

Summary
Therapeutic inhibition of mTOR in cancer is complicated by the existence of a negative feedback loop linking mTOR to the PI3K-Akt pathway. Thus, mTOR inhibition by Rapamycin or TSC1/2 results in increased PI3K-Akt activation. The death domain kinase receptor interacting protein 1 (RIP, RIP1), plays a key role in NF-κB activation and also activates the PI3K-Akt pathway through unknown mechanisms. RIP1 has recently been found to be overexpressed in glioblastoma (GBM), the most common adult primary malignant brain tumor, but not in Grade II-III glioma. Our data suggest that RIP1 activates PI3K-Akt using dual mechanisms by removing the two major brakes on PI3K-Akt activity. Firstly, increased expression of RIP1 activates PI3K-Akt by interrupting the mTOR negative feedback loop. However, unlike other signals which regulate mTOR activity without affecting its level, RIP1 negatively regulates mTOR transcription via a NF-κB dependent mechanism. The second mechanism used by RIP1 to activate PI3K-Akt is downregulation of cellular PTEN levels which appears to be independent of NF-κB activation. The clinical relevance of these findings is highlighted by the demonstration that RIP1 levels correlate with activation of Akt in GBM. Thus, our study shows that RIP1 regulates key components of the PTEN-PI3K-Akt-mTOR pathway and elucidates a novel negative regulation of mTOR signaling at the transcriptional level by the NF-κB pathway. Our data suggest that the RIP1-NF-κB status of tumors may influence response to treatments targeting the PTEN-PI3K-mTOR signaling axis.
Keywords: PI3K-Akt, mammalian target of rapamycin, receptor interacting protein, NF-kappa B, negative feedback
Introduction
Activation of mammalian target of rapamycin (mTOR) may play an important role in cancer (1). The PI3K-Akt signaling pathway plays an essential role in the activation of mTOR. PTEN is a lipid phosphatase that is commonly mutated in human cancer and is the key negative regulator of PI3K-Akt activity upstream of Akt (2). mTOR is also involved in a negative feedback regulation of PI3K-Akt (3). Thus, exposure of cells to rapamycin or silencing mTOR with siRNA leads to PI3K-Akt activation (3, 4). An important mechanism mediating this negative feedback loop is via p70S6K-IRS1 (3, 5). Increased activation of mTOR results in activation of p70S6K, which phosphorylates IRS1 resulting in inhibition of IRS1. The IGF/Insulin and IRS-1 signaling pathways are major activators of the PI3K-Akt pathway. Thus, mTOR-p70S6K mediated inhibition of IRS1 results in decreased PI3K-Akt activation. Aberrant activation of the PI3K-Akt-mTOR axis is well documented in cancer and has led to efforts to target mTOR. This feedback loop may explain the limited efficacy of mTOR inhibition in certain cancers (6), since increased activation of PI3K-Akt could activate mTOR independent oncogenic signaling pathways downstream of Akt.
The receptor interacting protein (RIP, RIP1) is involved in the activation of two key pro-survival pathways, the NF-κB (7, 8) and PI3K-Akt pathways (9, 10). The mechanism used by RIP1 to activate PI3K-Akt is unknown. RIP1 is widely expressed and is a member of a kinase family that mediates cellular response to inflammation and stress (7, 11, 12). RIP1 is expressed both constitutively and inducibly in response to inflammatory cytokines (7, 11, 13-15). We have recently shown that RIP1 levels are increased in about 30% of patients with glioblastoma (GBM), the most common primary brain tumor in adults, and confers a worse prognosis in this disease (16).
In this study, we show that RIP1 negatively activated PI3K-Alt by regulating two key inhibitors of the PI3K-Akt pathway, upstream and downstream of Akt, and show that RIP1 links NF-κB activation to the mTOR-PI3K-Akt negative feedback loop.
Materials and Methods
Antibodies
IκBα antibodies were obtained from Santa Cruz Biotechnology, RIP antibody from BD Biosciences for both WB and IHC, FLAG, mTOR, pAkt S473 (#9271) for both Western blot and IHC, PTEN, pp70S6K Thr 389, p70S6K, phospho-IRS-1 Ser636/639 from Cell Signaling Technology, IRS-1 antibodies from Upstate/Millipore, and Actin antibodies were from Sigma.
