

Abstract
The enteric bacterium Escherichia coli is the most extensively used prokaryotic organism for production of proteins of therapeutic or commercial interest. However, it is common that heterologous over-expressed recombinant proteins fail to properly fold resulting in formation of insoluble aggregates known as inclusion bodies. Complex systems have been developed that employ simultaneous over-expression of chaperone proteins to aid proper folding and solubility during bacterial expression. Here we describe a simple method whereby a protein of interest, when fused in frame to the E. coli chaperones DnaK or GroEL, is readily expressed in large amounts in a soluble form. This system was tested using expression of the mouse prion protein PrP, which is normally insoluble in bacteria. We show that while in trans over-expression of the chaperone DnaK failed to alter partitioning of PrP from the insoluble inclusion body fraction to the soluble cytosol, expression of a DnaK-PrP fusion protein yielded large amounts of soluble protein. Similar results were achieved with a fragment of insoluble Varicella Zoster virus protein ORF21p. In theory this approach could be applied to any protein that partitions with inclusion bodies to render it soluble for production in E. coli.
Keywords: DnaK, GroEL, PrP, DnaJ/GrpE, GroES
1. Introduction
Demand for proteins required for various molecular biology and biotechnology applications cannot be met from natural reservoirs. Fortunately, the universality of the genetic code enables protein expression in convenient organisms. The enterobacterium Escherichia coli (E. coli) is frequently a first choice for production of recombinant proteins because of its ease of culture, rapid growth on inexpensive carbon sources, amenability to high cell density fermentations and the extensive knowledge of its genetic and physiological processes (Gold, 1990; Hodgson, 1993; Olins and Lee, 1993; Hockney, 1994; Makrides, 1996). However, despite its popularity, not every protein can be efficiently expressed in this organism. Production of large amounts of recombinant proteins in E. coli is often burdened by a number of problems, including stability and translation efficiency of mRNA, degradation of the protein by host cell proteases and product-induced toxicity (Makrides, 1996). The most common problem, however, is that many recombinant proteins fail to reach and maintain their native conformation when overproduced in E. coli (Makrides, 1996). Accordingly, the yield of these recombinant proteins is low.
Over-expression of recombinant proteins in the crowded milieu of the E. coli cytoplasm, where transcription and translation are tightly coupled, makes folding an extraordinary challenge. Although small, single domain proteins often spontaneously reach their native conformation, expression of more complex and heterologous proteins frequently results in kinetically trapped, slow-folding, nonproductive intermediates that are prone to aggregate (Baneyx and Mujacic, 2004). The latter results from interaction of exposed hydrophobic surfaces within the proteins’ partially unfolded structure. These regions are normally buried inside the core of a properly folded native protein. However, failure to correctly fold recombinant proteins results in their exposure and free interaction with other hydrophobic constituents of the crowded intracellular environment (Wetzel, 1994; Baneyx and Mujacic, 2004; Fahnert et al., 2004). Subsequently, these misfolded proteins are usually deposited as dense refractile particles called inclusion bodies or degraded by bacterial proteolytic systems (Wetzel, 1994; Baneyx and Mujacic, 2004; Fahnert et al., 2004). Proteins deposited into inclusions bodies require complex solubilization and refolding procedures to gain biological activity (Fahnert et al., 2004).
Bacterial chaperone proteins facilitate conformational processing of a significant fraction of de novo synthesized polypeptides by preventing nonproductive hydrophobic interactions and by helping them acquire their correct tertiary conformation (Hartl, 1996; Fink, 1999; Hartl and Hayer-Hartl, 2002; Young et al., 2004). E. coli employs two major chaperone systems, the DnaK (Hsp70) system, composed of DnaK and its co-chaperones DnaJ and GrpE, and the GroEL (Hsp60)/GroES system (Hartl, 1996; Fink, 1999; Hartl and Hayer-Hartl, 2002; Young et al., 2004). Expression of these proteins is positively regulated under elevated temperatures or other forms of cellular stress that affect protein conformation (Lemaux et al., 1978; Hoffmann and Rinas, 2004a; Hoffmann and Rinas, 2004b). Chaperones are protein machines that bind to nonnative proteins and rely on ATP-driven conformational changes to mediate refolding/unfolding of their substrates (Hartl, 1996; Fink, 1999; Hartl and Hayer-Hartl, 2002; Young et al., 2004).
