

Abstract
MED1 is a base excision repair enzyme that interacts with the mismatch repair protein MLH1 and maintains genomic integrity by binding methylated DNA and repairing spontaneous deamination events. MED1 mutations have been associated with microsatellite instability and accelerated colorectal cancer (CRC) tumorigenesis. We propose that promoter methylation may serve as an alternative epigenetic mechanism for MED1 gene suppression during sporadic CRC tumorigenesis. Methylation status of the MED1 promoter was investigated in a panel of ovarian and colorectal cancer cell lines. The MED1 promoter region was sequenced following bisulfite treatment and sequence analysis identified a CpG island within the MED1 promoter which is frequently and preferentially methylated (≥ 50%) in ovarian and colorectal cancer cell lines with low/reduced MED1 expression. In vitro reversal of methylation restored MED1 expression. In colorectal cancer patients, when MED1 methylation was present, both tumor and matched mucosa were affected equally (mean frequency of methylation 24%) and there was no correlation between methylation and tumor stage. Patients without history of CRC showed significantly lower frequency of methylation (mean 14%, p<0.05). Decreased MED1 transcript levels were observed in matched normal mucosa when compared to controls (median fold difference 8.0). Additional decreased expression was seen between mucosa and matched tumor (median fold decrease 4.4). Thus, MED1 promoter methylation and gene silencing occur in sporadic CRC patients and represent an early event in CRC tumorigenesis. Detection of MED1 methylation and gene suppression in normal colon mucosa may contribute to identifying patients at higher risk of developing CRC during screening procedures.
Keywords: MED1, colorectal cancer, ovarian cancer, methylation
INTRODUCTION
Tumors of epithelial cell origin, or carcinomas, comprise approximately 85% of all diagnosed human cancers. Two of the most frequent carcinomas are colorectal adenocarcinoma and epithelial ovarian cancer. The Centers for Disease Control report colorectal cancer (CRC) as the second leading cancer cause of death.1 It is the third most common cancer in both men and women in the United States. In 2007 it is estimated that over 150,000 cases of CRC will be diagnosed and over 52,000 deaths will be attributed to the disease in an estimated US population of 301.6 million. Ovarian cancer also plays a significant role in cancer deaths for women. Nearly 15,000 deaths from ovarian cancer were recorded for the year 2003 with an estimated US female population of 147.7 million and it is predicted to be the fifth leading cause of cancer death in women for 2007 with over 22,000 new cases predicted to be diagnosed with an estimated US female population of 152.9 million.2
The causes of both CRC and ovarian cancer are multifactorial but inactivating mutations in tumor suppressor genes such as MLH1 and BRCA1 leading to tumorigenesis are well established.3,4 Epigenetic modifications of DNA have also become an important mechanism in tumorigenesis. Epigenetic changes are heritable changes that affect DNA expression without changing the DNA sequence per se. Epigenetic changes include DNA methylation, histone modifications, and chromatin remodeling that can either activate or silence genes. When present, these changes result in altered chromatin structure and a subsequent silencing of genes due to the resulting condensed chromatin.5 In this article, we use the term epigenetic downregulation to refer to the silencing effects of DNA methylation. Importantly, gene inactivation through DNA methylation has been implicated in the development of cancer.6 This epigenetic modification of the genome generally occurs at CpG islands located in gene promoters. DNA methylation is associated with alterations of the chromatin structure which ultimately leads to transcriptional silencing of the gene. Although promoter methylation may be physiologic as seen with imprinted genes and the inactive X chromosome,7,8 it is more frequently associated with tumor suppressor genes and the development of cancers including renal, hematologic, ovarian, and colorectal.8–10