Plasmids
Genomic DNA was extracted from U87MG cell line and a part of the promoter region of human mTOR (from − 2450 to − 495) was amplified by PCR and cloned into pGL3 basic vector to generate the wild type mTOR-LUC construct. Two putative NF-κB binding sites in the promoter region of human mTOR located at −1450 to −1440 (GGGATTTTCC) and −748 to − 740 (GGGAATTT) were disrupted by converting GGGATTTCC to AAATATTCC and GGGGAATTT to GAATAATTT using by QuikChange Multi-site directed mutagenesis kit (Stratagene). PTEN-LUC wild type and NF-κB mutant have been described previously (17).
Quantitative real-time PCR
After extracting RNA, complementary DNA was synthesized and PCR reactions were performed using a SYBR PCR master kit (AB Biosystems, Inc.). GADPH was used as an internal control between samples. The PCR reactions were performed in triplicates for each gene being validated. Primer sequences will be provided upon request.
Luciferase assays
Were conducted as we have described previously (9).
Production of Adenovirus expressing RIP
Preparation of adenovirus expressing RIP1 has been described previously (9). Adenovirus-null or Adenovirus-IκBαM were obtained from Vector Biolabs.
RIP1 siRNA knockdown
RIP1 siRNA were obtained from Dharmacon. Three sequences directed against the RIP1 mRNA were used. A negative control siRNA (non sequence) was obtained from Ambion. siRNA was performed according to the manufacterer's protocol using Lipofectamine 2000 reagent (Invitrogen).
Immunohistochemistry
All studies were conducted according to IRB approved protocols. Paraffin-embedded GBM sections were exposed to the mouse monoclonal RIP antibody or rabbit polyclonal pAkt antibodies. Semiquantitative evaluation of immunohistochemical signal was performed on coded slides in a blinded fashion by a neuropathologist (KJH). We used a previously described semiquantitative method to evaluate pAkt staining (18). In this grading scheme, 0−1 is considered negative and 2−3 is positive. RIP1 was graded we have previously described (16). Briefly, the IHC staining intensity for RIP1 was scored semiquantitatively as 0, + (1), ++ (2), and +++ (3) with 0 representing lack of RIP1 staining and +++ (3) the strongest staining.
Results and Discussion
RIP1 regulates PTEN levels
Since NF-κB activation can negatively regulate PTEN at a transcriptional level (17), we sought to determine whether RIP1 influences PTEN levels to activate Akt. Introduction of an adenoviral RIP1 into LN229 cells (wild type PTEN), resulted in a downregulation of PTEN control adenovirus null had no effect (Figure 1A). As expected, RIP1 mediated downregulation of PTEN is accompanied by activation of Akt. We confirmed the effect of RIP1 on PTEN in LN18 cells (Supplemental Figure 1A). RIP1 mediated activation of Akt is blocked by the PI3K inhibitor LY290042 (Supplemental Figure 1B) and by tetracycline induced expression of PTEN in U251MG cells (PTEN null) as shown in Figure 1B, supporting regulation at the level of PI3K.
Figure 1. RIP1 regulates PTEN levels.
