

Abstract
Background
Infantile Pompe disease (glycogen storage disease type 2) is a fatal disorder caused by deficiency of acid α-glucosidase. This deficiency results in glycogen accumulation in the lysosomes of many tissues including cardiac muscle. The disease is characterized by profound hypotonia, poor growth, organomegaly, and cardiomegaly. Severe hypertrophic cardiomyopathy often is present in early infancy, and most patients die of cardiac or respiratory failure in the first year of life. This report describes the cardiac response of infants with Pompe disease to a phase 2 trial of enzyme replacement therapy (ERT).
Methods
Eight patients with classical infantile Pompe disease were given intravenous recombinant human GAA (rhGAA) for 1 year. Cardiac monitoring included echocardiography, electrocardiograms (ECGs), chest radiographs, and clinical cardiac evaluation at 4, 8, 12, 24, 36, and 52 weeks. At 52 weeks, 6 patients were alive.
Results
Most of the treated patients had rapid regression of ventricular hypertrophy in response to ERT, with near normalization of posterior wall thickness, ventricular mass, and ventricular size. Systolic ventricular function was preserved despite rapid changes in ventricular mass and size. Concomitantly, ECGs documented lengthening of the PR interval and decreased ventricular voltages, whereas chest radiographs documented a decreased cardiothoracic ratio. Symptoms of pulmonary congestion were diminished, and survival was improved.
Conclusion
The cardiovascular system responds quickly and strikingly to ERT with rhGAA, suggesting rapid reversal of excessive glycogen storage in cardiac muscle cells. Changes in ventricular mass and function are maintained throughout 1 year of follow-up evaluation and associated with decreased morbidity and prolonged survival.
Keywords: Hypertrophy, Cardiomyopathy, Pediatrics, Genetics, Trials
Pompe disease, also known as glycogen storage disease type 2 (GSD-II) or acid maltase deficiency, is a rare autosomal recessive disorder caused by a deficiency of acid α-glucosidase (GAA), a glycolytic lysosomal enzyme. The enzyme deficiency results in glycogen accumulation in the lysosomes of multiple tissue types including cardiac, skeletal, and smooth muscle cells [1]. Glycogen accumulation damages cellular function by causing both cellular hypertrophy and lysosomal rupture [2].
The combined incidence of all Pompe disease forms is estimated to be 1 in 40,000 live births [3]. The infantile form, originally described by Pompe in 1932, is caused by complete deficiency of GAA and typically is associated with the most severe symptoms [4]. Partial enzyme deficiency results in a milder late-onset phenotype. Studies investigating the natural history of untreated infantile-onset Pompe disease show that 92% to 95% of infants with the disorder die within the first year of life at a median age of 6 to 8.7 months [4, 5].
With the infantile form of Pompe disease, symptoms typically are evident by 6 months of age including hypotonia, loss or delay of motor milestones, and symptoms of cardiac/respiratory compromise. The effect of glycogen storage on the cardiovascular system includes cardiomegaly, hypertrophic cardiomyopathy, and variable left ventricular outflow tract obstruction. These changes in turn produce symptoms of both systolic and diastolic dysfunction.
In the past 5 years, phase 1 and 2 trials of enzyme replacement therapy (ERT) have been promising, [6–10]. Recombinant human acid α-glucosidase (rhGAA) produced in Chinese hamster ovary (CHO) cell cultures or transgenic mouse and rabbit milk has been shown to have physiologic activity in both animal disease models and several small clinical trials, leading to decreased morbidity and prolonged survival [6–10].
These results prompted this multicenter phase 2 trial to evaluate the safety and efficacy of ERT in a larger group of infants with the most severe form of Pompe disease. Eight patients with infantile Pompe disease were enrolled in a phase 2 open-label trial of ERT with CHO-derived rhGAA. This is the initial report on the detailed cardiac surveillance in this cohort.
Methods
This study, designed as an open-label multicenter trial, was conducted at one center in the United States (Duke University Medical Center, Durham, NC) and two centers in Europe (Pediatrique Hôpital Debrousse, Lyon, France and Klinik und Poliklinik-und Jungenmedizin Universitatsk-linikum, Essen, Germany). The trial evaluated the safety and efficacy of rhGAA therapy administered for 52 weeks to naive patients with early-onset infantile Pompe disease. The results were compared with those for a large group of historical control subjects. Because of the improved survival in preliminary phase 1 studies, it was decided that a placebo control group would be unethical.
The study was approved by the institutional review boards and ethics committees of all the participating sites. Written informed consent was obtained from parents or guardians. Patients still living after the 52-week initial phase were enrolled in an extension protocol.
Eight patients met the criteria for inclusion in the study, which required endogenous GAA activity in skin fibroblasts less than 1% of normal (assayed against 4-methylumbelliferyl-alpha-D glucopyranoside), a cardio-thoracic ratio exceeding 0.5 by chest X-ray, and an indexed left ventricular mass of 65 g/m2 or more or greater than two standard deviations above the normal mean. Patients were excluded if they had respiratory insufficiency at presentation, a history of ventilator dependence, a cardiac ejection fraction less than 40%, or an additional major congenital abnormality or significant organic disease likely to decrease survival.
