamiltonian cycles A, B, and C. Along these modifications, we maintain a subset of arcs called supplementary arcs, noted S. Before any transformation, S is empty. The goal is to maintain the property that there exists a cycle covering all vertices of G except p and q and not using supplementary arcs if and only if there is a hamiltonian cycle in G0. As already remarked, the property is true at the beginning. Choose X ∈ {A, B, C} such that X is not a hamiltonian cycle in G (it is a set of disjoint paths). Choose two vertices a and b such that adding the arc ab to G and X would either give a graph in which X is a hamiltonian cycle, or a set of disjoint paths with fewer components. Then choose any vertex x of G different from a and b. Perform the following transformation illustrated in Figure 4: add two new vertices y and z. For each arc xw of G, replace it by the arc zw, and add zw to Y ∈ {A, B, C} whenever xw ∈ Y. Add arcs xy, yz, xz, ay, yb to G. Add xz, ay, yb to X. Also add xz, ay, yb to S, and add xy, yz to all {A, B, C}\X. Clearly, the property that there exists a cycle covering all vertices of G except p and q and not using supplementary arcs if and only if there is a hamiltonian cycle in G0 is still true after this transformation.
Figure 4.

Reduction of hamiltonian cycle in directed graph to breakpoint median for linear genomes. This figure is redrawn from [21]. Vertex a has no outgoing arc with X = A in its label set, and b has no incoming arc with A in its label set. We choose a, b such that adding arc ab to G[A] would not give a non-Hamiltonian circuit. We choose an another vertex x and insert two new vertices y and z. The incoming arcs of x in the right hand graph are the same as in the left hand graph. The outgoing arcs of z are the same as the incoming edges of x in the left hand graph. The remaining edges reduce the number of components in G[A] but leave the same number of components in G[B] and G[C].
Repeat this process until A, B and C are all hamiltonian cycles in the resulting graph, which we call G'. The weight of an arc of G' is the number of hamiltonian cycles among A, B and C which contain this arc. Note that G' has only weight 1 and 2 arcs.
Let v be an arbitrary vertex of G, different from p and q. Let = V(G')\{v} ∪ {v1, v2} be a set of genes (v1 and v2 are two new genes). For every X ∈ {A, B, C}, construct a genome ΠX on such that xhyt is an adjacency in genome X if xy is an arc of X in G' and x, y are different from v; If xv and vy are the arcs of X covering v, add the adjacencies 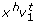 and
and 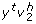 . This gives three linear unichromosomal genomes on with the same pair of telomeres (
. This gives three linear unichromosomal genomes on with the same pair of telomeres (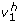 and
and 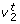 ), thus an instance of the 2-chromosome breakpoint median problem. For any genome M on , the weight of an adjacency xy is the number of genomes, among ΠA, ΠB and ΠC, which contain this adjacency. Adjacencies uhwt in M such that uw is an arc in S are called supplementary adjacencies.
), thus an instance of the 2-chromosome breakpoint median problem. For any genome M on , the weight of an adjacency xy is the number of genomes, among ΠA, ΠB and ΠC, which contain this adjacency. Adjacencies uhwt in M such that uw is an arc in S are called supplementary adjacencies.
Let λi be the number of arcs of weight i in the graph G', for each 0 ≤ i ≤ 3. For any genome M on , note s(M) = d(ΠA, M) + d(ΠB, M) + d(ΠC, M). The following is inspired by a result from [9] used in [21].
Claim 4. Let n = |V (G')|. A genome M with NM linear chromosomes on satisfies s(M) ≥ 2n - 1 + NM - λ2, where equality holds if and only if M contains all adjacencies of weight 2, and no adjacency of weight 0.
Indeed, for a genome M, denote by w(xy) the weight of the adjacency xy, and li = |xy adjacency of M: w(xy) = i|, for each 0 ≤ i ≤ 3. Let t1 = 1 if M has as a telomere, and t1 = 0 otherwise, and t2 = 1 if M has as a telomere, and t2 = 0 otherwise. Then we may write 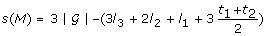 . As genome M has NM chromosomes, we have l3 + l2 + l1 + l0 = ||NM and || = n + 1, so
. As genome M has NM chromosomes, we have l3 + l2 + l1 + l0 = ||NM and || = n + 1, so 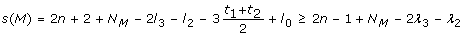 . As by construction no arc of G' has weight 3, we may write s(M) ≥ 2n - 1 + NM - λ2. Equality holds if and only if l2 = λ2, l0 = 0, and
. As by construction no arc of G' has weight 3, we may write s(M) ≥ 2n - 1 + NM - λ2. Equality holds if and only if l2 = λ2, l0 = 0, and 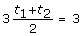 , that is, if and only if M contains all adjacencies of weight at least 2, and no adjacency of weight 0, because l0 = 0 implies that and are telomeres of M, thus .