The importance of DNA methylation with subsequent gene silencing in CRC is well described.11–16 Methylation of multiple tumor suppressor genes has been identified in CRC as part of a “CpG island methylator phenotype”.13 The DNA mismatch repair (MMR) gene MLH1 is one of several genes observed to be methylated in CRC but has also been shown to be methylated in ovarian cancer.17 MLH1 methylation and silencing has specifically been associated with the development of microsatellite instability (MSI) in sporadic CRC.11,16
The base excision repair (BER) enzyme MED1, also known as MBD4, has been shown to interact with the MMR protein MLH1 and bind methylated DNA.18,19 In addition to BER, MED1 plays multiple functions in cellular processes including DNA damage response, apoptosis, and transcriptional repression.20–24 The protein structure of MED1 reveals an amino-terminal 5-methylcytosine binding domain (MBD) and a carboxy terminus with DNA N-glycosylase and MLH1 complex formation capabilities.19 Specifically, MED1 acts as a thymine and uracil glycosylase that recognizes and repairs G:T and G:U mismatches originating from spontaneous deamination events of 5-methylcytosine and cytosine at CpG sites.18,25 If not repaired, these mismatches would cause CpG to CpA or CpG to TpG transition mutations.23,26
MED1 mutations that affect its BER function have been described in CRC. Mutations observed in MSI+ CRC result in a truncated protein predicted to retain the MBD but not the glycosylase domain.27,28 The known mutations in MED1 have been related to MSI+ tumors only. Alternative mechanisms of gene inactivation, such as silencing by promoter methylation, have not been described thus far for MED1. By analogy with other tumor suppressor genes of the caretaker type, we propose promoter methylation as a plausible mechanism leading to MED1 gene silencing in sporadic colorectal cancers. The specific comparison to MLH1 is based on the interactions between the two proteins as well as the caretaker roles these two proteins play in CRC.11,19,29 Methylation of the MED1 promoter may potentially lead to decreased gene expression causing an increase in CpG site mutations, decreased apoptosis and accelerated tumorigenesis similar to murine knockout models.30 Here we report identification of CpG islands within the MED1 promoter and increased methylation with reduced expression in both CRC and ovarian cancer.
MATERIALS AND METHODS
Cell lines
A panel of colorectal cancer cell lines (SW403, DLD1, CaCo2, SW480, HT29, RKO, SW948, HCT116, and WiDr) and a panel of ovarian cancer cell lines (OVCAR-2, OVCAR-3, OVCAR-4, OVCAR-8, OVCAR-10, and OVCAR-13) were obtained from ATCC and cultured according to their guidelines. Cells were incubated in a humidified atmosphere at 37°C with 5% CO2.
Cloning of the MED1 promoter CpG island
A human genomic lambda bacteriophage library (Stratagene) was screened with an exon 1 probe derived from MED1 cDNA, as previously described.31 Clones that hybridized to the probe were identified, subcloned, and sequenced. The MED1 gene sequence was later confirmed by comparison with human genome project data. Location of the CpG sites was done with the program Map of the Genetics Computer Group (GCG) Software Package (Wisconsin Package Version 9.1).
Clinical specimens
Patient samples were procured from the Department of Surgery at UAB, following IRB-approved methods and informed consent. Thirty-nine human tissue specimens were collected at time of surgery. Samples included matched normal colon mucosa and tumor from each patient. Five specimens had synchronous colon adenomas that were also evaluated. Three patients had only adenomatous disease and no colorectal cancer was identified. Tissues were macrodissected, snap frozen in liquid nitrogen, and stored at −80°C. Normal colonic mucosa was obtained in the operating room from fresh, surgically resected colon specimens. Mucosa was harvested from the clinically negative margin of the specimens and macro-dissected from the colonic basement membrane prior to the preservation process. Tumor and polyp tissue were also harvested in the operating room from the clinical specimens. The tumors were grossly incised to obtain tissue. The amount of stroma tissue was not quantified in our specimens; it is known that colon adenocarcinomas are constituted by a majority of epithelial cells. Patients’ age ranged from 25 to 87 years old (mean age 61.0, median age 66). The group included 54% males and 46% females. Eighty percent of the patients were Caucasian and 20% were African-American (Table 1). Control tissue of normal colon mucosa was obtained from six patients with no history of CRC or polyps (Table 1).
Table 1.