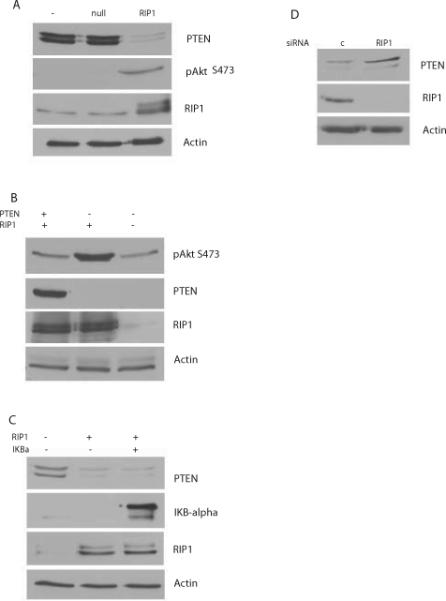
LN229 cells were infected with RIP1 adenovirus (MOI 50) or adenovirus null followed by preparation of lysates after 24h. Increased RIP1 downregulates PTEN levels and activates Akt as detected by phosphorylation of Akt at S473. B. U251MG cells (which are PTEN null) transfected with a tet inducible PTEN were exposed to tetracycline for 24h, followed by infection with RIP1 adenovirus. C. Inhibition of NF-κB using an IKBαM fails to inhibit RIP1 mediated downregulation of PTEN. LN229 cells were infected with adenovirus null or adenovirus IKBαM for 24h, followed by infection with RIP1 adenovirus for an additional 24h. D. Silencing RIP1 in LN229 cells results in an upregulation of PTEN levels. LN229 cells were transfected with RIP1 siRNA or control siRNA followed by preparation of lysates after 72h.
Since previous studies have shown a role for NF-κB activation in regulation of PTEN (17) and RIP1 expression is sufficient for NF-κB activation, we investigated whether a dominant negative IKBα mutant (IKBαM, S32/S36 super repressor) would inhibit RIP1 mediated PI3K-Akt activation. Surprisingly, introduction of IKBαM fails to block RIP1 mediated PTEN downregulation (Figure 1C), suggesting an NF-κB independent effect. Activation of NF-κB by RIP1 is demonstrated by downregulation of endogenous IKBα (Figure 1C). siRNA knockdown of RIP1 in LN229 cells resulted in increased levels of PTEN (Figure 1D). Quantitative PCR analysis of RIP1 mRNA levels in LN229 cells demonstrates that increased RIP1 expression downregulates PTEN mRNA (Supplemental Figure 1C). Furthermore, RIP1 inhibits activity of both a wild type and a NF-κB deleted PTEN-LUC reporter in LN229 cells (Supplemental Figure 2A and B). These experiments demonstrate that increased RIP1 levels downregulate PTEN through NF-κB independent pathways, while loss of RIP1 induces an increase in PTEN levels.
RIP1 regulates mTOR levels
Expression of RIP1 in U87MG cells (PTEN null) also results in Akt activation (Figure 2A). We also find that RIP1 expression downregulates mTOR levels in both PTEN null (U87MG) and PTEN wt (LN229) cells resulting in a decreased phosphorylation of p70S6K on Thr 389 (Figure 2 A and B). mTOR is a component of both mTORC1 and mTORC2. While mTORC1 activity inhibits the activation of PI3K-Akt, mTORC2 has the opposite effect and phosphorylates Akt on serine 473. In the glioma cell lines studied here RIP1 mediated mTOR downregulation is associated with increased PI3K-Akt activation suggesting that the inhibitory effect on mTORC1 dominates.
Figure 2.

RIP1 regulates mTOR levels in GBM cells. A. Ad-RIP1 or adenovirus null were introduced into cells for 24h followed by lysate preparation and Western blotting for mTOR, phospho-p70S6K, p70S6K, pAkt, Akt, phospho-IRS-1m IRS-1, RIP1 and Actin. B. a similar experiment was conducted in LN229 cells. C and D. Western blots showing that siRNA knockdown of RIP1 results in an increase in mTOR levels in U87MG and LN229 cells compared to control (non sequence) siRNA.
RIP1 interrupts the mTOR-p70S6K-IRS1 negative feedback loop
Increased expression of RIP1 results in a decrease in inhibitory phosphorylation of IRS-1 (Figure 2A and B) in both LN229 and U87MG cells. These experiments demonstrate an interruption of the mTOR-S6-IRS1 inhibitory loop by RIP1 in glioma cells regardless of PTEN status. Quantitative PCR analysis of mTOR levels in U87MG and LN229 cells shows that RIP1 downregulates mTOR mRNA levels (Supplemental Figure 3A and B). Importantly, siRNA knockdown of RIP1 results in an increase in mTOR levels in LN229 and U87MG cells (Figure 2C and D).