, that is, if and only if M contains all adjacencies of weight at least 2, and no adjacency of weight 0, because l0 = 0 implies that and are telomeres of M, thus .
Claim 5. There is a linear genome M on with at most two chromosomes, with s(M) = 2n - 1 + NM - λ2 if and only if there is a hamiltonian cycle in G0.
(⇒) Suppose there is a linear genome M on with at most two chromosomes, with s(M) = 2n - 1 + NM - λ2. This implies by Claim 4 that M contains all adjacencies of weight 2, no adjacency of weight 0, and that and are telomeres of M. From the construction of G', M cannot contain any supplementary adjacency, since the extremities of supplementary adjacencies all are also extremities of weight two adjacencies, which are all contained in M. Note that in G', paths between vertices p and q to other vertices of the graph necessarily contain supplementary arcs. This yields that M has two chromosomes, one containing gene extremities from p and q, and the other containing the gene extremities from the other vertices, with telomeres and . Let H ⊆ E(G') contain the arcs xy such that xhyt is an adjacency in M, plus the arcs xv and vy for adjacencies of type and in M.
H ∩ E(G0) yields a hamiltonian cycle in G0.
(⇐) Suppose there is a hamiltonian cycle in G0. Then there is a cycle H covering all vertices of G' except p and q. Construct genome M on by adding (1) adjacencies xhyt whenever xy is in H and x, y are different from v (2) adjacencies and 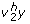 whenever xv or vy are arcs of H, (3) all weight two adjacencies, and (4) the adjacency ptqh. It is easy to check that M is a genome on , and by construction it contains all weight 2 adjacencies and no weight 0 adjacencies.
whenever xv or vy are arcs of H, (3) all weight two adjacencies, and (4) the adjacency ptqh. It is easy to check that M is a genome on , and by construction it contains all weight 2 adjacencies and no weight 0 adjacencies.
This proves that it is NP-complete to decide if a median genome reaches the lower bound of Claim 4 for its score, thus computing the minimum score median genome is NP-hard. □
A byproduct of this proof is the NP-hardness of the 2-chromosome breakpoint median problem. The result implies NP-completeness of the general case, where the genomes in the instance do not necessarily have the same pairs of telomeres. A consequence is that for any fixed k, it is also NP-hard to compute a best linear median genome with at most k chromosomes.
Halving
Surprisingly, this problem has not been treated in the literature. We conjecture it has a polynomial solution, because the halving problem for all other rearrangement distances is polynomial. Constructing a solution is beyond the scope of this paper, and the problem remains open.
Guided Halving
This problem is NP-hard, as proved in [27], using the NP-completeness result for the median proved just above in this section.
DCJ distance, general case
In this section, d = dDCJ. Genomes can have several chromosomes, circular or linear. This is the most general context in which the DCJ distance has been explicitly formulated [3]. Genomes rarely contain both circular and linear chromosomes; eukaryote nuclear genomes contain multiple linear chromosomes while prokaryotes generally contain one large circular chromosome, sometimes with additional plasmids.
Nevertheless, the simplicity of the computational framework where both genomes may contain both circular and linear chromosomes makes it attractive to mathematical study.
Note that the complexity of the median problem is not established by the work of Caprara [22], who proved the unichromosomal result only. We show the NP-hardness of the multichromosomal case here. Alekseyev and Pevzner [16] mention that the complexity of the double distance problem in the context of circular genomes is open; we show here that it is NP-hard as well.
Distance
There is an easy linear solution, both for the distance and the scenario computation [3,12]. We briefly recall the formula for computing the distance, because the underlying principle will be used in our proofs later on in this paper.
The breakpoint graph of two genomes Π and Γ on a gene set , denoted by BP(Π, Γ), is the graph whose vertex set is the set of extremities of the genes in , where there is an edge between two vertices x and y if xy is an adjacency in either Π (these are Π-edges) or Γ (Γ-edges). Note that we do not invoke any ∘ symbols in the construction of the breakpoint graph. Vertices in this graph have degree zero, one or two, so that the graph is a set of paths (possibly including some with no edges) and cycles. It is also the line-graph of the adjacency graph, an alternate representation in [3]. Figure 5 shows an example of a breakpoint graph. Theorem 3 shows how to obtain the distance directly from the graph. The formula is presented in [3] with the cycles and odd paths of the adjacency graph. This corresponds to cycles and even paths of the breakpoint graph, as it is the line-graph of the adjacency graph.
Figure 5.

A Breakpoint Graph. The breakpoint graph of the genomes Π (see Figure 1) and Γ, given by the union of C1 = {T 12t, 12h14h, 14t7h, 7t4t, 4h1h, 1t8t, 8h2t, 2h6t, 6hT} and C2 = {T 9t, 9h3t, 3h10t, 10h5t, 5h11h, 11t13h, 13tT}. Π-edges are dotted lines, and Γ-edges are plain lines.