Two preparations (CHO-1 and CHO-2) of the investigational product, rhGAA, were purified from two different CHO cell lines transfected with the cDNA for human GAA. The protein sequences of the rhGAAs produced from each preparation were identical to each other and to a commonly occurring form of human GAA with a calculated protein mass of 99.4 kDa.
During the 52-week trial, all the patients were started on weekly intravenous (IV) infusions of CHO-1 at a dose of 10 mg/kg. The dose for patient H was increased to 20 mg/kg weekly after week 43 of the initial phase and maintained at that level for the further doses. All doses of rhGAA were administered in 3-h infusions (2 mg/kg over 30 min and the remaining dose over 2.5 h). An independent safety monitoring board reviewed safety data. Details of the study design and the overall results have been reported previously [11]. Baseline cardiac laboratory assessments including standard two-dimensional echocardiography, Doppler examination, 12-lead electrocardiograms (ECGs), and chest radiograph were performed within 7 days before the first infusion of rhGAA and repeated at 4, 8, 12, 24, 36, and 52 weeks.
The noncardiac outcome parameters included growth, ventilator dependence, motor development (as measured by the Alberta Infant Motor Scale), cognitive function (as measured by the Modified Bayley Scales of Infant Development, 2nd ed), hearing, muscle biopsy to assess GAA activity and glycogen content, and blood anti-rhGAA antibody. Safety was monitored with routine complete blood counts, blood chemistries, urine analysis, and physical examination. Additional data included gene mutation analysis and the recording of adverse events.
Echocardiograms
Left ventricular (LV) length was measured from the apical view, whereas LV area and wall thickness were measured from the parasternal short axis view. From these measurements, LV volume, ejection fraction, and mass were calculated using the methods outlined in Sluysman and Colan [12]. Systolic and diastolic measurements were performed at end systole and end diastole from the same cardiac cycle. M-mode was used for the calculation of shortening fraction [12]. When M-mode was technically inadequate, the shortening fraction was based on ventricular diameters taken directly from the parasternal short axis image. Doppler evaluation of the LV outflow tract was performed from the apical view.
All measurements used in this analysis were made offline by a single investigator not involved in data acquisition (J.C.L.). This reader was blinded to clinical condition but not to patient identification and date of study. Subsequent phase 3 trials are being conducted with de-identified data. Congenital heart disease that might contribute to changes in LV function, mass, or both was excluded at the initial echocardiographic study. All z-scores were calculated using the normative database at the Children’s Hospital of Boston [12].
Electrocardiograms
Standard 12-lead ECGs (10 mm/mV and 25 mm/s) were obtained using Hewlett-Packard Page Writer Xli recorders (Hewlett-Packard, PaloAlto, CA, USA) and read by two blinded observers. From three consecutive heartbeats, QT and RR intervals were obtained. Measurement of the QT was obtained in lead II or lead V5 because these leads are unlikely to have a U-wave, which can obscure the downslope of the T-wave. In the presence of a U-wave, the downslope was extrapolated to the baseline. The QTc interval was obtained using Bazett’s formula (QT interval divided by the square root of the preceding R-R interval). The left ventricular voltage, expressed in millivolts, was determined by the sum of the S-wave in V1 and the R wave in V6.
Chest X-Ray
Standard anteroposterior and lateral chest X-rays were performed. The studies were assessed for cardiothoracic ratio (maximum heart width: chest width) and the presence or absence of pulmonary edema. Cardiomegaly was defined as a cardiothoracic ratio greater than 0.6. Chest X-rays of the five U.S. patients were de-identified and interpreted by a single radiologist who was not a study investigator. The chest X-rays of the three European patients were interpreted at their respective institutions.
Cardiac Medications
The use of cardiac medications including diuretics, spironolactone, digoxin, angiotensin-converting enzyme (ACE) inhibitors, and intravenous inotropes were tracked as a measure of clinical congestive heart failure. Because of the skeletal muscular disease, the assessment of congestive heart symptoms was difficult. Although imperfect, surrogates for the usual signs and symptoms were needed. To get some idea of clinical response, we examined the presence/absence of pulmonary edema and the patient’s dependence on cardiac medications to stay free of ventilator support.
Statistical Analysis
Because of the small sample size, most of the data evaluation was descriptive. Statistical significance was examined for a few key variables. All p values were derived using the Wilcoxin rank sum method. A nonparametric test was used because of the small sample.
Results
Eight patients met the entry criteria and were enrolled over a 6-month period (Table 1). Their ages ranged from 1.8 to 6.5 months at the time of diagnosis and from 2.7 to 14.6 months at the time of enrollment. The median gestational age at birth was 37.5 weeks (range, 33–40 weeks), and the median weight at enrollment was 6.7 kg (range, 4.4–9.0 kg). Patients D, E, and H had siblings with infantile Pompe disease, all of whom died of cardiorespiratory failure before the age of 1 year.
Table 1.
