

Abstract
Vascular endothelial growth factor (VEGF) is required for vasculogenesis and angiogenesis during embryonic and early postnatal life. However the organ-specific functional role of VEGF in adult life, particularly in skeletal muscle, is less clear. To explore this issue, we engineered skeletal muscle-targeted VEGF deficient mice (mVEGF−/−) by crossbreeding mice that selectively express Cre recombinase in skeletal muscle under the control of the muscle creatine kinase promoter (MCKcre mice) with mice having a floxed VEGF gene (VEGFLoxP mice). We hypothesized that VEGF is necessary for regulating both cardiac and skeletal muscle capillarity, and that a reduced number of VEGF-dependent muscle capillaries would limit aerobic exercise capacity. In adult mVEGF−/− mice, VEGF protein levels were reduced by 90 and 80% in skeletal muscle (gastrocnemius) and cardiac muscle, respectively, compared to control mice (P < 0.01). This was accompanied by a 48% (P < 0.05) and 39% (P < 0.05) decreases in the capillary-to-fibre ratio and capillary density, respectively, in the gastrocnemius and a 61% decrease in cardiac muscle capillary density (P < 0.05). Hindlimb muscle oxidative (citrate synthase, 21%; β-HAD, 32%) and glycolytic (PFK, 18%) regulatory enzymes were also increased in mVEGF−/− mice. However, this limited adaptation to reduced muscle VEGF was insufficient to maintain aerobic exercise capacity, and maximal running speed and endurance running capacity were reduced by 34% and 81%, respectively, in mVEGF−/− mice compared to control mice (P < 0.05). Moreover, basal and dobutamine-stimulated cardiac function, measured by transthoracic echocardiography and left ventricular micromanomtery, showed only a minimal reduction of contractility (peak +dP/dt) and relaxation (peak –dP/dt, τE). Collectively these data suggests adequate locomotor muscle capillary number is important for achieving full exercise capacity. Furthermore, VEGF is essential in regulating postnatal muscle capillarity, and that adult mice, deficient in cardiac and skeletal muscle VEGF, exhibit a major intolerance to aerobic exercise.
Vascular endothelial growth factor (VEGF) is a potent angiogenic mitogen known to increase vascular permeability and promote angiogenesis (Folkman, 1976; Ferrara, 1999). VEGF is required for vascular development during the early phases of life (i.e. embryonic growth and postnatal development), and attempts to generate traditional global VEGF knock-out mice result in embryonic lethality (Ferrara et al. 1996; Gerber et al. 1999). Remarkably, the loss of even a single VEGF allele (i.e. a heterozygous defect) results in embryonic death due to a poorly organized and/or malformed cardiovascular network (Carmeliet et al. 1996). The role and importance of VEGF in the control of normal postnatal vessel regulation is less well known. In many but not all mature organs, capillary networks are stable or maintained independent of VEGF signalling (Benjamin et al. 1999; Baffert et al. 2004; Kamba et al. 2006). However, in skeletal muscle, Tang et al. (2004), utilizing a Cre/loxP strategy in adult mice, have shown that selective deletion of VEGF within a small localized region in a single gastrocnemius muscle results in a capillary regression. Similarly, Giordano et al. (2001) found that cardiac-specific VEGF gene deletion results in fewer coronary microvessels, thinned ventricular walls, and impaired contractile function in young adult mice. The data from Tang et al. (2004) and Giordano et al. (2001) imply that the myocyte may be an important paracrine source of VEGF for regulating muscle microvasculature and contractile function.
Skeletal muscle vasculature is well known to exhibit a high degree of plasticity (Holloszy, 1976; Holloszy & Coyle, 1984). For instance, numerous studies have implicated VEGF in regulating exercise-induced skeletal muscle angiogenesis in both man and animals (Breen et al. 1996; Gustafsson et al. 1999; Richardson et al. 1999; Olfert et al. 2001; Prior et al. 2003, 2004). Capillary rarefaction in chronic pathological conditions, such as COPD and diabetes (Barreiro et al. 2008; Kivela et al. 2008), suggests that VEGF is not only important in de novo vessel formation (i.e. vasculogenesis) but also in the maintenance of adult skeletal muscle microvasculature. Yet, it has been difficult to evaluate the adverse physiological consequences of reduced muscle capillarity in the adult, given high variability of individual exercise performance capabilities and that compensatory responses (i.e. changes in fibre type composition, muscle atrophy, metabolic adaptations) are frequently found. Thus, this new transgenic model allows the consequences of reduced VEGF-dependent muscle capillarity to be assessed using an integrated physiological approach.
We report here the physiological consequences and exercise responses of combined skeletal and cardiac muscle-specific VEGF gene deletion (mVEGF−/−) in mice. We hypothesized that if VEGF is vital to the maintenance and formation of skeletal muscle capillaries during adult life, there would be (1) fewer capillaries in mVEGF−/− skeletal muscle and heart, and (2) impaired muscle O2 transport, as evidenced by reduction in maximal and/or endurance exercise capacity. We also questioned, to the extent possible, the relative importance of skeletal versus cardiac muscle dysfunction in explaining changes in exercise performance. Finally, we queried whether skeletal muscle compensatory mechanisms to increase oxygen availability are invoked to mitigate the loss of VEGF. Possibilities examined included increased haemoglobin concentration and haematocrit, altered muscle fibre type distribution, and/or increased metabolic enzyme activities.