It has been speculated that overproduction of a slow-folding recombinant protein in E. coli may overwhelm the cell’s chaperones, leading to accumulation of aggregates as inclusion bodies (Georgiou and Valax, 1996). Expression of large amounts of chaperones can overcome this limitation and benefit folding of recombinant proteins (Georgiou and Valax, 1996; Schlieker et al., 2002). This idea was first demonstrated by increasing the solubility of Rubisco, a large oligomeric protein, in cells that over-expressed GroEL and GroES (Goloubinoff et al., 1989). Subsequently, a number of increasingly elaborate plasmid systems were developed that permit co-expression of various combinations of chaperones, or other facilitators of protein folding to allow production of large amounts of proteins of interest (Perez-Perez et al., 1995; Nishihara et al., 1998; Vonrhein et al., 1999; Yanase et al., 2002; Stevens et al., 2003; Xu et al., 2005; Schlapschy et al., 2006; de Marco et al., 2007). Although over-expression of substrate-specific chaperone and co-chaperone combinations improves folding of some recombinant proteins, this sometimes results in bacterial toxicity and leads to decreased protein yield (Blum et al., 1992).
Here we describe development of a system that utilizes bacterial chaperones to facilitate production of large amounts of soluble recombinant proteins. This system utilizes a vector where the sequence of a target polypeptide is placed in frame at the carboxy-terminus of either DnaK or GroEL. A poly-histidine carboxy-terminal tag is present for rapid affinity purification of the fusion protein and a thrombin cleavage site is placed at the fusion junction to liberate the protein of interest from its chaperone partner. The system was evaluated using the mouse prion protein (PrP), which is involved in the protein folding disease spongiform encephalopathy, and Varicella Zoster virus (VZV) ORF21p as targets. Both proteins are normally insoluble when expressed in bacteria. We demonstrate that the chaperone fusions are soluble and when these fusion proteins are co-expressed with their cognate co-chaperone solubility is further increased.
2. Materials and Methods
2.1. Bacterial strains
E. coli strains used in this study were N99, Top10F′ and BL21[DE3]. All strains were grown at 37°C on 2xYT agar plates or with aeration in 2xYT broth, supplemented with 150μg/ml Ampicillin, 50μg/ml Kanamycin and/or 25μg/ml Chloramphenicol as necessary.
2.2. Isolation of bacterial genomic DNA
Pellets from overnight bacterial cultures were suspended in TE buffer (10mM TrisHCl pH8.0 and 1mM EDTA). Sodium dodecyl sulfate (SDS) was added to a final concentration of 0.6% w/v and the solution was incubated with Proteinase K (0.12mg/ml) for 1 hr at 37°C. Nucleic acids were extracted with phenol/chroroform and precipitated following addition of isopropanol, before treatment with RNaseA. DNA was re-extracted, precipitated, dried and suspended in TE.
2.3 Isolation of Varicella Zoster Virus DNA
Virus DNA was prepared using purified nucleocapsids as previously described (Kyratsous and Silverstein, 2007).
2.4. Plasmid construction