Baseline demographics
| Patient | Sex | Ethnicity | Age (mo) |
||
|---|---|---|---|---|---|
| First symptoms | Diagnosis | First infusion | |||
| A201 | M | Asian | 1.6 | 3.8 | 4.6 |
| B202 | M | Asian | 1.4 | 4.7 | 4.8 |
| C203 | M | Caucasian | 2.0 | 6.5 | 8.0 |
| D204 | F | Caucasian | 4.8 | 5.4 | 8.4 |
| E205 | M | Asian Indian | 0.4 | 2.5 | 3.1 |
| F206 | F | Caucasian | 1.4 | 1.8 | 2.9 |
| G207 | F | Caucasian | 3.3 | 4.6 | 14.6 |
| H208 | F | Arab | NA | NA | 2.7 |
The clinical symptoms at the time of enrollment varied. Four patients (B, C, D, G) were profoundly hypotonic with minimal use of the lower extremities. Three patients (C, D, G) required tube feedings, and four patients (B, C, F, G) required supplemental oxygen.
At baseline, one patient (A) had recurrent supraventricular tachycardia (SVT) requiring propranolol therapy. An additional patient (F) was receiving propranolol for left ventricular outflow tract obstruction. Six patients (B, C, D, F, G, H) had symptoms of congestive heart failure requiring diuretics (B, C, D, F, G, H), captopril (B, G) digoxin (G), or spironolactone (B, G). All medications were prescribed or discontinued by the primary cardiologists caring for the patients, some of whom also were study investigators.
LV Mass and Volume
Figure 1 shows that baseline LV mass was severely increased at study entry. The baseline indexed LV mass ranged from 157 to 565 gm/m2 and fell to a range of 54 to 124 gm/m2 at study end (p < 0.001). For children with a body surface area of 0.2 to 0.6 m2, the upper limits of normal (z-score of 2) for indexed LV mass is 70 to 75 gm/m2 in our normative database. All eight patients achieved a decrease in LV mass, with some patients achieving a mass within the normal range. The baseline hypertrophy was fairly concentric, although the ventricular septal wall was somewhat thicker on the average than the posterior wall (Fig. 2).
Fig. 1.

Indexed left ventricular (LV) mass versus time. The graph shows the change in mass during the 52 weeks of therapy (p < 0.001). Note the rapid change in the indexed mass between the baseline and week 8 measurements. All eight patients are represented, but not all patients have a calculated mass for all seven time points due to technical limitations. The dotted line delineates the upper limits of normal in the normative database used for this analysis for children with a body surface area ranging from 0.2 to 0.6 m2. The patient whose data ends at 12 weeks (inverted triangles) died 14 weeks into treatment
Fig. 2.

Ventricular wall diameter versus time. Composite of the septal and posterior wall diameters during the 52 weeks of therapy. (a) The change in septal wall z-score. (b) The corresponding change in posterior wall z-score. Some data points are missing due to technical limitations. All five U.S. patients are represented in this data set, but only one of the European patients. The patient whose data ends at 12 weeks (inverted triangles) died 14 weeks into treatment
The septal wall diameters ranged from 14 to 21 mm (z-scores of 4.9–9.5), whereas the posterior wall diameters ranged from 8.7 to 14.7 mm (z-scores of 5.5–9.5). With treatment, both the septal and posterior wall thicknesses declined rapidly, with some patients showing a response as early as week 4, and most patients showing at least some response by week 8 of therapy. By study end, six of seven patients with reliable measurements had a septal wall thickness less than 1 cm, and four measurements were within the normal range. Six of seven patients with good quality measurements had a posterior wall thickness less than 1 cm, all of which were within the normal range.
The overall size of the LV was assessed by calculating the LV epicardial cross-sectional area in the parasternal short axis view, defined as all the LV within the epicardial surface at end diastole, including the ventricular septum. Unfortunately, apical images were not of adequate quality to calculate epicardial volumes for most patients. This hampered the derivation of LV mass based on volume data.
At baseline, the LV epicardial area was increased in all five of the patients for whom it could be calculated, corresponding to the increased size of the cardiac silhouette on chest radiograph. With treatment, overall LV size decreased dramatically, at about the same rate that the ventricular walls thinned. Over that same period, the intracavitary diameter increased by only a small amount. Figure 3a shows that the LV epicardial cross-sectional area dropped very quickly after treatment began. Figure 3b shows the concomitant change in the internal LV cavity diameter at end diastole. Some patients had an initial increase in LV internal diameter as the walls thinned, but the diameter adjusted downward again fairly quickly as the overall LV size became smaller.
Fig. 3.

Epicardial cross-sectional area and end diastolic diameter versus time. (a) The change in epicardial cross-sectional area measured at end diastole. Only the five U.S. patients are represented in this figure. (b) The change in intracavitary diameter at end diastole, measured from the M-mode data. All eight patients are represented in this graph. The patient whose data ends at 12 weeks died 14 weeks into treatment
The best illustration of how these variables interact during cardiac remodeling is seen in the graph of LV mass-to-volume ratio over time (Fig. 4). Because the intracavitary volume increases only by a small amount compared with the dramatic drop in LV mass, the mass-to-volume ratio falls precipitously in most patients.
Fig. 4.