Demographics of control and adenoma/CRC patients
| Control individuals | Adenoma/CRC patients | ||||||
|---|---|---|---|---|---|---|---|
| Age | <40 | 1 | 16.7% | Age | <40 | 7 | 17.9% |
| 40–60 | 3 | 50.0% | 40–60 | 8 | 20.5% | ||
| 60–80 | 2 | 33.3% | 60–80 | 21 | 53.8% | ||
| >80 | 0 | 0.0% | >80 | 3 | 7.7% | ||
| N= | 6 | 100.0% | N= | 39 | 100.0% | ||
| Sex | Male | 3 | 50.0% | Sex | Male | 21 | 53.8% |
| Female | 3 | 50.0% | Female | 18 | 46.2% | ||
| N= | 6 | 100.0% | N= | 39 | 100.0% | ||
| Race | Caucasian | 5 | 83.3% | Race | Caucasian | 31 | 79.5% |
| African American | 1 | 16.7% | African American | 8 | 20.5% | ||
| N= | 6 | 100.0% | N= | 39 | 100.0% | ||
| Stage | 0 | 2 | 5.1% | ||||
| I | 5 | 12.8% | |||||
| II | 9 | 23.1% | |||||
| III | 12 | 30.8% | |||||
| IV | 11 | 28.2% | |||||
| N= | 39 | 100.0% | |||||
| Tumor | |||||||
| Location | Ascending Colon | 17 | 43.6% | ||||
| Transverse Colon | 1 | 2.6% | |||||
| Descending Colon | 7 | 17.9% | |||||
| Rectum | 11 | 28.2% | |||||
| n/a | 3 | 7.7% | |||||
| N= | 39 | 100.0% | |||||
Identification and analysis of MED1 CpG island methylation
DNA was extracted from ovarian cancer cell lines, colorectal cancer cell lines, and clinical specimens using AquaPure Genomic DNA Isolation Kit (Bio-Rad, Hercules, CA). DNA from all specimens was subjected to a bisulfite reaction32 using the EZ DNA Methylation kit following the manufacturer’s recommendations (Zymo Research, Orange, CA). Products from the bisulfite reactions were subjected to PCR using the following primers designed to amplify a region of the MED1 promoter upon bisulfite conversion: 5′-GTTATTTTATAAGTTATTTTGGTTATT-3′ and 5′-CACCAATCAAATCCATTCTC-3′. PCR reactions contained 1x PCR buffer (Denville Scientific, Inc, Metuchen, NJ), 0.5mM dNTP mix, 0.5µM of each primer, and 0.04U/µL of Choice Taq Blue DNA Polymerase (Denville Scientific). After an initial denaturation step of 95°C for five minutes, DNA was amplified through 35 cycles of 30 seconds denaturing at 95°C, 30 seconds annealing at 54°C, and 30 seconds extension at 72°C. The products were resolved on a 1.6% agarose gel in TAE buffer and visualized by UV light after staining with ethidium bromide. PCR products were purified using PureLink PCR Purification Kit (Invitrogen, Carlsbad, CA). After purification, amplified products of the promoter region of MED1 were subcloned into pGEM-T Easy or pCR 2.1 vectors (the latter using TOPO TA cloning kit, Invitrogen), to produce PCR fragment libraries of mucosa and tumor for each matched specimen. Individual inserts were PCR amplified using M13 primers flanking the polylinker sites vectors to verify a size-appropriate insert. Correct size inserts were identified as described above and plasmids were directly sequenced using M13 (−20) reverse primer.
MED1 promoter demethylation in ovarian and CRC cell lines
OVCAR-4 and DLD-1 cells were grown to confluence, split 1:4 and treated with a 5µM final concentration of the demethylating agent 5-azacytidine in culture media (Sigma, St. Louis, MO) for five days.33 Every 24 hours the culture media was changed and supplemented with fresh medium containing 5-azacytidine. During the final 12 hours, cells were also treated with 500 nM trichostatin A (TSA) (Sigma) suspended in 100% EtOH. An equivalent amount of 100% EtOH was added to the control cells. Cells were viable at the end of the treatment cycle. DNA and RNA were extracted from both treated and control cell lines. Cytotoxicity assays for 5-azacytidine and TSA were not performed.
MED1 Gene Expression
RNA was extracted from specimens using Trizol reagent (Invitrogen) according to manufacturer’s protocol. cDNA was synthesized using High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). For mRNA expression analysis by quantitative real time PCR, TaqMan® Gene Expression assays (Applied Biosystems, Foster City, CA) were used for MED1 (Hs00187498_m1) and ribosomal S9 (forward-5′-ATCCGCCAGCGCCATA-3′, reverse-5′-TCAATGTGCTTCTGGGAATCC-3′, and probe-5′-6FAMAGCAGGTGGTGAACATCCCGTCCTTTAMRA-3′). Twenty-five microliter reactions contained 1x TaqMan® Universal PCR Master Mix, 1x expression assay, and 2µL of cDNA. S9 was used as the endogenous control for gene expression in specimens.34 A cDNA control stock was made using equal amounts of RNA from the six control patients: 500 ng of RNA from each patient were combined and cDNA was synthesized as described. This stock solution was used as a normal MED1 expression value for colon mucosa. MED1 and S9 expression were determined by measuring PCR product fluorescence compared to cycle number to determine CT values. Relative MED1 expression for each tissue sample (ΔCT = MED1 CT – S9 CT) was calculated so that matched specimens could be compared. The change in MED1 expression (ΔMED1) between normal and tumor specimens was calculated with the formula (ΔMED1) = 2^−(ΔNormal CT−ΔTumor CT). The change in MED1 expression was also evaluated between control mucosa and morphologically normal colon mucosa from cancer patients using a similar equation (ΔMED1) = 2^−(ΔControl CT−ΔNormal CT). All gene expression studies were performed in triplicate and repeated.