(i) DnaK plasmids: The dnaK coding sequence was amplified from E. coli, strain N99 genomic DNA using the oligonucleotide primers DnaK-FW: 5′ – GGCATATGGGTAAAATAATTGGTATCG – 3′ and DnaK-RV: 5′ – GGGGGATCCGAGCCGCGTGGGACTAATTCAAATTCAGCGTAGACAAC – 3′. The PCR product was cloned into pDrive (Qiagen) to yield pD-DnaK. The DnaK gene was released with NdeI/BamHI and cloned into NdeI/BamHI digested pET-21a(+) to produce pXCK-K. (ii) GroEL plasmids: GroEL was amplified from pUCE using the oligonucleotide primers GroEL-FW: 5′ – GGCATATGGCAGCTAAAGACGTA – 3′ and GroEL_RV: 5′ – GGGAGATCTGAGCCACGAGGGACTAAGTCAGCTGCATCGTT – 3′. The PCR product was cloned into EcoRV digested pZero2.1 (Invitrogen) to yield pZ-GroEL. pXCK-EL was constructed by cloning the NdeI/BglII digestion fragment from pZ-GroEL into NdeI/BamHI digested pET-21a(+). (iii) PrP plasmids: The gene encoding murine PrP was amplified from pVPmPrP14 (a gift from Dr Sklaviadis, Aristotle University, Thessaloniki, Greece) using mPrP-FW: 5′ – GGGCTAGCAAAAAGCGGCCAAAGCCT – 3′ and mPrP-RV: 5′ – GGCGCGGCCGCGGATCTTCTCCCGTCGTA – 3′ and cloned into EcoRV digested pZero2.1 to yield pZeTA-PrP. PrP sequences were released by NheI/NotI digestion and cloned into NheI/NotI digested pET-21a(+) to construct pX-PrP. pZ-PrP was made by cloning PCR amplified mouse PrP from pVPmPrP14, using as primers mPrP-FW2: 5′ – GGCAAGCTTAAAAAGCGGCCAAAGCCT – 3′ and mPrP-RV2: 5′ – GGCGCGGCCGCGGATCTTCTCCCGTCGTA – 3′, into EcoRV digested pZero2.1. pX-DnaK-PrP and pX-GroEL-PrP were constructed by releasing PrP from pZ-PrP following NotI/HindIII digestion and cloning into NotI/HindIII digested pXCK-K and pXCK-EL respectively. (iv) ORF21 plasmids: The sequence of VZV ORF21 encoding for aa878-1038 was amplified from virus DNA using ORF21for: 5′ – GGGAATTCGGGTCTTGCAAATGTAGAGATTT – 3′ and ORF21rev: 5′ – TGCGGCCGCAGGGTCACTCCCACTTGTAT – 3′. The PCR product was cloned into pCR2.1-TOPO to yield pTOPO-ORF21. The ORF21 coding sequence was released by digesting with EcoRI and NotI and cloned into EcoRI and NotI digested pALEX (Panagiotidis and Silverstein, 1995) to yield pALEX-ORF21. pDnaK-ORF21 was constructing by cloning the XhoI/HindIII fragment from pTOPO-ORF21 into the XhoI/HindIII digested pXCK-K. (v) co-chaperone plasmids: GrpE and DnaJ were amplified from N99 genomic DNA using the oligonucleotide primers GrpE-FW: 5′ – GCCCATGGGCGGAGAAATTCATGAGTA – 3′, GrpE-RV: 5′ – GGGCTAGCTACGAAAGCAGAAATTAAGC – 3′ and DnaJ-FW: 5′ – GGGCTAGCAAAAATAATCGCCCTATAAA – 3′, DnaJ-RV: 5′ – GGCTCGAGGGAGGTTAGCGGGTCA – 3′. The two PCR products were cloned into EcoRV digested pZero2.1 to yield pZeTA-GrpE and pZeTA-DnaJ respectively. pX-GrpE was constructed by cloning an NcoI/EcoRI fragment from pZeTA-GrpE into NcoI/EcoRI digested pET-21d(+). DnaJ was released from pZeTA-DnaJ with NheI/XhoI and cloned into NheI/XhoI digested pX-GrpE to yield pX-GrpE-DnaJ. The GrpE/DnaJ operon was released as an EcoRV/XhoI fragment from pX-GrpE-DnaJ and cloned into EcoRV/SalI digested pLysE to yield pXCK-E/J. GroES was amplified from N99 genomic DNA using the oligonucleotide primers: GroES-FW: 5′ – GGCCATGGCAAAGGAGAGTTATCAATG – 3′ and GroES-RV: 5′ – GGGCTAGCTTACGCTTCAACAATTGCC – 3′ and cloned into EcoRV digested pZero2.1 to yield pZeTA-GroES. GroES was released with NcoI/EcoRI and cloned into NcoI/EcoRI digested pET-21d(+) to yield pX-GroES. pXCK-ES was constructed by cloning a EcoRV/XhoI digestion fragment from pX-GroES into EcoRV/SalI digested pLysE.
2.5. Protein purification
Proteins tagged with 6His were purified using Ni-NTA (Qiagen) according to the manufacturer’s instruction.