Theorem 3. [3]For two genomes Π and Γ on a gene set of size n, let c(Π, Γ) be the number of cycles of the breakpoint graph BP(Π, Γ), and p(Π, Γ) be the number of paths with an even number of edges. Then
Note the similarity to the breakpoint distance formula in the background section on page 5. The number of genes n is the same in both formulae, the parameter c is related to parameter a in the breakpoint formula in that each common adjacency is a cycle of the breakpoint graph (with two parallel edges), and parameter p is related to parameter e, as each shared telomere is an even path (with no edge) in the breakpoint graph. Although these two measures of genomic distance were derived in different contexts and through different reasoning, their formulae show a remarkably similar form. They differ in that the DCJ formula also counts non-trivial cycles and paths, but for distant genomes, both measures tend to give similar values.
Double distance
The NP-completeness proof for the double distance problem follows the principles of Caprara's hardness proof for the median problem in the unichromosomal case [22].
Theorem 4. The DCJ double distance problem is NP-hard for multichromosomal mixed or circular genomes.
Proof. The reduction is from the breakpoint graph decomposition (BGD) problem (see [22]). A graph G is bicoloured if all its edges are coloured either red or blue; it is balanced if it has only degree 2 or degree 4 vertices, every vertex is incident to the same number of red and blue edges, and there is no cycle formed by only red or only blue edges. Given a balanced bicoloured graph G, the breakpoint graph decomposition problem is to find a partition of the edges of G into a maximum number of edge-disjoint cycles, each alternating between red and blue edges. Caprara [22] first proved the NP-hardness of this problem, and Berman and Karpinski [28] extended this by proving APX-hardness.
Let G be a balanced bicoloured graph on n vertices, defining an instance of the BGD problem. Let w2 be the number of degree 2 vertices of G, and w4 be the number of degree 4 vertices of G. Define the gene set as the vertex set of G. Construct an all-duplicates genome Δ and a genome Π on in the following way, as illustrated in Figure 6. First, for each gene X of , let XtXh be an adjacency in Π. Then, for every vertex X of G, let X1t, X1h, X2t and X2h, be the extremities of the duplicated gene X. If X has degree two in G, add the adjacency X1tX2h in Δ (if X has degree four, no adjacency is added at this point). Then for each blue edge XY in G, choose among X1h and X2h an extremitiy that is not yet involved in an adjacency, and another among Y1h and Y2h (arbitrarily if neither is involved in an adjacency yet). Add an adjacency between the two chosen extremities in Δ. Then for each red edge XY in G, choose among X1t and X2t an extremitiy that is not yet involved in an adjacency, and another among Y1t and Y2t (arbitrarily if neither is involved in an adjacency yet). Add an adjacency between the two chosen extremities in Δ.
Figure 6.

Reduction of BGD to DCJ double distance problem. The left hand graph is the balanced bicoloured graph G, and the right hand graph represents the adjacencies of the duplicated genomes Δ and Π ⊕ Π. In the case of a degree 2 vertex in G, the adjacencies of Π ⊕ Π are determined, as one solution gives more cycles. In the case of a degree 4 vertex in G, the two possibilities for the adjacencies of Π ⊕ Π are shown (Π ⊕ Π contains either the vertical or horizontal dotted adjacencies).
We then have an all-duplicates genome Δ, and a genome Π. Note that Π is composed of n circular chromosomes, one for each gene, and that neither Π nor Δ have telomeres.
Claim 6. The maximum number of edge-disjoint alternating cycles in G is equal to 2n - d(Δ, Π) - w2.
(This claim implies the theorem).
We first prove that the maximum number of edge-disjoint alternating cycles in G is at least 2n - d(Δ, Π) - w2. Let Π ⊕ Π be the doubled genome such that d(Δ, Π ⊕ Π) = d(Δ, Π). As no genome has a telomere, by Theorem 3, d(Δ, Π ⊕ Π) = 2n - c(Δ, Π ⊕ Π). Therefore there are c(Δ, Π ⊕ Π) edge-disjoint cycles in BG(Δ, Π ⊕ Π) alternating between Π ⊕ Π-edges and Δ-edges. Among them, w2 cycles are containing only two edges: if a vertex X of G has degree 2, then Π ⊕ Π has the adjacency X1tX2h and X2tX1h because the other possibility systematically has one cycle less in BG(Δ, Π ⊕ Π). The Δ edges of all the other cycles are the edges of an alternating blue-red cycle in G. Indeed, every blue edge defines an adjacency in Δ containing two gene heads, and every red edge defines an adjacency containing two gene tails. The Π ⊕ Π-edges all join one tail and one head, so two consecutive Δ-edges in a cycle of BG(Δ, Π ⊕ Π) have different colours. This means there are at least 2n - d(Δ, Π) - w2 alternating cycles in G.