MED1 mutational analysis
Primers were designed to amplify exon 3 of MED1 including an A10 track representing codons 310–313 and three A6 tracks representing codons 247–248, 280–282, and 327–329. DNA was amplified using MED1 primers forward-5′-AAAGGTTAGAAAGCCCAAAGG-3′ and reverse 5′-GCAAATGTTCTTTCCTTTCCA-3′ following previously described conditions,28 purified with PureLink PCR Purification Kit (Invitrogen) and sequenced. Colorectal cancer cell line HCT116 was used as a positive control for screening of mutations at the A10 motif due to its previously described point deletion at this site.27,28
Statistical analysis
SPSS for Windows version 15.0 (SPSS Inc., Chicago, IL) was used for statistical analysis of patient data. Patient age, relative MED1 expression, and change in MED1 expression were given as medians (range) or means (standard deviation). Differences between control individual and cancer patients were compared using a t-test, Fischer’s exact test, or chi-squared test, as appropriate. Correlations for MED1 expression, methylation status, cancer stage, age, race, tumor histology and tumor location were performed using Spearman’s rho. Survival was evaluated using Kaplan-Meier plots. Significance was set to p < 0.05.
RESULTS
Analysis of MED1 gene expression using quantitative real time-PCR
MED1 expression was compared in normal mucosa and matched tumor from individual cancer patients to identify changes in gene expression associated with the development of sporadic CRC. Colorectal adenocarcinoma tissue showed decreased MED1 expression when compared to matched normal colon mucosa (Fig. 1). The median change in expression was −8.00 fold (+0.84 to −52.71). Following the paradigm of the adenoma-carcinoma sequence, we evaluated gene expression in adenomatous tissue to identify when these changes occur in CRC tumorigenesis. MED1 expression was decreased in colorectal adenomas (n = 8) when compared to matched normal mucosa. The median change in adenoma expression was −4.44 fold (−1.29 to −7.49). MED1 suppression was not limited to adenomatous tissue and tumor. A decrease in MED1 expression was also observed in normal mucosa from the cancer specimens when compared to normal mucosa from control patients (Fig. 2). The median change observed was −3.9 fold (+3.87 to −30.55). MED1 expression consistently decreased in colorectal cancer specimens as they progressed from normal mucosa to the invasive carcinoma phenotype.
Figure 1.

Relative expression of MED1 in adenomatous polyp or colorectal cancer specimens (N=39) compared to MED1 expression in matched normal mucosa from the same individual. (− = polyp, ● = stage I, ■ = stage II, ◆ = stage III, ▲= stage IV)
Figure 2.

Relative expression of MED1 in morphologically normal mucosa from patients with polyps or cancer (N=39) compared to MED1 expression in mucosa from non-cancer control patients. (− = polyp, ● = stage I, ■ = stage II, ◆ = stage III, ▲ = stage IV)
Evaluation of clinical specimens for previously described MED1 mutations associated with MSI+ tumors
To account for alternative mechanisms of reduced gene expression, DNA from clinical specimens and CRC cell lines DLD1 and HCT116 were evaluated for previously described MED1 mutations in exon 3. Four poly-adenine sites have been documented as known sites for frameshift mutations resulting in a prematurely truncated protein.27,28 MED1 mutational analysis of all 39 CRC patients revealed no mutations in the four poly-A mutational hotspots within exon 3 of the MED1 gene (data not shown). The positive control CRC cell line HCT116 did show a loss of a single adenine in the A10 sequence as previously described.28
Identification of MED1 gene promoter CpG islands
In order to isolate the promoter region of MED1, a probe derived from exon 1 of MED1 was used to screen a human genomic library. Several clones were isolated and subcloned into smaller fragments. Subcloned fragments hybridizing with the probe were sequenced, which allowed assembling of the tentative promoter region of the gene. In order to determine whether the promoter region of MED1 may contain a CpG island, we mapped the CpG sites by sequence analysis. Sequences were later confirmed by comparison with the Human Genome Project. Sequence analysis revealed a concentration of approximately 74 CpG sites centered around the exon 1 transcription start site comprising a putative CpG island within the promoter (Fig. 3).