2.6. Solubility assay
Overnight bacterial cultures were used to inoculate 50ml of fresh media. When the cultures reached an OD≈0.5 protein expression was induced by adding Isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.5mM and then bacteria were harvested and washed twice with phosphate-buffered saline (PBS; 1mM KH2PO4, 10mM Na2HPO4, 137mM NaCl, 2.7mM KCl, pH7.4). The bacterial pellets were suspended in PBS supplemented with 10mM β-mercaptoethanol, 1mM EDTA, 0.2mg/ml lysozyme and complete protease inhibitors (Roche). After stirring at 4°C for 30min Triton X-100 was added to a final concentration of 1% and stirring was continued for an additional 5min. The lysates were sonicated 3 times for 20sec each at 4°C and then total protein concentrations were normalized between the various preparations. A portion of each lysate (total lysate) was reserved for further analysis after addition of 5x SDS sample buffer (250mM TrisHCl pH 6.8, 500mM DTT, 10% SDS, 0.5% bromophenol blue, 50% glycerol) to a final concentration of 1.5x. A separate portion of each lysate was centrifuged at 12,000×g for 30min at 4°C. The supernatants (soluble lysates) were saved after addition of SDS sample buffer to 1.5x. The pellets (inclusion bodies) were suspended in an equivalent volume of 1.5x SDS sample buffer.
2.7 Antibodies and western blotting
Mouse monoclonal anti-His antibody was purchased from Sigma. Proteins analyzed by SDS-PAGE were transferred to nitrocellulose membranes before western blotting as previously described (Kyratsous and Silverstein, 2009).
2.8. Autophosphorylation assay
Autophosphorylation of DnaK and the DnaK-PrP fusion were performed as described (Panagiotidis et al., 1994).
3. Results and Discussion
3.1. Construction of fusion vectors
DNA vectors expressing either DnaK or GroEL and compatible plasmids expressing their cognate co-chaperones (DnaJ, GrpE and GroES) were constructed as described in Materials and Methods. mRNA sequences encoding the chaperone proteins specified by these DNA constructs (pXCK-K and pXCK-EL) are transcribed from an inducible T7 promoter. The resulting chaperone proteins have a thrombin cleavage site and a 6His tag at their carboxy termini (Fig. 1A, B and C). A multiple cloning site is present between the DNA sequences encoding a thrombin cleavage site and the 6His tag (Fig. 1A, B and C). The thrombin site should permit release of a protein of interest from the fusion and the 6His tag is there to facilitate purification. DnaK and GroEL were expresssed from identical plasmid vectors to minimize any effects from expression in different backgrounds (Fig. 1A). Each DNA construct was analyzed for its ability to direct synthesis of the cloned chaperone or co-chaperone following induction. Cell lysates were prepared and subjected to SDS-polyacrylamide gel electrophoresis. Following staining with Coomassie Brilliant Blue abundant bands corresponding to each purported protein were detected (Fig. 1D). With the exception of DnaJ, all of the proteins accumulated in large amounts and migrated with the expected mobility. Thus, inducible DNA constructs expressing the chaperones DnaJ and GroEL and their cognate co-chaperones were constructed.
Figure 1. DNA vectors expressing either DnaK or GroEL and their cognate co-chaperone partners.

(A) Plasmid maps of DNA constructs expressing DnaK (pXCK-K), GroEL (pXCK-EL), DnaJ and GrpE (pXCK-E/J) and GroES (pXCK-ES). DnaK and GroEL were cloned into identical plasmid vectors to minimize any possible effects from expression in different backgrounds. (B) Schematic diagram of the heat shock fusion protein showing the position of the thrombin cleavage site and 6His tag. (C) DNA sequence of the multiple-cloning restriction sites between the sequence encoding the thrombin cleavage site and the 6His tag. (D) BL21[DE3] cells were transformed with the indicated plasmids and protein expression was induced with IPTG. Cells were suspended in SDS-lysis buffer and proteins were analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue.
3.2. Fusion of PrP to DnaK and GroEL results in expression and accumulation of soluble PrP
Development of a useful expression system requires accumulation of soluble protein that is otherwise normally found only within inclusion bodies. To test our system the insoluble mouse prion protein (PrP) was expressed in E. coli on its own or as a fusion with DnaK and GroEL with in trans expression of cognate and unrelated co-chaperones. In each case the abundance of PrP in the total, soluble and insoluble (inclusion bodies) fractions was evaluated. Induced expression of each construct resulted in accumulation of significant levels of PrP, PrP fused to DnaK and PrP fused to GroEL. (Fig. 2A). Importantly, co-expression of the cognate or unrelated in trans co-chaperone did not significantly alter fusion protein levels (Fig 2A). While large amounts of PrP accumulate in induced cells (Fig. 2A) the protein does not partition with the soluble fraction (Fig. 2B), but is exclusively found in insoluble inclusion bodies (Fig. 2C). In contrast, when this protein was fused to DnaK or GroEL soluble proteins were readily detected (Fig. 2B). Fusion of PrP to DnaK consistently increased protein recovery in the soluble fraction when compared to the GroEL fusion (Fig. 2B and data not shown). Co-expression of DnaJ and GrpE resulted in less insoluble DnaK fusion protein, whereas expression of GroES had no affect (Fig. 2C). Similarly, co-expression of GroEL-PrP with GroES increased its solubility but co-expression with DnaJ/GrpE had no affect (Fig. 2B). These observations suggested that increased solubility of chaperone fusions was a result of their chaperone activity, which is enhanced by simultaneous expression of their cognate partners, but unaffected by non-cognate co-chaperones. Finally, we note that while PrP was exclusively found associated with inclusion bodies only a fraction of this protein remained insoluble after fusion to DnaK or GroEL (Fig. 2B and C). Thus fusion of PrP to DnaK or GroEL resulted in accumulation of large amounts of a soluble form of PrP. Furthermore, because DnaK fusion protein was more soluble than GroEL fusion protein we concentrated our studies on DnaK as the fusion partner.