Conversely, if there are k edge-disjoint alternating cycles in G, then d(Δ, Π) ≤ 2n - k - w2. Indeed, let C be any cycle of this partition. For every covered vertex X of degree 4 in G, let e and f be two consecutive edges of C (say e is blue and f is red) which are both incident to X. If e defines an adjacency in Δ which contains X2h and f defines an adjacency which contains X2t, choose X1hX1t and X2hX2t as adjacencies for Π ⊕ Π. If e defines an adjacency which contains X2h (or X1h) and f defines an adjacency which contains X1t (or X2t), choose X1hX2t and X2hX1t as adjacencies for Π ⊕ Π. For vertices of degree 2, always choose X1hX2t and X2hX1t as adjacencies for Π ⊕ Π. In this construction, each red-blue alternating cycle in G is a Π ⊕ Π-Δ alternating cycle in BP(Π ⊕ Π, Δ) that has at least k cycles. And there are w2 additional length 2 cycles at each degree 2 vertex. So d(Δ, Π) ≤ 2n - k - w2. □
Median
Though effective exact algorithms [29] and heuristics [30,31] are available, we have:
Theorem 5. The DCJ median problem for multichromosomal genomes is NP-hard, even for circular genomes.
Proof. We use a reduction from the breakpoint graph decomposition defined in the proof of Theorem 4, in a way very similar to part of Caprara's proof [22] for the unichromosomal case.
Let G be a balanced bicoloured graph on n vertices. Define the gene set as a set containing one gene X for every degree 2 vertex of G, and two genes X and Y for every degree 4 vertex of G.
Then construct the genomes Π1, Π2, Π3 in the following way, which is similar to the transformation in [22], as illustrated in Figure 7.
Figure 7.

Reduction of BGD to DCJ median problem. The left hand graph is the balanced bicoloured graph G, and the right hand graph represents the adjacencies of the genomes Π1, Π2 and Π3. Again, in the case of a degree 2 vertex in G, the choice for M is determined. But in the case of a degree 4 vertex in G, either the edges of Π1 or Π2 can be chosen for the median M.
For each degree 4 vertex v of G, add the two adjacencies XtXh and YtYh to Π1, and the two adjacencies XtYh and YtXh to Π2.
Now fo every blue edge vw in G, add to Π3 an adjacency between heads of genes contructed from v and w (choose one duplicated gene head arbitrarily if v or w have degree 4). And for every red edge vu in G, add to Π3 an adjacency between tails of genes contructed from v and u (choose one duplicated gene tail arbitrarily if v or u have degree 4).
It is easy to see that Π1, Π2, and Π3 define genomes on the set of genes , and they have no telomeres. Let w2 be the number of degree 2 vertices of G, and w4 be the number of degree 4 vertices of G.
Claim 7. There exists a genome M on such that d(M, Π1) + d(M, Π2) + d(M, Π3) ≤ w2 + 3w4 - k if and only if there exists at least k edge-disjoint alternating cycles in G.
(This claim implies the theorem.)
(⇐): Suppose there are k edge-disjoint alternating cycles in G. We will construct a median genome M such that d(Π1, M) + d(Π2, M) + d(Π3, M) = w2 + 2w4 - k. First, for each degree 2 vertex v of G, let XtXh be in M. Then, let v be a degree 4 vertex in G, and vw be a blue edge incident to v. In an alternating cycle, edge vw is consecutive with a red edge, say uv. To vw is associated a constructed Π3 adjacency, say XhWh. Then either the Π3 adjacency associated to uv contains the gene extremity Xt, or it contains the extremity Yt. In the first case, let XhXt and YhYt be in M, and in the second case, let XhYt and XtY h be in M. The matching M defines the adjacencies of a circular genome on , that we also call M. There are w2 + 2w4 genes, so by Theorem 3, d(Π1, M) + d(Π2, M) + d(Π3, M) = 3(w2 + 2w4) - (c(Π1, M) + c(Π2, M) + c(Π3, M)). By construction, we have c(M, Π1) + c(M, Π2) = 2w2 + 3w4, and c(M, Π3) = k, so d(Π1, M) + d(Π2, M) + d(Π3, M) = w2 + 3w4 - k.
(⇒): Suppose M is a genome such that d(Π1, M) + d(Π2, M) + d(Π3, M) ≤ w2 + 3w4 - k. Suppose M is chosen such that d(Π1, M) + d(Π2, M) + d(Π3, M) is minimum, and among all such genomes, choose M with a maximum number of edges parallel to Π1-edges or Π2-edges. A circular genome is said to be canonical if it only has adjacencies that belong to Π1 or Π2. We will prove that M is canonical.
Subclaim. M is canonical.
Suppose M is not canonical. Suppose first that there is a degree 2 vertex v in G, such that M does not contain the adjacency XtXh. Suppose M contains adjacencies Xta and Xhb, where a and b are gene extremities or ∘ symbols if Xt or Xh is a telomere in M. Then replace Xta and Xhb by XhXt and ab (simply XhXt if both Xt and Xh are telomeres in M). By this operation, c(M, Π1) and c(M, Π2) both increase by at least 1, and c(M, Π3) decreases by at most 1, so d(Π1, M) + d(Π2, M) + d(Π3, M) decreases by one, contradicting the hypothesis.