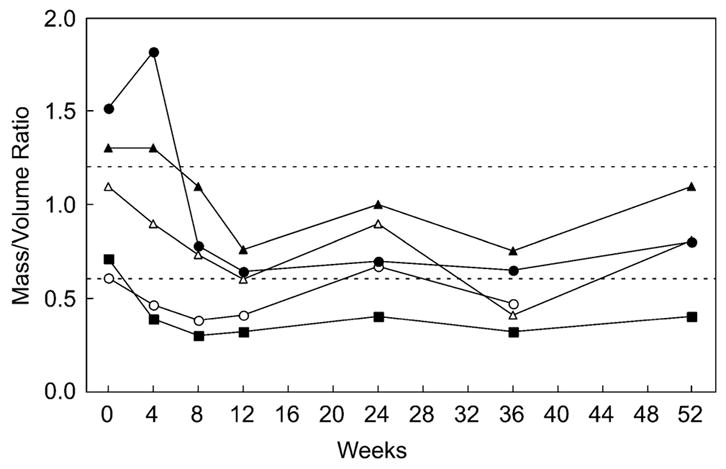
Left ventricular (LV) mass-to-volume ratio versus time. The change in LV mass-to-volume ratio. Only the five U.S. patients are represented in this figure. The dotted lines outline the approximate normal range for this parameter in children whose body surface area ranges from 0.2 to 0.6 m2. The patient whose data ends at the 36-week time point died before the final data collection time point (open circles)
By the 12th week of ERT, all five patients had a mass-to-volume ratio less than 1. Thereafter, there was some variability, with a final range of 0.4 to 1.1. Figure 5 illustrates the two-dimensional echocardiographic appearance of the ventricle (end diastole) over the year of therapy in one of the patients.
Fig. 5.
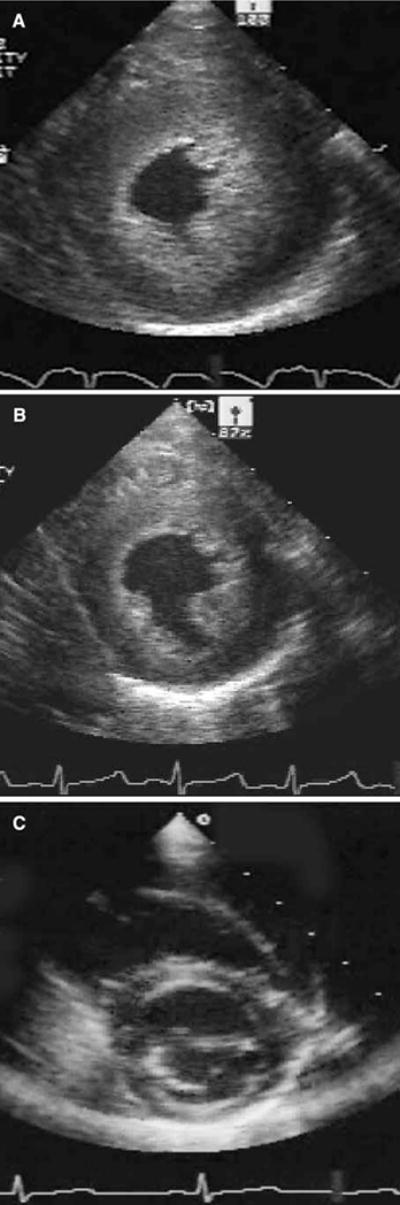
Echocardiographic appearance of the left ventricle. (a) Image of the left ventricle at end diastole before the start of treatment. (b) Image from the same patient after 4 weeks of therapy. (c) Image of the same patient after 52 weeks of therapy
Ventricular Function
The ejection fraction and the shortening fraction were used as indices of systolic function. The shortening fraction was calculated for all eight patients, but due to technical limitations in the data, the ejection fraction could be calculated only for the five U.S. patients. Figure 6 shows the ejection fraction data for the five U.S. patients and the shortening fraction for all eight patients. Six patients had normal indices of systolic function at study entry, whereas two had mildly depressed systolic function. With the start of therapy, five patients experienced a decline in indices of systolic function. The patient with the largest change experienced a cardiac arrest before the week 4 measurement. It is likely that this acute event contributed to the transiently poor function in this patient. All the patients had normal indices of systolic function by 36 weeks into the study period (p = 0.84).
Fig. 6.