Figure 2. PrP partitions in the soluble fraction when expressed as a chaperone fusion.
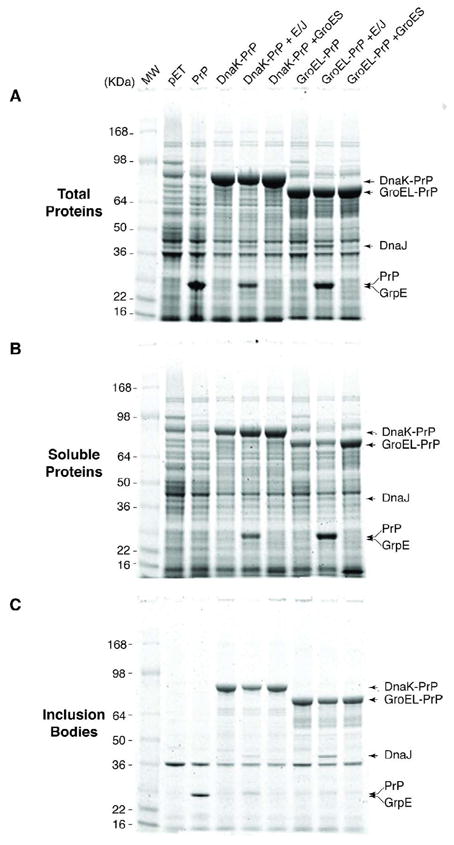
BL21[DE3] cells were transformed with the indicated plasmids and protein expression was induced with 1mM IPTG. Cell lysates were prepared and proteins were fractionated, analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue. (A) Total cell extract, (B) Soluble fraction and (C) Inclusion bodies.
3.3. VZV ORF21 accumulates as soluble protein when expressed a DnaK fusion
To test the generality of this method, we evaluated the solubility of a fragment of VZV ORF21p expressed as a fusion to DnaK. In this experiment, we also compared the efficiency of the DnaK fusion with a glutathione S-transferase (GST) fusion, another common expression and affinity purification tag (Smith and Johnson, 1988). ORF21p was expressed in E. coli as a fusion with GST and DnaK and the abundance of the fusion proteins in the total, soluble and insoluble fractions was measured (Fig. 3). Although large amounts of GST-ORF21p accumulated as evidenced by its level in the total extract, only very small amounts of the protein were soluble. In contrast, the DnaK fusion to the same protein accumulated in high levels as soluble protein. This experiment clearly demonstrates that increased solubility of DnaK fusions is not a unique property of PrP. Importantly; it also extends the utility of this method to proteins that when over-expressed in bacteria precipitated as an inclusion body when using common expression tags such as GST.
Figure 3. VZV ORF21p partitions in the soluble fraction when expressed as a chaperone fusion.

BL21[DE3] cells were transformed with the indicated plasmids and protein expression was induced with 1mM IPTG. Cell lysates were prepared and proteins were fractionated and analyzed by western blotting.
3.4. The thrombin site in a DnaK-PrP fusion protein is available for cleavage
The availability of a thrombin cleavage site at the DnaK-PrP border would be useful for preparing PrP free of its fusion partner. Therefore we examined accessibility of DnaK-PrP to cleavage by thrombin as a function of time. DnaK-PrP protein was purified by affinity chromatography using its 6His-tag. Purified protein was then incubated with thrombin and aliquots were withdrawn over time for analysis by SDS-PAGE (Fig. 4A). This analysis revealed that by 30 min more than 50% of the fusion protein was cleaved resulting in accumulation of DnaK and PrP as unique species. Cleavage was essentially complete after 3 hrs. Interestingly, cleaved PrP remained in solution and did not precipitate over time, suggesting that expression as a fusion to DnaK enabled it to attain its native hydrophilic conformation.