Now suppose that there is a degree 4 vertex in G, such that M does not contain any of the adjacencies XhXt, YhYt, XhYt, YhXt. Say it contains adjacencies Xha, Xtb, Y hc, Ytd, where a, b, c, d may be null symbols if any of Xh, Xt, Y h, Yt is a telomere in M. Then replace Xha, Xtb, Yhc, Ytd by XhXt, YhY t, and either ab, cd, or ac, bd, or ad, bc, according to the combination that creates the largest number of cycles in BP(M, Π3). Suppose now that M contains only one among the adjacencies XhXt, YhY t, XhYt, YhXt, say XhXt, and M has adjacencies Ytb and Y hc. Then replace edges Ytb and Y hc by YhYt and bc. All these operations decrease d(Π1, M) + d(Π2, M) + d(Π3, M) or maintain it constant, while increasing the number of edges parallel to Π1 and Π2, contradicting the hypothesis. So the subclaim is proved.
Now, since M is canonical, there are c(Π3, M) edge-disjoint alternating cycles in G, since an adjacency of M always joins a head and a tail, so the corresponding edge in G is adjacent to one red edge at one of its vertices and one blue edge at the other. By Theorem 3, c(Π3, M) = 3(w2 + 2w4) - (d(Π1, M) + d(Π2, M) + d(Π3, M) + c(Π1, M) + c(Π2, M)) and, by hypothesis, c(Π3, M) ≥ 3(w2 + 2w4) - (w2 + 2w4 - k + 2w2 + 3w4), that is, c(Π3, M) ≥ k, which proves the claim. □
Halving
This problem has a polynomial solution, as recently stated for unichromosomal genomes by [16] and in the general case by [4,5]. All these algorithms are simplified versions of the algorithm by El-Mabrouk and Sankoff [32], developed for the RT rearrangement distance, which allows reversals, translocations, fusions and fissions, but not the other DCJ operations.
Guided Halving
Theorem 6. The DCJ guided halving problem is NP-complete for multichromosomal genomes.
Proof. Again, we use a reduction of the breakpoint graph decomposition problem, as in the proofs of Theorems 4 and 5.
Let G be a balanced bicoloured graph on n vertices. Define the gene set as a set containing one gene X for every degree 2 vertex of G, and two genes X and Y for every degree 4 vertex of G. From G, we define one genome Π and one all-duplicates genome Δ on as illustrated in Figure 8.
Figure 8.

Reduction of BGD to DCJ guided halving problem. The left hand graph is the balanced bicoloured graph G, and the right hand graph represents the adjacencies of the genomes Δ and Π. Adjacencies of Π are doubled in the drawing to be presented with the doubled genes.
For every degree 2 vertex v of G, let X be the corresponding gene in and Xt and Xh its extremities. For every degree 4 vertex v of G, let X and Y be the two genes in , and Xt and Xh, Y t and Yh their extremities. For every blue edge uv in G, construct an adjacency in Π between the heads of the genes constructed from u and v, choosing arbitrarily between the heads of vertices X and Y if u or v have degree 4, in such a way that no two adjacencies share an extremity (choose a different head for the two blue edges incident to a degree 4 vertex). For every red edge tv in G, construct an adjacency in Π between the tails of the genes constructed from t and v, again choosing arbitrarily between the tails of vertices X and Y if t or v have degree 4. This defines a genome Π on that has no telomere.
Now, define the genome Δ in the following way. For each degree 2 vertex of G, Δ contains the adjacencies X1hX1t and X2hX2t defined by the extremities of the two copies of gene X. For each degree 4 vertex in G, Δ contains the adjacencies X1hX1t, Y2hY2t, X2hY1t, and X2tY1h defined by the extremities of the two copies of the two genes X and Y. This defines an all-duplicates genome Δ on that has no telomere. Let w2 be the number of degree 2 vertices of G, and w4 be the number of degree 4 vertices of G.
Claim. There exists a genome M such that d(M, Π) + d(M, Δ) ≤ w2 + 3w4 - k if and only if there exists at least k edge-disjoint alternating cycles in G. (This claim implies the theorem.)
(⇐): Suppose there are k edge-disjoint alternating cycles in G. We will construct a genome M and a doubled genome M ⊕ M such that d(M, Π) + d(M, Δ) ≤ w2 + 3w4 - k. First, for each degree 2 vertex v of G, let XtXh be in M, and M ⊕ M be constructed so that X1tX1h and X2tX2h are in M ⊕ M. Then, for every vertex v of degree 4 of G, either the blue edge incident to Xh is followed by the red edge incident to Xt in one alternating cycle, or it is followed by the red edge incident to Yt. In the first case, let XhXt and Y hYt be in M, and X1hX1t, X2hX2t, Y1hY1t, Y2hY2t be in M ⊕M. In the second case, let XhYt and XtYh be in M, and X2hY1t, X1hY2t, X1tY2h, X2tY1h be in M ⊕ M.