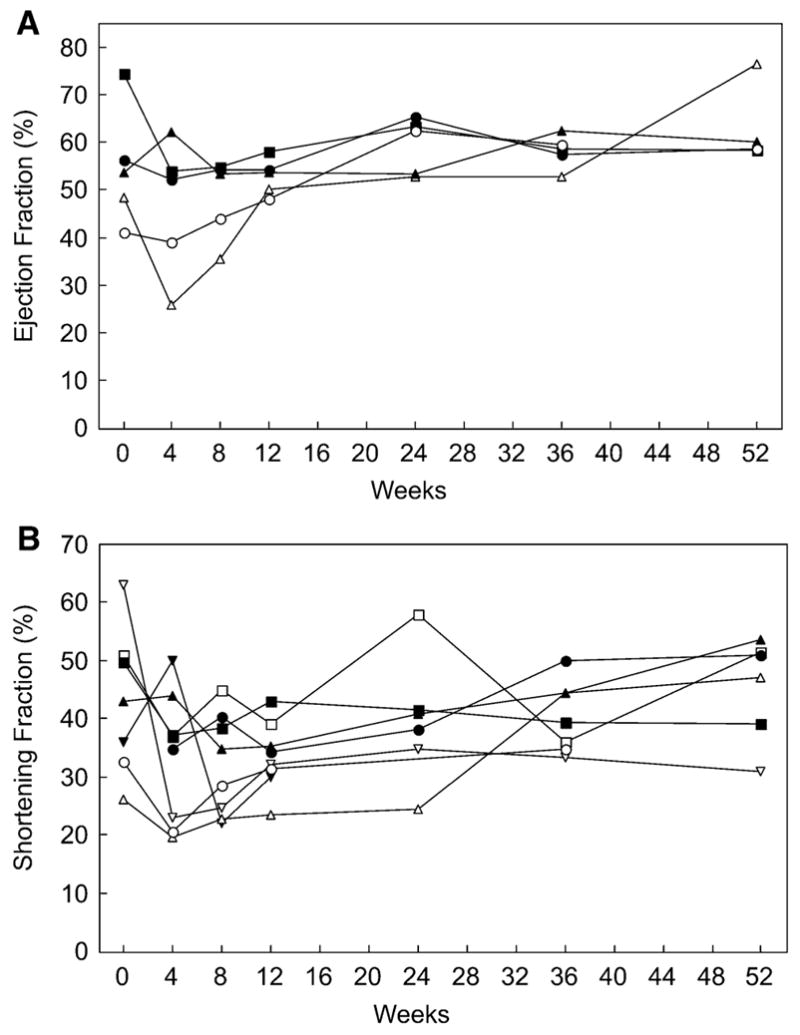
Systolic function versus time. (a) Ejection fraction. Only the five U.S. patients are represented. The patient whose data ends at 36 weeks (open circle in both graphs) died before the 52-week data collection. (Normal range of ejection fraction is approximately 60% to 75% depending on age at the time of data collection). (b) Shortening fraction. All eight patients are represented in this graph, although not all time points are available for each patient due to technical limitations. The patient whose data ends at the 12-week time point (inverted triangle) died 14 weeks into treatment. (Normal range of the shortening fraction is approximately 32% to 45% depending on age at the time of data collection.) There is no significant change in the indices of systolic function between the start of therapy and the 52-week time point (p = 0.84)
Detailed diastolic function variables were not collected as part of this initial study, but E- to A-wave (E/A) ratios were assessed when the data were adequate. Because of high heart rates in this group, the E- and A-waves were frequently summed, and a ratio could not be obtained. When the E/A ratio was available, it usually was normal, although occasionally a reversed relationship with a taller A-wave was identified.
Additional Data
Left ventricular outflow tract obstruction was present in three of eight patients at baseline, with peak gradients of 36 to 60 mmHg at rest (mean gradients could not be calculated). Obstruction was caused by hypertrophy of the ventricular septum and never by aortic valve disease. Some of the patients had associated systolic anterior motion of the mitral valve, but none had more than mild mitral regurgitation. Left ventricular outflow tract obstruction resolved by 8 weeks into therapy for all three patients.
Right ventricular pressure could not be accurately calculated for most of the patients. The data from the three European patients were less complete and therefore not included in some of the graphs.
Chest Radiograph
All the patients had cardiomegaly in the baseline study. Patients B, C, and F had pulmonary edema at baseline, which resolved by week 8. The baseline cardiothoracic ratio had a median value of 0.70 (range, 0.60–0.73). At 52 weeks, there was a trend toward a decrease in median cardiothoracic ratio to 0.57 (range, 0.55–0.60; p = 0.06). Four of the six surviving patients had a cardiothoracic ratio below 0.6 at 52 weeks, suggesting that they no longer met the criteria for cardiomegaly.
ECGs
All patients had a short PR interval and large precordial voltages on 12-lead electrocardiogram. At baseline, the median PR interval was 90 ms (range, 80–100 ms), and the LV voltage (SV1 + RV6) was 54 mV (range, 40–72 mV). At 52 weeks, there was a significant increase in the PR interval to a median value of 120 (range, 100–140; p < 0.001) and a significant decrease in LV voltage to a median value of 36 mV (range, 18–60 mV; p = 0.02) (Table 2). One patient (A) had Wolff-Parkinson-White syndrome, according to baseline electrocardiogram, and recurrent episodes of SVT. He received a successful radiofrequency ablation at the age of 2 years.
Table 2.
Median laboratory parameters
| Parameter | Baseline (n = 8) | 52 weeks (n = 6) | p value |
|---|---|---|---|
| Range | Range | ||
| LV mass by 2D echo (g/m2) | 191 (157–565) | 87 (53–124) | <0.001 |
| PR interval on ECG (ms) | 90 (80–100) | 120 (100–140) | <0.001 |
| Sv1 + RV6 on ECG (mV) | 54 (40–71) | 36 (18–60) | 0.02 |
| Cardiothoracic ratio on CXR | 0.70 (0.60–0.73) | 0.57 (0.55–0.60) | 0.06 |
LV, left ventricular; PR, 2D, two-dimensional; ECG, electrocardiogram; CXR, chest X-ray
Congestive Heart Failure
Six patients (B, C, D, F, G, H) required ongoing therapy with furosemide during the initial weeks of ERT. Two of these six patients also were maintained on captopril (B, G), and two patients were receiving spironolactone (B, G) One patient (G) was additionally maintained using digoxin. At a median time of 8 weeks (5–12 weeks) after the initiation of ERT, all medications started for the treatment of congestive heart failure symptoms had been discontinued.