Figure 3.

Schematic of MED1 promoter with transcription start site indicated by rightward arrow. Enlarged sequence represents amplified region of promoter with vertical hash marks signifying individual CpG sites. Preferentially methylated sites 14 and 16 are in bold.
Identification of MED1 promoter methylation
In the absence of mutations, promoter methylation was evaluated as a cause of MED1 downregulation in our clinical specimens. We focused on the 22 CpG sites most proximal to the transcription start site. To determine whether these CpG sites in the proximal region of the MED1 promoter were methylated, genomic DNA was first treated with sodium bisulfite, a reaction that exclusively converts unmethylated cytosines into uracils. Methylated cytosines are protected from the reaction allowing identification of methylated sites.32 Analysis of DNA methylation in the MED1 promoter from our six ovarian cell lines, nine colorectal cell lines and 39 patient specimens revealed preferential methylation of CpG sites 14 and 16 of the 22 sites evaluated from the MED1 promoter region. DLD1 and OVCAR-4 were the only cell lines that consistently showed methylation of the MED1 promoter. Methylation was not restricted to tumor tissue in patients with CRC. When methylation was present in clinical subjects, methylation was observed in both normal mucosa and matched colorectal adenocarcinoma (Fig. 4). Methylation of the MED1 promoter was consistently detected in both cell lines and clinical specimens.
Figure 4.
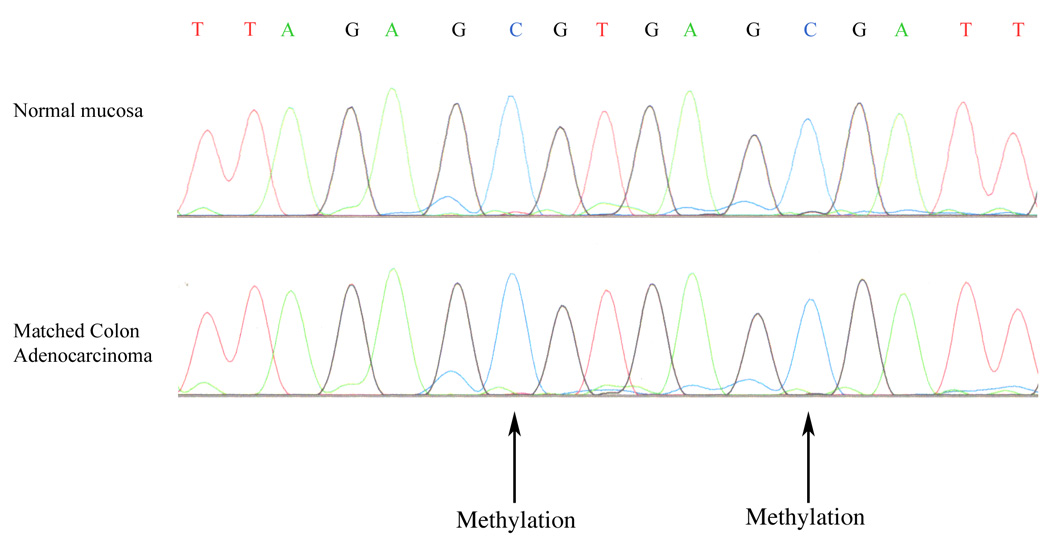
Examples of methylation of the MED1 promoter in normal colon mucosa and matched colon adenocarcinoma. Methylation of CpG sites 14 and 16 (arrows) is indicated by preservation of cytosine nucleotides in PCR sequences of both normal mucosa and tumor from the same patient.