The matching M defines a genome without telomeres, and M ⊕ M is one of its doubled genomes. There are w2 + 2w4 genes, so by Theorem 3, d(Π, M) = w2 + 2w4 - c(Π, M) = w2 + 2w4 - k; and d(Δ, M ⊕ M) = 2(w2 + 2w4) - (2 × w2 + 3 × w4). So d(Π, M) + d(Δ, M) = w2 + 3w4 - k.
(⇒): Suppose M is a genome such that d(Π, M) + d(Δ, M) ≤ w2 + 3w4 - k. Suppose M is chosen such that d(Π, M) + d(Δ, M) is minimum, and among all such genomes, choose M with a maximum number of edges of type XtXh for a degree 2 vertex in G, or XhXt, YhYt, XhYt and XtYh for degree 4 vertices of G. A genome is said to be canonical if it has no telomeres and has only edges of type XtXh for a degree 2 vertex in G, or XhXt, YhYt, XhYt and XtYh for degree 4 vertices of G. We will prove that M is canonical.
Subclaim. M is canonical.
Suppose M is not canonical. Suppose first that there is a degree 2 vertex v in G, such that M does not contain the edge XtXh. Suppose M contains edges Xtu and Xhv, where u and v are gene extremities or ∘ symbols if Xt or Xh is a telomere in M. Then replace Xtu and Xhv by XhXt and uv (simply XhXt is both Xt and Xh are telomeres in M). By this operation, c(M, Π) decreases by at most one, while defining M ⊕ M as containing X1hX1t and X2hX2t makes c(M ⊕ M, Δ) increase by at least 2. This contradicts the hypothesis.
Now suppose that there is a degree 4 vertex in G, such that M does not contain any of the edges XhXt, YhYt, XhYt, YhXt. Say it contains edges Xht, Xtu, Yhv, Ytw, where t, u, v, w may be null symbols if any of Xh, Xt, Yh, Yt is a telomere in M. Then replace Xht, Xtu, Yhv, Ytw by Xh, Xt, YhYt, tu, vw or XhYt, tw, XtYh, uv, depending on the cycles in c(M ⊕ M, Δ).
Suppose now that M contains only one among the edges XhXt, YhYt, XhYt, YhXt say XhXt, and M has edges Ytu and Yhv. Then replace edges Ytu and Yhv by YhYt and uv. All these operations decrease d(Π, M) + d(Δ, M) or maintain it constant, while increasing the number of edges of type XtXh for a degree 2 vertex in G, or XhXt, YhYt, XhY t and XtYh for degree 4 vertices of G, contradicting the hypothesis. At the end of this process, M is canonical, so the subclaim is proved.
Now, since M is canonical, there are c(Π, M) edge-disjoint alternating cycles in G, since an edge of M always joins a head and a tail, so that it is adjacent to one red and one blue edge. By Theorem 3, c(Π, M) = 3(w2 + 2w4) - (d(Π, M) + d(Δ, M ⊕ M) + c(Δ, M ⊕ M)), and by hypothesis, c(Π3, M) ≥ 3(w2 + 2w4) - (w2 + 3w4 - k + 2w2 + 3w4)), that is, c(Π3, M) ≥ k, which proves the claim. □
DCJ distance, linear chromosomes
In the original formulation of the DCJ distance [12], it was shown that there is a solution where each excision of a circular intermediate could be followed directly by its reinsertion. Thus the median and halving problems can be stated in terms of exclusively linear chromosomes in both the data genomes and the reconstructed ancestor. They all remain open.
Reversal/Translocation distance
Hannenhalli and Pevzner proposed a polynomial-time algorithm for calculating dRT (Π, Γ) for two genomes Π and Γ [17], after solving the problem for unichromsomal genomes [39]. This was reformulated in [33], minor corrections were added in [34] and [35], and Bergeron et al. simplified the formula [14] and investigated the relations between dRT and dDCJ.
A polynomial time genome halving algorithm was given in [32]. Though the constrained DCJ distance in the previous section is arguably just as realistic, because of the long history of dRT, effective heuristics for RT have been developed and applied for the double distance [18,36], median [31,37] and guided halving problems [18,19,36], but their complexities remain open questions. Note that [38] gives an NP-completeness result on a problem which slightly generalizes the reversal double-distance probem on unichromosomal genomes.
Discussion and conclusion
Table 1 summarises the current knowledge of the complexity of the five genome rearrangement problems, including the new results in this paper. Note that all the results on general multichromosomal genome (that is, circular or linear) also hold for exclusively circular genomes, as the polynomial algorithms can always provide a circular solution to a circular instance, and all NP-completeness proofs are constructed with circular chromosomes.