Noncardiac Outcome
The noncardiac findings and the safety data have been detailed in a prior publication [11]. In summary, ERT was found to be safe and generally well tolerated. Patients B and G died during the study after 43 and 16 weeks of treatment, respectively. Patient B died of respiratory complications at the age of 14.7 months. This patient had a profile and response to treatment similar to those for the rest of the cohort. Patient G began ERT at the late age of 14.6 months and had very severe disease at initiation of the study. This patient died just 16 weeks into treatment during an episode of pneumonia complicated by cardiac arrest. This patient’s late age at the initiation of treatment may well have contributed to the poor outcome. Notably, both patients who died responded to treatment with a marked decrease in LV mass, similar to the other subjects.
No patient had to discontinue therapy because of adverse side effects. On the average, this cohort had better growth, neurologic status, and motor skills than a cohort of historical controls [11]. The response of the skeletal muscle was much more variable than that of the cardiac muscle. Overall, however, there was a marked increase in muscle GAA activity and a significant decrease in glycogen content on quadriceps muscle biopsy. Neurologic improvement also was variable, with some patients having a much better performance on neurologic testing than others. It was encouraging to note that most subjects exhibited some improvement in test parameters, with some subjects showing nearly normal skills for age.
In addition to improved symptoms, this cohort had significantly improved survival and ventilator-free survival. A large retrospective study suggests that less than 10% of infants with Pompe disease survive beyond the first year of life [4, 5]. Six of the eight patients in this cohort were alive at 18 months. Four of the eight were alive at 24 months, and at this writing, two of the eight are alive and ambulatory.
Extension Phase
At the conclusion of the original 52-week protocol, the patients were offered the opportunity to continue treatment as part of an extension protocol, which included continued regular monitoring of all parameters including the cardiac parameters. Six of the eight patients were enrolled for the extended protocol. All six showed sustained improvement in LV mass compared with baseline, and some patients had continued improvement during the extension phase. The longest follow-up period at this writing is more than 3 years since the institution of drug therapy. The patients also continued to show gains in growth and motor milestones during the extension phase of the protocol.
Four patients died during the extension phase. Therefore, only two of the original eight patients are still alive at this writing. Patient C died at 33.8 months of pneumonia complications. Patient D died of pneumonia at 24.8 months (the family declined invasive ventilation). Patient F died unexpectedly at 32 months, also of symptoms associated with a respiratory infection. Finally, Patient H became ventilator dependent after pneumonia at 18.5 months of age. Invasive treatment was discontinued in accordance with the family’s wishes, and the patient died 5 months later.
Altogether, six of the eight patients died, two during the phase 2 trial and four during the extension phase. Although these patients lived well beyond the usual life span for an infant affected with Pompe disease, it is disappointing that this cohort lost so many subjects in early childhood. The reasons why these patients remained vulnerable to respiratory disease are unclear. The cardiac data show no feature that separates those who survived from those who did not. One could speculate that the variability in the skeletal muscle GAA activity identified may be a clue. It certainly is possible that the muscles of respiration had the same variable response and therefore left some patients with diminished ability to handle respiratory distress. Whether different treatment regimens or different dosing might affect this response or not is still speculative.
Discussion
Despite the small number of patients in this study, it is clear that IV administration of rhGAA can reverse glycogen accumulation in cardiac muscle cells when administered to infants with Pompe disease. In the small study group, ERT led to a decrease in ventricular hypertrophy, reversal of LV outflow tract obstruction, normalization of the conduction system as assessed by ECG (except for the patient with Wolff-Parkinson-White syndrome), and a decreased need for medication to control symptoms of pulmonary congestion. Some patients experienced complete normalization of LV mass. The rapidity with which the effect of ERT could be measured was striking. Changes in the heart were evident in some infants as soon as 4 weeks after the initiation of therapy, and improvements continued or were maintained throughout the first year of therapy. All patients in this small group experienced regression of LV hypertrophy regardless of age or the stage of disease at entry. Although the extent of the response varied, the pattern of remodeling was remarkably uniform, much more so than the skeletal muscle response, which varied markedly between patients, as evidenced by both clinical response and skeletal muscle biopsy [11].
The pattern of remodeling in these patients was interesting. With thinning of the walls, we expected to see one of three initial responses: the heart size would remain stable and the intracavitary volume would increase as the walls thinned; the heart size would shrink and the intracavitary volume would remain stable; or some combination of the two would occur. In this group, the response was a combination of the two, although it was closer to the second scenario in that overall heart size decreased, with only a small increase in intracavitary volume.
The effect of these rapid changes on ventricular function was noteworthy. With the early and sudden increase in intracavitary volume, most patients had a transient drop in shortening fraction and ejection fraction. However, as LV cavity size normalized, so did the indices of systolic function. Left ventricular cavity size and indices of systolic function were generally within the normal range by 12 weeks into therapy.