Figure 4. (A) The DnaK-PrP is efficiently cleaved by thrombin.

DnaK and DnaK-PrP were affinity purified using Ni-NTA chromatography. DnaK-PrP was incubated with thrombin for the indicated times. The proteins were analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue. (B) DnaK has enzymatic activity when fused to PrP. DnaK and DnaK-PrP were affinity purified by Ni-NTA chromatography. Equal amounts of each protein were used in autophosphorylation reactions for the indicated times. The products of the reactions were analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue (top panel). The dried gel was exposed to a phosphorimager cassette to assess 32P incorporation (bottom panel).
The components of the protein mix following thrombin cleavage are DnaK and PrP-6His (Fig. 4A). The properties of each protein are useful for separation of the two constituents. PrP-6His can be purified using a second Ni-NTA affinity column and because DnaK binds ATP, it can be removed by affinity purification on an ATP-agarose column (Welch and Feramisco, 1985; Zylicz et al., 1987). We noted that thrombin cleavage of the DnaK-PrP fusion protein on the Ni-NTA column prior to elution was inefficient and significantly reduced the yield of recombinant PrP (data not shown).
Therefore this expression system is useful for production and purification of a normally insoluble protein as a soluble protein, free of any fusion partners that might burden further functional analysis of the recombinant protein of interest.
3.5. Preservation of DnaK enzymatic activity in a DnaK-PrP fusion protein
The observations that DnaK-PrP was soluble led us to ask if the chaperone retained biologic activity. DnaK possesses ATPase activity and it can also autophosphorylate (Zylicz et al., 1983). We used autophosphorylation of the protein as a surrogate marker for activity. DnaK and DnaK-PrP were purified using Ni-NTA chromatography. Equal amounts of protein were then incubated with [γ-32P]ATP and at the indicated times the reactions were halted and subjected to SDS-PAGE analysis. Staining of the gel with Coomassie Brilliant Blue verified equal loading and demonstrated that both proteins were stable during the 30 min incubation (Fig. 4B top panel). The gel was then dried and exposed to a phosphorimager cassette. Analysis of the image revealed that the DnaK-PrP fusion was labeled with 32P to levels similar to DnaK alone, (Fig. 4B bottom panel). This assay demonstrated that the DnaK portion of the fusion protein retained its enzymatic activity, further supporting our hypothesis that it retains its chaperone activity even as a fusion protein.
3.6. Co-expression of chaperones and co-chaperones with PrP is not sufficient to solubilize PrP
As described in the introduction, several proteins are expressed in a soluble form in E. coli when chaperone proteins are simultaneously over-expressed. Our observation that translational fusions of DnaK or GroEL to PrP resulted in production of a soluble chimeric protein led us to ask what effect over-expression of DnaK and its cognate or unrelated co-chaperones would have on accumulation and solubility of PrP. Accordingly, cells containing a PrP expression vector and a plasmid expressing DnaK alone, DnaK and DnaJ/GrpE or DnaK and GroES were induced to express these proteins. Cell lysates were prepared, separated into total, soluble and insoluble (inclusion bodies) fractions and subjected to SDS-PAGE. While PrP and each of the chaperones and co-chaperones were detected in total lysates only very little PrP partitioned with the soluble fraction. Instead, PrP was found almost exclusively in inclusion bodies (Fig. 5). This is in contrast to what we described for the chaperone fusions (Fig. 2). Although co-expression of the DnaK co-chaperone partners DnaJ/GrpE reduced the amount of PrP detected with inclusion bodies, its co-expression failed to increase the amount of soluble protein. Thus, over-expression of DnaK alone is insufficient to result in soluble PrP whereas fusion with DnaK or GroEL permits PrP to partition to the soluble fraction.
Figure 5. Over-expression of DnaK and co-chaperone proteins is unable to direct PrP to the soluble fraction.

BL21[DE3] cells were transformed with the indicated plasmids and protein expression was induced with IPTG. Proteins were fractionated as described in materials and methods, analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue.