Authors' contributions
ET, CZ and DS have elaborated the definition and conjectures, proved the results and written the paper.
Acknowledgments
Acknowledgements
Research supported in part by a grant to DS and a doctoral fellowship to CZ from the Natural Sciences and Engineering Research Council of Canada (NSERC). ET is funded by the Agence Nationale pour la Recherche (ANR-08-GENM-036-01 and NT05-3 45205) and the Centre National de la Recherche Scientifique (CNRS). DS holds the Canada Research Chair in Mathematical Genomics. Figures 1, 2, 3 and 5 are the work of Sèverine Bérard, used with her kind permission.
Contributor Information
Eric Tannier, Email: Eric.Tannier@inria.fr.
Chunfang Zheng, Email: czhen033@uottawa.ca.
David Sankoff, Email: sankoff@uottawa.ca.
References
- Fertin G, Labarre A, Rusu I, Tannier E, Vialette S. Combinatorics of Genome Rearrangements. MIT Press; 2009. [Google Scholar]
- Tannier E, Zheng C, Sankoff D. Multichromosomal genome median and halving problems. Algorithms in Bioinformatics, proceedings of WABI'08, of Lecture Notes in Bioinformatics. 2008;5251:1–13. [Google Scholar]
- Bergeron A, Mixtacki J, Stoye J. A unifying view of genome rearrangements. Algorithms in Bioinformatics, proceedings of WABI'06, of Lecture Notes in Computer Science. 2006;4175:163–173. [Google Scholar]
- Mixtacki J. Genome Halving under DCJ revisited. Proceedings of COCOON'08, Lecture Notes in Computer Science. 2008.
- Warren R, Sankoff D. Genome halving with double cut and join. Proceedings of the 6th Asia-Pacific Bioinformatics Conference, of Advances in Bioinformatics and Computational Biology. 2008;6:231–240. doi: 10.1142/s0219720009004102. [DOI] [PubMed] [Google Scholar]
- Aury J, Jaillon O, Duret L, Noel B, Jubin C, Porcel B, Ségurens B, Daubin V, Anthouard V, Aiach N, Arnaiz O, Billaut A, Beisson J, Blanc I, Bouhouche K, Câmara F, Duharcourt S, Guigó R, Gogendeau D, Katinka M, Keller A, Kissmehl R, Klotz C, Koll F, Mouël AL, Lepère G, Malinsky S, Nowacki M, Nowak J, Plattner H, Poulain J, Ruiz F, Serrano V, Zagulski M, Dessen P, Bétermier M, Weissenbach J, Scarpelli C, Schächter V, Sperling L, Meyer E, Cohen J, Wincker P. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature. 2006;444:171–178. doi: 10.1038/nature05230. [DOI] [PubMed] [Google Scholar]
- Otto S, Whitton J. Polyploid incidence and evolution. Annual Review of Genetics. 2000;24:401–437. doi: 10.1146/annurev.genet.34.1.401. [DOI] [PubMed] [Google Scholar]
- Watterson G, Ewens W, Hall T, Morgan A. The chromosome inversion problem. Journal of Theoretical Biology. 1982;99:1–7. [Google Scholar]
- Sankoff D, Blanchette M. The median problem for breakpoints in comparative genomics. Proceedings of the Third International Computing and Combinatorics Conference COCOON'97, of Lecture Notes in Computer Science. 1997;1276:251–263. [Google Scholar]
- Pevzner P, Tesler G. Transforming men into mice: the Nadeau-Taylor chromosomal breakage model revisited. Proceedings of the seventh annual international conference on Research in computational molecular biology RECOMB'03. 2003. pp. 247–256.
- Bergeron A, Mixtacki J, Stoye J. Proceedings of Recomb Workshop on Comparative Genomics, Lecture Notes in Bioinformatics. Springer-Verlag; 2008. On computing the breakpoint reuse rate in rearrangement scenarios. [Google Scholar]
- Yancopoulos S, Attie O, Friedberg R. Efficient sorting of genomic permutations by translocation, inversion and block interchange. Bioinformatics. 2005;21:3340–3346. doi: 10.1093/bioinformatics/bti535. [DOI] [PubMed] [Google Scholar]
- Lin Y, Lu C, Chang HY, Tang C. An efficient algorithm for sorting by block-interchange and its application to the evolution of vibrio species. Journal of Computational Biology. 2005;12:102–112. doi: 10.1089/cmb.2005.12.102. [DOI] [PubMed] [Google Scholar]
- Bergeron A, Mixtacki J, Stoye J. HP distance via Double Cut and Join distance. Combinatorial Pattern Matching, proceedings of CPM'08, of Lecture Notes in Computer Science. 2008;5029 [Google Scholar]
- Lin YC, Lu CL, Liu YC, Tang CY. SPRING: a tool for the analysis of genome rearrangement using reversals and block-interchanges. Nucleic Acids Res. 2006;34:W696–W699. doi: 10.1093/nar/gkl169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyev M, Pevzner PA. Colored de Bruijn graphs and the genome halving problem. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2008;4:98–107. doi: 10.1109/TCBB.2007.1002. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S, Pevzner P. Transforming men into mice (polynomial algorithm for genomic distance problem) Proceedings of the 36th Annual Symposium on Foundations of Computer Science FOCS'95) 1995. pp. 581–592.