Frequency of methylation in cell lines and clinical specimens
In order to derive semiquantitative measurements of the extent of DNA methylation of the MED1 promoter, PCR fragments after bisulfite treatment were subcloned in plasmids and used to transform bacteria. Independent bacterial colonies (ranging in number from 5 to 26) from each sample were sequenced, thus allowing an estimation of the frequency of methylation. Frequency of methylation revealed a wide range in cell lines and patients. Promoter methylation in ovarian cell lines ranged from 22% to 75%, and in colon cancer clinical specimens ranged from 0% to 100%. The average frequency of methylation in ovarian cell lines was 50% (± 18%) and in colon cancer patients was 24% (± 19%). Colon mucosa from the six control patients with no history of cancer was evaluated and consistently showed low frequency of methylation. The average frequency of methylation for these control patients was 14% (± 9%). When frequency of methylation was compared between cancer patients and control patients by t-test, these frequencies were significantly different (p<0.001).
Evaluation of demethylation on MED1 gene expression
As mentioned, MED1 methylation was detected both in cell lines and in clinical specimens. To determine the effect of this epigenetic change on the MED1 transcription, experiments were conducted to demethylate the promoter and subsequently evaluate gene expression. Cells were treated with the demethylating agent 5-azacytidine (AZA) and the potent histone deacetylase inhibitor trichostatin A (TSA). The addition of TSA has been shown to act synergistically with DNA demethylating agents in reactivating gene expression.35 Sequencing of ovarian cell line OVCAR-4 and CRC cell line DLD-1 had previously revealed methylation at sites 14 and 16. Treatment of these two cell lines with AZA and TSA resulted in a 2.5 and 7.8 fold increase, respectively, in MED1 expression when compared to baseline levels.
Statistical analysis
Correlations were performed in CRC patients between MED1 expression, frequency of methylation, and clinico-pathological variables, such as age, sex, tumor stage, race, anatomic location, lymph node status, and survival. Statistical analysis revealed a trend towards more advanced tumor stage and decreased MED1 expression, though this did not reach statistical significance (p=0.081). Frequency of methylation negatively correlated with patient age (p<0.05). No correlation with tumor location was detected.
DISCUSSION
Our findings demonstrate that expression of the tumor suppressor gene MED1 is decreased in sporadic colorectal cancer and that it can be attributed to promoter methylation. We suggest that MED1 is suppressed in a large proportion of sporadic colorectal cancers in the absence of known MED1 mutations. Additionally, there is a significantly lower incidence of MED1 promoter methylation in normal control patients when compared to patients with colorectal cancer. Our in vitro studies show that reversal of MED1 methylation leads to increased gene expression, suggesting that methylation does contribute to gene suppression. The importance of these findings stem from the fact that MED1 methylation and gene suppression seem to develop in normal colon mucosa prior to the development of the adenoma-carcinoma sequence. The epigenetic regulation of the tumor suppressor gene MED1 thus seems to be associated with tumor initiation rather than tumor progression. This is corroborated by the lack of a significant decrease in MED1 expression correlated to more advanced tumor stages that would imply a role in tumor progression. Gene suppression can be detected in the normal colon mucosa of tumor-bearing patients as well as in polyps and carcinomas from matched specimens.
MED1 promoter methylation observed in tumor tissue is also present in adjacent, matched normal mucosa. This phenomenon of methylated tumor suppressor genes in tumor and adjacent normal mucosa has previously been reported in sporadic CRC and may reflect a “field effect” involving the mucosal lining of CRC patients. Epigenetic changes have been implicated as an early event in CRC tumorigenesis.12,36 We propose that a similar mechanism involving MED1 plays a role in sporadic CRC tumor initiation. The resulting CRC patient decrease in MED1 gene expression can be readily detected in the associated normal mucosa when compared to control samples. Normal colonic mucosa MED1 gene methylation and suppression has also been reported for the MLH1 promoter.37 The 56% increase in the amount of MED1 promoter methylation observed in mucosa from cancer patients further supports the role of promoter methylation as a plausible cause of MED1 suppression in these patients.