RIP1 regulates mTOR via a NF-κB dependent pathway
An extensive cross-talk has been described between the PI3K-Akt and NF-κB signaling networks (19, 20). Next, we investigated if NF-κB activation is required for RIP1 mediated downregulation of mTOR. We introduced adenoviral IKBαM or adenovirus null along with RIP1 into glioma cell lines and examined the level of mTOR. Inhibition of NF-κB blocks RIP1 mediated downregulation of mTOR in U87MG and U251MG cells (Figure 3 A and B) but not in LN229 cells (Supplemental Figure 4). This result suggests that depending on the cell type RIP1 downregulates mTOR via NF-κB dependent or independent mechanisms.
Figure 3.

A dominant-negative IκBα super-repressor mutant blocks the effect of RIP1 on mTOR. A. U87MG cells were infected with Ad-RIP1+Ad-null or Ad RIP1+ Ad- IκBαM for 24h. B. shows the same experiment in U251MG cells. C. U87 cells were transfected with the putative mTOR promoter linked to luciferase (mTOR-LUC) and co-transfected with FLAG-RIP1 plasmid or empty vector. Introduction of RIP1 results in a significant suppression of mTOR-LUC. Expression of FLAG-RIP1 was confirmed by Western blot with FLAG antibodies. D. this experiment is similar to C except that a mTOR-LUC reporter with mutated NF-κB binding sites was used.
RIP1 inhibits mTOR transcription
Introduction of RIP1 into U87MG cells resulted in decreased activity of a mTOR-LUC reporter construct, suggesting that RIP1 affects mTOR transcription (Figure 3C). Mutation of two putative NF-κB binding sites in the mTOR promoter reverses the effect of RIP1 in reporter assays (Figure 3D). Indeed RIP1 seems to have a stimulatory effect on the mutant mTOR promoter through unknown mechanisms.
RIP1 level correlates with phosphorylation of Akt in GBM
Next, we examined whether RIP1 expression correlated with activation of Akt in GBM. We performed immunohistochemistry for RIP1 and pAkt in a set of 10 GBM tumors. IHC grading for RIP1 and pAkt is described in the methods section. As can be seen in Figure 4, the RIP1 expression correlates with pAkt and the correlation is statistically significant (p=0.006 by Spearman's rho statistic), suggesting that increased RIP1 may activate Akt in GBM.
Figure 4. Correlation between RIP1 and pAkt in GBM.
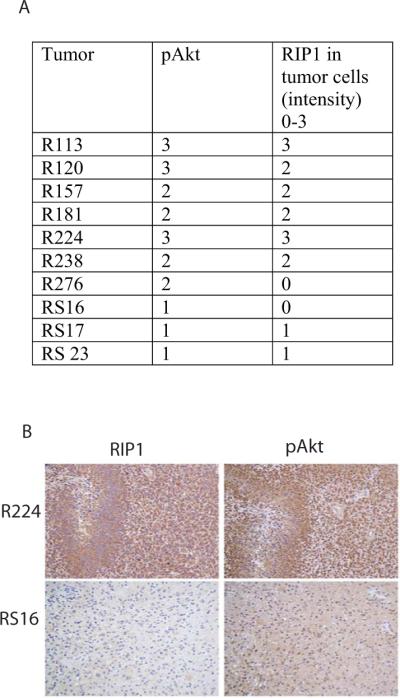
A. The table shows shows the immunohistochemistry (IHC) results for pAkt and RIP1 proteins in tumor cells. The pAkt (s473) was scored from 1 to 3 and the RIP1 was scored from 0 to 3. Spearman correlation between pAkt and RIP1 equaled 0.80 with p-value 0.006 (by Spearman's rho statistic), indicating significant correlation between pAkt and RIP1 intensities.
B. Immunohistochemistry (IHC) shows RIP1 level correlates with pAkt in GBM. IHC of a high RIP1 and high pAkt (S473) staining GBM (R224) and a low RIP1 and low pAkt GBM (RS-16).