- Zheng C, Zhu Q, Sankoff D. Descendants of whole genome duplication within gene order phylogeny. Journal of Computational Biology. 2008;15:947–964. doi: 10.1089/cmb.2008.0118. [DOI] [PubMed] [Google Scholar]
- Zheng C, Zhu Q, Sankoff D. Genome halving with an outgroup. Evolutionary Bioinformatics. 2006;2:319–326. [PMC free article] [PubMed] [Google Scholar]
- Pe'er I, Shamir R. The median problems for breakpoints are NP-complete. Electronic Colloquium on Computational Complexity. 1998;5 [Google Scholar]
- Bryant D. Tech Rep CRM-2579. Centre de recherches mathématiques, Université de Montréal; 1998. The complexity of the breakpoint median problem. [Google Scholar]
- Caprara A. The reversal median problem. INFORMS Journal on Computing. 2003;15:93–113. [Google Scholar]
- Ohlebusch E, Abouelhoda MI, Hockel K. A linear time algorithm for the inversion median problem in circular bacterial genomes. J of Discrete Algorithms. 2007;5:637–646. [Google Scholar]
- Bernt M, Merkle D, Middendorf M. Solving the Preserving Reversal Median Problem. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2008;5:332–347. doi: 10.1109/TCBB.2008.39. [DOI] [PubMed] [Google Scholar]
- Lovasz L, Plummer MD. Matching Theory, of Annals of Discrete Mathematics Amsterdam: North Holland. 1986.
- Garey MR, Johnson DS. Computers and intractability A guide to the theory of NP-completness. New York: W. H. Freeman; 1979. [Google Scholar]
- Zheng C, Zhu Q, Adam Z, Sankoff D. Guided genome halving: hardness, heuristics and the history of the Hemiascomycetes. Bioinformatics. 2008;24:i96–104. doi: 10.1093/bioinformatics/btn146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman P, Karpinski M. On some tighter inapproximability results. Automata, Languages and Programming, of Lecture Notes In Computer Science. 1999;1644:705. [Google Scholar]
- Xu W, Sankoff D. Decompositions of Multiple Breakpoint Graphs and Rapid Exact Solutions to the Median Problem. Algorithms in Bioinformatics, proceedings of WABI'08, Lecture Notes in Bioinformatics. 2008.
- Adam Z, Sankoff D. The ABCs of MGR with DCJ. Evol Bioinform Online. 2008;4:69–74. [PMC free article] [PubMed] [Google Scholar]
- Lenne R, Solnon C, Stützle T, Tannier E, Birattari M. Reactive stochastic local search algorithms for the genomic median problem. Proceedings of EvoCOP'08, of Lecture Notes in Computer Science. 2008;4972:266–276. [Google Scholar]
- El-Mabrouk N, Sankoff D. The reconstruction of doubled genomes. SIAM Journal of Computing. 2003;32:754–792. [Google Scholar]
- Tesler G. Efficient algorithms for multichromosomal genome rearrangements. Journal of Computer and System Sciences. 2002;65:587–609. [Google Scholar]
- Ozery-Flato M, Shamir R. Two notes on genome rearrangement. Journal of Bioinformatics and Computational Biology. 2003;1:71–94. doi: 10.1142/s0219720003000198. [DOI] [PubMed] [Google Scholar]
- Jean G, Nikolski M. Genome rearrangements: a correct algorithm for optimal capping. Information Processing Letters. 2007;104:14–20. [Google Scholar]
- Zheng C, Wall PK, Leebens-Mack J, de Pamphilis C, Albert V, Sankoff D. The effect of massive gene loss following whole genome duplication on the algorithmic reconstruction of the ancestral Populus diploid. Proceedings of CSB'08. 2008. [PubMed]
- Bourque G, Pevzner P. Genome-scale evolution: Reconstructing gene orders in the ancestral species. Genome Research. 2002;12:26–36. [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zheng J, Fu Z, Nan P, Zhong Y, T Jiang SL. Assignement of orthologous genes via genome rearrangement. IEEE/ACM Transactions on Computational Biology and Bioinformatics. 2005;2:302–315. doi: 10.1109/TCBB.2005.48. [DOI] [PubMed] [Google Scholar]
- Hannenhalli S, Pevzner P. Transforming Cabbage into Turnip: Polynomial Algorithm for Sorting Signed Permutations by Reversals. Journal of the ACM. 1999;46:1–27. [Google Scholar]