Our in vitro studies demonstrated the reversibility of MED1 promoter methylation restoring MED1 gene expression, also in concordance with the MLH1 methylation reports.38 The reversibility of MED1 expression in ovarian and CRC cell lines suggests that observed promoter methylation does affect gene expression in at least two different types of epithelial cancers. If tissue methylation causes an initial drop in MED1 expression within normal mucosa, it poses the question as to what additional changes are necessary to induce the downregulation of MED1 expression detected in carcinomas. As suggested by Jones and Laird, promoter methylation in MED1 may require a second “hit” to see the ultimate degree of gene suppression observed in tumors. We speculate that methylation may trigger the initial decrease in gene expression and this is followed by mutations or LOH causing greater gene silencing and the development of cancer. An initial decrease in MED1 expression is seen when DNA from normal mucosa is methylated. This process could enable subsequent genetic changes that would further decrease MED1 expression, as described in polyps and carcinomas.39
The importance of decreased MED1 in CRC tumorigenesis has been demonstrated in mouse models with MED1/MBD4 targeted gene inactivation.30,40 Mice with MED1 knockout in the setting of an Apc Min/+ background had a significantly reduced survival and increased CRC tumor burden compared to control mice.30,40 Work from our and other laboratories suggests an additional role for MED1 inactivation in tumorigenesis.20,21,41 These studies suggest that decreased MED1 protein levels have an inhibitory effect on apoptosis. These concepts support our findings. The early changes that we have observed in MED1 expression in colon mucosa from cancer patients suggest that either decreased genomic surveillance, decreased apoptosis, or both, could lead to initiation of tumorigenesis.
A high frequency of MED1 methylation in cancer patients and the ability to demethylate and restore gene expression in cell lines does implicate a role for methylation in the silencing of this putative tumor suppressor gene. Further, comparison of frequently methylated DNA from CRC patients with DNA from non-diseased patients and their consistently low frequency of methylation status suggest that hypermethylation of the MED1 promoter is an abnormal occurrence in CRC tumorigenesis. The significance of this finding is that it appears to be an early occurrence in the development of CRC similar to other genes involved in the development of sporadic colorectal cancer.12
In conclusion, two observations suggest that MED1 promoter methylation and gene suppression are important in CRC tumorigenesis. First, methylation occurs in cancer patients at higher frequencies than those found in patients without cancer. Second, and perhaps most importantly, MED1 suppression seems to occur first at the level of the normal mucosa. The suppression of MED1 follows the adenoma-carcinoma sequence and is observed in normal mucosa, adenomas, and adenocarcinoma of the colon suggesting an early and detectable change. Suppression of MED1 expression and early detection of promoter methylation may necessitate a more frequent and rigorous screening repertoire in this population of patients leading to earlier detection of malignant changes.
Acknowledgments
We thank S. Cortellino and M. Sannai for critical reading of the manuscript; P. Barth and J. Doss for help with DNA sequencing; and R. Sonlin for secretarial assistance. We thank the following core facilities and personnel at the Fox Chase Cancer Center: Cell Culture (S. Howard), Sequencing (A. Cywinski), and the Fannie E. Rippel Biotechnology Facility (P. Do, G. Miller and A. Yeung). This work was supported by NIH grants CA78412 and CA06927, the Robert E. Reed Gastrointestinal Oncology Research Foundation and an appropriation from the Commonwealth of Pennsylvania to the Fox Chase Cancer Center.
REFERENCES
- 1.United States Cancer Statistics: 2003 Incidence and Mortality (preliminary data) US Cancer Statistics Working Group; 2006. [Google Scholar]
- 2. www.census.gov.
- 3.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 4.Pal T, Permuth-Wey J, Betts JA, Krischer JP, Fiorica J, Arango H, LaPolla J, Hoffman M, Martino MA, Wakeley K, Wilbanks G, Nicosia S, Cantor A, Sutphen R. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104:2807–2816. doi: 10.1002/cncr.21536. [DOI] [PubMed] [Google Scholar]
- 5.Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341–348. doi: 10.1503/cmaj.050774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 7.Singer-Sam J, Riggs AD. X chromosome inactivation and DNA methylation. Exs. 1993;64:358–384. doi: 10.1007/978-3-0348-9118-9_16. [DOI] [PubMed] [Google Scholar]
- 8.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 9.Makarla PB, Saboorian MH, Ashfaq R, Toyooka KO, Toyooka S, Minna JD, Gazdar AF, Schorge JO. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res. 2005;11:5365–5369. doi: 10.1158/1078-0432.CCR-04-2455. [DOI] [PubMed] [Google Scholar]