This remodeling process illustrates an impressive ability of the cardiovascular system to compensate quickly for rapid changes in wall thickness and compliance, similar in some ways to the process noted after aortic valve replacement for severe aortic regurgitation. With resolution of severe aortic regurgitation, a rapid decrease in ventricular cavity size usually causes the LV to appear hypertrophied as mass is conserved. Because of the Frank-Starling mechanism, this change in cavity size also may lead to diminished indices of systolic function. The cardiovascular system adjusts rapidly by decreasing wall thickness, thus normalizing cavity size and leading to improved wall stress and cardiac output.
The patients in this trial illustrate a similar process but in the opposite direction. Instead of thick walls and a smaller ventricular cavity, the treatment resulted in a rapid thinning of the walls and a larger ventricular cavity. In this setting, the cardiovascular system normalized wall stress by decreasing LV end diastolic diameter. As the cavity size and mass-to-volume ratio normalized, the indices of systolic function improved.
Unfortunately, diastolic function in these patients was not studied in detail. Presumably, LV compliance was diminished at baseline and improved with a decrease in glycogen storage. The best evidence for this is the decreased pulmonary edema shown on the chest X-ray and the decreased dependence on diuretics during the trial. The majority of the patients enrolled required daily medication for symptoms of congestive heart failure using a combination of diuretics, angiotensin-converting enzyme inhibitors, digoxin, and spironolactone. It is likely that edema and diuretic dependence were secondary to poor compliance as a result of LV hypertrophy. All medications could be discontinued at a median time of 8 weeks after initiation of therapy except in the case of one patient maintained on propanolol for SVT. The improvement in these surrogates for congestive failure symptoms correlated with the rapid drop in the LV mass and likely resulted from improved LV compliance.
With ERT, the ECG parameters became more normal at about the same rate as the echo parameters. We have previously reported ECG changes in a heterogeneous group of patients with Pompe disease who received ERT under a different protocol [13]. This study confirms these findings with a larger group of patients studied over a longer period. The PR interval is characteristically short in patients with Pompe disease due to glycogen accumulation and larger cell size in the conduction system [14]. With ERT, the PR interval lengthened, another indicator that the cardiac cells were able to decrease their glycogen content. A similar change in PR interval has been noted after ERT for Fabry’s disease [15].
The ECGs at baseline also showed the high-voltage QRS complexes commonly associated with Pompe disease [16]. With treatment, the ECG correlates of LVH (increased S voltage in V1 and R voltage in V6) decreased rapidly, mirroring the decrease in LV mass by echocardiography and the improvement in clinical status.
All the patients responded to enzyme replacement by decreasing ventricular hypertrophy, although the degree of improvement varied. It is unclear at this writing whether the variable response to treatment was related to differences among the patients in either the number or type of myofiber cell surface receptors or whether perhaps an immune-mediated response prevented a uniform availability of drug to the cell surface once it was administered.
Another unanswered question is why the response of skeletal muscle and the change in the noncardiac clinical parameters were so much more variable than the response of the cardiac muscle. There clearly are more GAA receptors on the cardiac cell surface then on the skeletal muscle cell surface [17]. This may explain why the heart muscle was more responsive to therapy and raises the question of whether an increased dosage would have produced a more robust response in the skeletal muscle or not. Data from ongoing trials suggest that it would not. Other possible explanations include the presence of different receptor-mediated uptake of GAA in the two different muscle types or a different ability of skeletal and cardiac muscle cells to clear glycogen once it is present to this degree [17, 18]. Ongoing studies of this cohort and others should help us to understand this mechanism better.
Study Limitations
It would have been helpful to have more information about the cardiac response including intracardiac pressures, pathology of the cardiac muscle, and noninvasive assessment of diastolic function. At the time the study was initiated, diastolic assessment was not practical for the group as a whole. Trials currently in progress will include additional data on diastolic function. Invasive data were not part of the protocol because severe clinical symptoms made any procedure requiring sedation too risky to consider [19]. However, as we begin to study the less severe forms of infantile Pompe disease, it may be reasonable to incorporate other methods of investigating the heart including cardiac magnetic resonance imaging (MRI) and perhaps cardiac catheterization/biopsy. These methods have the potential to increase greatly our understanding of the mechanisms underlying the response of the heart to ERT in Pompe disease.
Summary
The cardiac response to rhGAA therapy of patients with classical infantile Pompe disease is dramatic, as evidenced by improvements in clinical symptoms, echocardiographic parameters, ECG parameters, and cardiac size on chest radiograph. It is likely that a decrease in ventricular hypertrophy or its resolution improves both systolic and diastolic cardiac function and decreases the risk of early death due to cardiovascular and pulmonary complications.
This is the first detailed report of the cardiac response of infants with the severe form of Pompe disease to ERT. Long-term study investigating this group of patients continues, and new trials are underway to evaluate the effect of ERT on less severe forms of the disease. The ongoing studies have been designed to include multiple noninvasive measures of both diastolic and systolic function in an effort to increase our understanding of the cardiac response to ERT.