Concluding Remarks
In this study, we demonstrate that RIP1 regulates the PI3K-Akt pathway at two key nodes in the pathway. Supplemental Figure 5 shows a schematic of our model. RIP1 downregulates PTEN and also suppresses the mTOR-PI3K inhibitory feedback loop to activate PI3K-Akt. Such dual regulation may lead to a synergistic and potent activation of the PI3K-Akt pathway. Aberrant activity of the PTEN-PI3K-Akt-mTOR axis is common in glioblastoma (GBM). RIP1 level is commonly increased in GBM and correlates with pAkt in GBMs (16), suggesting RIP1 may drive the PI3K-akt pathway in GBM.
Our study also highlights a novel regulation of mTOR at the transcriptional level by RIP1 mediated, at least in part, by an activation of NF-κB. The effect of RIP1 is in contrast to other components of the NF-κB signaling network such as IKKα and IKKβ, which tend to activate mTOR by phosphorylating and inactivating TSC1/2. Regulation of the PTEN-PI3K-Akt-mTOR by RIP1- NF-κB may impact treatments targeting the PI3K pathway in cancer.
Supplementary Material
Acknowledgments
This work was supported in part by NIH CTSA Grant UL1 RR024982. We thank Dr Guanghua Xiao for help with statistical analysis, Drs. Deepti Ramnarain and Alfred Yung for LN229 and LN18 cells and Dr. James Brugarolas for helpful discussions. This work was also supported in part by DOE grant #DE-FG02-06ER64186 to D.A.B.
References
- 1.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 3.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 4.McDonald PC, Oloumi A, Mills J, et al. Rictor and integrin-linked kinase interact and regulate Akt phosphorylation and cancer cell survival. Cancer Res. 2008;68:1618–24. doi: 10.1158/0008-5472.CAN-07-5869. [DOI] [PubMed] [Google Scholar]
- 5.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–83. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 6.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008;5:e8. doi: 10.1371/journal.pmed.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–23. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 8.Grimm S, Stanger BZ, Leder P. RIP and FADD: two ”death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc Natl Acad Sci U S A. 1996;93:10923–7. doi: 10.1073/pnas.93.20.10923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park S, Ramnarain DB, Hatanpaa KJ, et al. The death domain-containing kinase RIP1 regulates p27(Kip1) levels through the PI3K-Akt-forkhead pathway. EMBO Rep. 2008;9:766–73. doi: 10.1038/embor.2008.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivarelli MS, McDonald D, Miller M, Cusson N, Kelliher M, Geha RS. RIP links TLR4 to Akt and is essential for cell survival in response to LPS stimulation. J Exp Med. 2004;200:399–404. doi: 10.1084/jem.20040446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30:151–9. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. RIP1, a kinase on the crossroads of a cell's decision to live or die. Cell Death Differ. 2007;14:400–10. doi: 10.1038/sj.cdd.4402085. [DOI] [PubMed] [Google Scholar]
- 13.Pimentel-Muinos FX, Seed B. Regulated commitment of TNF receptor signaling: a molecular switch for death or activation. Immunity. 1999;11:783–93. doi: 10.1016/s1074-7613(00)80152-1. [DOI] [PubMed] [Google Scholar]
- 14.Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–13. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 15.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Park SHK, Xie Y, Mickey BE, Madden CJ, Raisanen J, Ramnarain DB, Xiao Guanghua, Saha D, Boothman DA, Zhao D, Bachoo RM, Pieper RO, Habib AA. The receptor interacting protein 1 inhibits p53 induction through Nuclear factor-kB activation and confers a worse prognosis in glioblastoma. Cancer Research. 2009 doi: 10.1158/0008-5472.CAN-08-4079. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-kappa B prevents apoptosis. Mol Cell Biol. 2004;24:1007–21. doi: 10.1128/MCB.24.3.1007-1021.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL. Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol. 2006;65:1181–8. doi: 10.1097/01.jnen.0000248549.14962.b2. [DOI] [PubMed] [Google Scholar]
- 19.Dan HC, Baldwin AS. Differential involvement of IkappaB kinases alpha and beta in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J Immunol. 2008;180:7582–9. doi: 10.4049/jimmunol.180.11.7582. [DOI] [PubMed] [Google Scholar]
- 20.Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell. 2007;130:440–55. doi: 10.1016/j.cell.2007.05.058. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.