- 10.Jones PALP. Cancer Epigenetics Comes of Age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 11.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HC, Roh SA, Ga IH, Kim JS, Yu CS, Kim JC. CpG island methylation as an early event during adenoma progression in carcinogenesis of sporadic colorectal cancer. J Gastroenterol Hepatol. 2005;20:1920–1926. doi: 10.1111/j.1440-1746.2005.03943.x. [DOI] [PubMed] [Google Scholar]
- 13.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci U S A. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Rijnsoever M, Elsaleh H, Joseph D, McCaul K, Iacopetta B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 16.Bai AH, Tong JH, To KF, Chan MW, Man EP, Lo KW, Lee JF, Sung JJ, Leung WK. Promoter hypermethylation of tumor-related genes in the progression of colorectal neoplasia. Int J Cancer. 2004;112:846–853. doi: 10.1002/ijc.20485. [DOI] [PubMed] [Google Scholar]
- 17.Geisler JP, Goodheart MJ, Sood AK, Holmes RJ, Hatterman-Zogg MA, Buller RE. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer. 2003;98:2199–2206. doi: 10.1002/cncr.11770. [DOI] [PubMed] [Google Scholar]
- 18.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 19.Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999;96:3969–3974. doi: 10.1073/pnas.96.7.3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, Clarke AR, Frisch SM. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proc Natl Acad Sci U S A. 2003;100:5211–5216. doi: 10.1073/pnas.0431215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005;25:4388–4396. doi: 10.1128/MCB.25.11.4388-4396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parsons BL. MED1: a central molecule for maintenance of genome integrity and response to DNA damage. Proc Natl Acad Sci U S A. 2003;100:14601–14602. doi: 10.1073/pnas.2637169100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sansom OJ, Bishop SM, Bird A, Clarke AR. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene. 2004;23:5693–5696. doi: 10.1038/sj.onc.1207767. [DOI] [PubMed] [Google Scholar]
- 25.Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000;275:32422–32429. doi: 10.1074/jbc.M004535200. [DOI] [PubMed] [Google Scholar]
- 26.Bellacosa A. Role of MED1 (MBD4) Gene in DNA repair and human cancer. J Cell Physiol. 2001;187:137–144. doi: 10.1002/jcp.1064. [DOI] [PubMed] [Google Scholar]
- 27.Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, Wyllie A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene. 1999;18:8044–8047. doi: 10.1038/sj.onc.1203229. [DOI] [PubMed] [Google Scholar]
- 28.Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, Ruggeri BA, Klein-Szanto AJ, Testa JR, Neri G, Bellacosa A. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet. 1999;23:266–268. doi: 10.1038/15443. [DOI] [PubMed] [Google Scholar]
- 29.Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton SR, Vogelstein B, Kinzler KW. Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med. 1996;2:169–174. doi: 10.1038/nm0296-169. [DOI] [PubMed] [Google Scholar]
- 30.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 31.Bellacosa A, Datta K, Bear SE, Patriotis C, Lazo PA, Copeland NG, Jenkins NA, Tsichlis PN. Effects of provirus integration in the Tpl-1/Ets-1 locus in Moloney murine leukemia virus-induced rat T-cell lymphomas: levels of expression, polyadenylation, transcriptional initiation, and differential splicing of the Ets-1 mRNA. J Virol. 1994;68:2320–2330. doi: 10.1128/jvi.68.4.2320-2330.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 34.Mukherjee S, Frolova N, Sadlonova A, Novak Z, Steg A, Page GP, Welch DR, Lobo-Ruppert SM, Ruppert JM, Johnson MR, Frost AR. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol Ther. 2006;5:674–683. doi: 10.4161/cbt.5.6.2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 36.Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Grieu F, Watanabe G, Iacopetta B. DNA hypermethylation in the normal colonic mucosa of patients with colorectal cancer. Br J Cancer. 2006;94:593–598. doi: 10.1038/sj.bjc.6602940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nuovo GJ, Nakagawa H, Sotamaa K, Chapelle Ade L. Hypermethylation of the MLH1 promoter with concomitant absence of transcript and protein occurs in small patches of crypt cells in unaffected mucosa from sporadic colorectal carcinoma. Diagn Mol Pathol. 2006;15:17–23. doi: 10.1097/00019606-200603000-00003. [DOI] [PubMed] [Google Scholar]
- 38.Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S, Li GM, Drummond J, Modrich PL, Sedwick WD, Markowitz SD. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 40.Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, Edelmann W. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci U S A. 2002;99:14937–14942. doi: 10.1073/pnas.232579299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR. MBD4 deficiency reduces the apoptotic response to DNA-damaging agents in the murine small intestine. Oncogene. 2003;22:7130–7136. doi: 10.1038/sj.onc.1206850. [DOI] [PubMed] [Google Scholar]