In conclusion, we developed a new method for preparing soluble proteins in E. coli. The method uses a plasmid based vector where the protein of interest is fused in fame to DnaK or GroEL. The resulting soluble protein is readily purified using affinity chromatography and the protein of interest can be freed of its chaperone tag after cleavage with thrombin. The system was developed and tested using murine PrP an verified by demonstrating solubility of an otherwise insolube fragment of VZV ORF21p. Parenthetically, we have used fusion of DnaK to other proteins including herpes simplex virus ICP0 and VZV ORF29p. In each of these instances the majority of the protein of interest partitioned with inclusion bodies when expressed on its own or as a GST-fusion protein. In contrast the DnaK fusion proteins were soluble and easily purified.
Acknowledgments
The authors thank Dr Daniel Wolf for critical reading of the manuscript and Dr. T. Sklaviadis, Aristotle University, Thessaloniki, Greece for the gift of plasmid pVPmPrP14. This study was funded by an award from EPEAEK II (Greek National Ministry for Education) and PEP (Region of Central Macedonia) and by core funding of the Aristotle University of Thessaloniki to CAP and a grant from the Public Health Service AI-024021 to SJS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christos A. Kyratsous, Email: cak2113@columbia.edu.
Saul J. Silverstein, Email: sjs6@columbia.edu.
Christine R. DeLong, Email: crd2105@columbia.edu.
Christos A. Panagiotidis, Email: pchristo@pharm.auth.gr.
References
- Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- Blum P, Ory J, Bauernfeind J, Krska J. Physiological consequences of DnaK and DnaJ overproduction in Escherichia coli. J Bacteriol. 1992;174:7436–44. doi: 10.1128/jb.174.22.7436-7444.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marco A, Deuerling E, Mogk A, Tomoyasu T, Bukau B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007;7:32. doi: 10.1186/1472-6750-7-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahnert B, Lilie H, Neubauer P. Inclusion bodies: formation and utilisation. Adv Biochem Eng Biotechnol. 2004;89:93–142. doi: 10.1007/b93995. [DOI] [PubMed] [Google Scholar]
- Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79:425–49. doi: 10.1152/physrev.1999.79.2.425. [DOI] [PubMed] [Google Scholar]
- Georgiou G, Valax P. Expression of correctly folded proteins in Escherichia coli. Curr Opin Biotechnol. 1996;7:190–7. doi: 10.1016/s0958-1669(96)80012-7. [DOI] [PubMed] [Google Scholar]
- Gold L. Expression of heterologous proteins in Escherichia coli. Methods Enzymol. 1990;185:11–4. doi: 10.1016/0076-6879(90)85004-8. [DOI] [PubMed] [Google Scholar]
- Goloubinoff P, Gatenby AA, Lorimer GH. GroE heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli. Nature. 1989;337:44–7. doi: 10.1038/337044a0. [DOI] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–9. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hockney RC. Recent developments in heterologous protein production in Escherichia coli. Trends Biotechnol. 1994;12:456–63. doi: 10.1016/0167-7799(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Hodgson J. Expression systems: a user’s guide. Emphasis has shifted from the vector construct to the host organism. Biotechnology (N Y) 1993;11:887–93. doi: 10.1038/nbt0893-887. [DOI] [PubMed] [Google Scholar]
- Hoffmann F, Rinas U. Roles of heat-shock chaperones in the production of recombinant proteins in Escherichia coli. Adv Biochem Eng Biotechnol. 2004a;89:143–61. doi: 10.1007/b93996. [DOI] [PubMed] [Google Scholar]
- Hoffmann F, Rinas U. Stress induced by recombinant protein production in Escherichia coli. Adv Biochem Eng Biotechnol. 2004b;89:73–92. doi: 10.1007/b93994. [DOI] [PubMed] [Google Scholar]
- Kyratsous CA, Silverstein SJ. BAG3, a host cochaperone, facilitates varicella-zoster virus replication. J Virol. 2007;81:7491–503. doi: 10.1128/JVI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyratsous CA, Silverstein SJ. Components of Nuclear Domain 10 bodies Regulate Varicella Zoster Virus Replication. J Virol. 2009 doi: 10.1128/JVI.00021-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaux PG, Herendeen SL, Bloch PL, Neidhardt FC. Transient rates of synthesis of individual polypeptides in E. coli following temperature shifts. Cell. 1978;13:427–34. doi: 10.1016/0092-8674(78)90317-3. [DOI] [PubMed] [Google Scholar]
- Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev. 1996;60:512–38. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T. Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl Environ Microbiol. 1998;64:1694–9. doi: 10.1128/aem.64.5.1694-1699.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olins PO, Lee SC. Recent advances in heterologous gene expression in Escherichia coli. Curr Opin Biotechnol. 1993;4:520–5. doi: 10.1016/0958-1669(93)90071-4. [DOI] [PubMed] [Google Scholar]
- Panagiotidis CA, Burkholder WF, Gaitanaris GA, Gragerov A, Gottesman ME, Silverstein SJ. Inhibition of DnaK autophosphorylation by heat shock proteins and polypeptide substrates. J Biol Chem. 1994;269:16643–7. [PubMed] [Google Scholar]
- Panagiotidis CA, Silverstein SJ. pALEX, a dual-tag prokaryotic expression vector for the purification of full-length proteins. Gene. 1995;164:45–7. doi: 10.1016/0378-1119(95)00417-5. [DOI] [PubMed] [Google Scholar]
- Perez-Perez J, Martinez-Caja C, Barbero JL, Gutierrez J. DnaK/DnaJ supplementation improves the periplasmic production of human granulocyte-colony stimulating factor in Escherichia coli. Biochem Biophys Res Commun. 1995;210:524–9. doi: 10.1006/bbrc.1995.1691. [DOI] [PubMed] [Google Scholar]
- Schlapschy M, Grimm S, Skerra A. A system for concomitant overexpression of four periplasmic folding catalysts to improve secretory protein production in Escherichia coli. Protein Eng Des Sel. 2006;19:385–90. doi: 10.1093/protein/gzl018. [DOI] [PubMed] [Google Scholar]
- Schlieker C, Bukau B, Mogk A. Prevention and reversion of protein aggregation by molecular chaperones in the E. coli cytosol: implications for their applicability in biotechnology. J Biotechnol. 2002;96:13–21. doi: 10.1016/s0168-1656(02)00033-0. [DOI] [PubMed] [Google Scholar]
- Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Stevens JM, Rao Saroja N, Jaouen M, Belghazi M, Schmitter JM, Mansuy D, Artaud I, Sari MA. Chaperone-assisted expression, purification, and characterization of recombinant nitrile hydratase NI1 from Comamonas testosteroni. Protein Expr Purif. 2003;29:70–6. doi: 10.1016/s1046-5928(03)00008-1. [DOI] [PubMed] [Google Scholar]
- Vonrhein C, Schmidt U, Ziegler GA, Schweiger S, Hanukoglu I, Schulz GE. Chaperone-assisted expression of authentic bovine adrenodoxin reductase in Escherichia coli. FEBS Lett. 1999;443:167–9. doi: 10.1016/s0014-5793(98)01714-1. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Feramisco JR. Rapid purification of mammalian 70,000-dalton stress proteins: affinity of the proteins for nucleotides. Mol Cell Biol. 1985;5:1229–37. doi: 10.1128/mcb.5.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel R. Mutations and off-pathway aggregation of proteins. Trends Biotechnol. 1994;12:193–8. doi: 10.1016/0167-7799(94)90082-5. [DOI] [PubMed] [Google Scholar]
- Xu Y, Weng CL, Narayanan N, Hsieh MY, Anderson WA, Scharer JM, Moo-Young M, Chou CP. Chaperone-mediated folding and maturation of the penicillin acylase precursor in the cytoplasm of Escherichia coli. Appl Environ Microbiol. 2005;71:6247–53. doi: 10.1128/AEM.71.10.6247-6253.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanase H, Moriya K, Mukai N, Kawata Y, Okamoto K, Kato N. Effects of GroESL coexpression on the folding of nicotinoprotein formaldehyde dismutase from Pseudomonas putida F61. Biosci Biotechnol Biochem. 2002;66:85–91. doi: 10.1271/bbb.66.85. [DOI] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–91. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zylicz M, Ang D, Georgopoulos C. The grpE protein of Escherichia coli. Purification and properties. J Biol Chem. 1987;262:17437–42. [PubMed] [Google Scholar]
- Zylicz M, LeBowitz JH, McMacken R, Georgopoulos C. The dnaK protein of Escherichia coli possesses an ATPase and autophosphorylating activity and is essential in an in vitro DNA replication system. Proc Natl Acad Sci U S A. 1983;80:6431–5. doi: 10.1073/pnas.80.21.6431. [DOI] [PMC free article] [PubMed] [Google Scholar]