Acknowledgments
We thank the study patients and their families for their participation in this clinical trial. In addition, we mention our great appreciation for the support and expertise of the physicians, study coordinators, and research assistants, without whom this study could not have been conducted. Finally, we acknowledge the tremendous input of the following individuals from the Genzyme Corporation, without whom this trial could not have been completed: Jennifer Hunt, Tara O’Meara, Florence Yong, MS PhD, and Deyanira Corzo, MD. This trial was supported by a grant from the Genzyme Corporation to the various sites at which patients were treated. Priya S. Kishnani and Y. T. Chen received research and grant support from the Genzyme Corp. Priya S. Kishnani is a member of the Pompe Disease Advisory Board for the Genzyme Corporation. Jami C. Levine and Y. T. Chen have served as consultants for the Genzyme Corporation. The U.S. FDA and the European Union have approved rhGAA, in the form of Genzyme’s product, Myozyme, as therapy for Pompe disease. Duke University and the inventors for the method of treatment and the predecessors of the cell lines used to generate the enzyme used in this clinical trial may benefit financially pursuant to the Duke University’s Policy on Inventions, Patents, and Technology Transfer. This study was supported in part by grants M01-RR30 and M01-RR01271 from the General Clinical Research Centers Program, Division of Research Resources, National Institutes of Health and by Genzyme Corporation.
References
- 1.Kishnani P, Howell RR. Pompe disease in infants and children. J Pediatrics. 2004;144:S35–S43. doi: 10.1016/j.jpeds.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 2.Griffin JL. Infantile acid maltase deficiency: muscle fiber hypertrophy and the ultrastructure of end-stage fibers. Virchows Arch Cell Pathol. 1984;45:37–50. [PubMed] [Google Scholar]
- 3.Martiniuk F, Chen A, Mack A, et al. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Gen. 1998;79:69–72. doi: 10.1002/(sici)1096-8628(19980827)79:1<69::aid-ajmg16>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 4.Van den Hout HM, Hop W, van Diggelen OP, et al. The natural course of infantile Pompe disease: 20 original cases compared with 133 cases from literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 5.Kishnani PS, Hwu WL, Mandel H, et al. A retrospective multinational multicenter study on the natural history of infantile-onset Pompe disease. J Pediatr. 2006;148:671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 6.Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med. 2001;3:132–138. doi: 10.109700125817-200103000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Van den Hout H, Reuser AJ, Vulto AG, et al. Recombinant human α-glucosidase 17 from rabbit milk in Pompe patients. Lancet. 2000;356:397–398. doi: 10.1016/s0140-6736(00)02533-2. [DOI] [PubMed] [Google Scholar]
- 8.Van den Hout JMP, Kamphoven JHJ, Winkel LPF, et al. Long-term intravenous treatment of Pompe disease with recombinant human α-glucosidase from milk. Pediatrics. 2004;113:e448–e457. doi: 10.1542/peds.113.5.e448. [DOI] [PubMed] [Google Scholar]
- 9.Kishnani P, Voit T, Nicolino M, et al. Enzyme replacement therapy with recombinant human α-glucosidase (rhGAA) in infantile Pompe disease: results form a phase 2 study. Pediatr Res. 2003;53:259A. [Google Scholar]
- 10.Klinge L, Straub V, Neudorf U, et al. Safety and efficacy of recombinant acid alpha-glucosidase (rhGAA) in patients with classical infantile Pompe disease: results of a phase II clinical trial. Neuromuscular Dis. 2005;15:24–31. doi: 10.1016/j.nmd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Kishnani P, Nicolino M, Voit T, et al. Chinese hamster ovary cell-derived recombinant human acid alpha-glucosidease in infantile-onset Pompe disease. J Pediatr. 2006;149:89–97. doi: 10.1016/j.jpeds.2006.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sluysman T, Colan SD. Theoretical and empirical derivation of cardiovascular allometric relationships in children. J Appl Physiol. 2005;99:445–457. doi: 10.1152/japplphysiol.01144.2004. [DOI] [PubMed] [Google Scholar]
- 13.Ansong AK, Li JS, Ing R, et al. Electrocardiographic changes in Pompe disease following enzyme replacement therapy for Pompe disease. Genet Med. 2006;8:297–301. doi: 10.1097/01.gim.0000195896.04069.5f. [DOI] [PubMed] [Google Scholar]
- 14.Bharati S, Serratto M, DuBrow I, et al. The conduction system in Pompe’s disease. Pediatr Cardiol. 1982;2:25–32. doi: 10.1007/BF02265613. [DOI] [PubMed] [Google Scholar]
- 15.Waldek S. PR interval and the response to enzyme-replacement therapy for Fabry’s disease. N Engl J Med. 2003;348:1186–1187. doi: 10.1056/NEJM200303203481224. [DOI] [PubMed] [Google Scholar]
- 16.Jacob JL, Leandro RL, Parro Junior A. Pompe’s disease or type IIa glycogenosis. Arq Bras Cardiol. 1999;73:435–440. doi: 10.1590/s0066-782x1999001100004. [DOI] [PubMed] [Google Scholar]
- 17.Raben N, Jatkar T, Lee A, et al. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther. 2002;6:601–608. [PubMed] [Google Scholar]
- 18.Raben N, Danon M, Gilbert AL, et al. Enzyme replacement therapy in the mouse model of Pompe disease. Mol Genet Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 19.Ing RJ, Cook DR, Bengur RA, et al. Anaesthetic management of infants with glycogen storage disease type II: a physiological approach. Paediatr Anaesth. 2004;14:514–519. doi: 10.1111/j.1460-9592.2004.01242.x. [DOI] [PubMed] [Google Scholar]