Methods
In this study we report on the morphometric and functional consequences of (combined skeletal and cardiac) muscle-specific VEGF gene deletion in 5-month-old mice (n= 29) compared to wild-type (WT, n= 25) sibling littermates expressing normal muscle VEGF levels. This study was approved by University of California, San Diego, Animal Care and Use Committee and conducted in accordance to guidelines outlined by the Guide for the Care and Use of Laboratory Animals (National Research Council). Mice were housed three to four per cage in the same room in a pathogen-free vivarium maintained on 12:12 h day–night cycle and were provided with standard chow (Harlan Tekland 8604, Madison, WI, USA) and tap water ad libitum. Upon conclusion of the respective experiments, mice were killed by surgical removal of the heart while under anaesthesia (sodium pentobarbital, 60 mg kg−1, i.p.)
Creation of the muscle VEGF-deficient (mVEGF−/−) mouse model
This mouse model was developed by crossbreeding two separate transgenic mouse lines, each on a C57BL background. VEGFLoxP mice, containing 34 bp loxP sequences flanking the exon 3 region in the VEGF gene, were kindly provided by Dr Neopolean Ferrara, Genentech, Inc. (San Francisco, CA, USA) (Gerber et al. 1999). Mice having muscle specific Cre recombinase expression, under the control of the muscle creatine kinase (MCK) promoter, i.e. MCK-cre transgenic mice, were kindly provided by Dr Randall Johnson, University of California San Diego (La Jolla, CA), with permission from Dr K. Ronald Kahn, Joslin Diabetes Center (Boston, MA, USA) (Bruning et al. 1998). In VEGFLoxP mice, the exon 3 region (flanked with loxP sites) gene provides coding common to all VEGF isoforms (i.e. VEGF120, VEGF164, VEGF188, VEGF205, including the less frequent VEGF144, VEGF182) (Robinson & Stringer, 2001; Ferrara et al. 2003) and therefore the VEGF gene mutation in our mouse model results in loss of all VEGF-A isoforms. Temporally, MCK mRNA levels are first detectable in the fetus on Day 16 (Trask & Billadello, 1990); thus inactivation of the VEGF gene in the myocyte in our model is expected to begin during late gestation and rapidly increases after birth, where MCK expression increases more than 5-fold by postnatal day 30 (Trask & Billadello, 1990).
Genotyping
Genotyping for identification and selection of breeding pairs was performed on DNA extracted (DNeasy Tissue Kit, Qiagen Inc., Valencia, CA, USA) from mouse tail sections. Later, DNA obtained from the gastrocnemius muscle was also used to verify animal genotype. Genotype determination was made by PCR analysis using TaqPro™ Red Complete DNA Polymerase Master Mix (Denville Scientific, Inc., Metuchen, NJ, USA) using the following probes: for VEGFLoxP, forward primer 5′-TCCGTACGACGCATTTCTAG-3′ and reverse primer 5′-CCTGGCCCTCAAGTACACCTT-3′; for Cre recombinase, forward primer 5′-CTAGAGCCTG-TTTTGCACGTTC-3′ and reverse primer 5′-TGCAA-GTTGAATAACCGGAAA-3′. PCR analysis was performed on a Robocycle Gradient 40 (Stratagene Inc., La Jolla, CA, USA) with the following conditions for both VEGFLoxP and MCK/Cre; one 2 min incubation at 95°C (polymerase activation), followed by 30 cycles of 75 s at 94°C (denaturation), 100 s of 53°C (annealing), and 170 s at 72°C (extending), followed by one 10 min period at 72°C. PCR product from each animal, along with positive and negative controls, was run on 2% ethidium bromide stained agarose gel (NuSieve GTG/SeaKem HGT agarose, Cambrex Bio Science Rockland, Inc., Rockland, ME, USA) in 1X TAE buffer.
The first generation (F1) offspring from VEGFLoxP× MCK/Cre mice were heterozygous for both genetic inclusions. Subsequent breeding of the heterozygous F1 generation mice yielded an F2 generation in which approximately one-quarter were homozygous for both transgenes (mVEGF−/−). Each F2 generation litter were normal size (6–8 pups) with no unexplained deaths. Control, i.e. WT, mice were selected from sibling F2 generation mice lacking either the LoxP or Cre sequence, or both. All mice were studied at 5 months of age.
Organ-specific VEGF, VEGF-R1 and VEGF-R2 levels
The gastrocnemius (medial and lateral heads) and cardiac muscle were homogenized separately in a lysis buffer containing 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 0.5% Triton X-100, and protease inhibitors (Complete™ Tablet, Roche Company, Mannheim, Germany). Homogenates were centrifuged at 4°C, 7200g, for 10 min and supernatants assayed for total protein concentration (Bio-Rad DC protein assay, Bio-Rad Laboratories, Hercules, CA, USA). VEGF protein levels were measured using enzyme-linked immunoassay kit for mouse (VEGF Mouse ELISA kit no. QIA52, Calbiochem, La Jolla, CA, USA) designed for detecting VEGF164 and VEGF120 isoforms of VEGF-A. VEGF receptor 1 (flt-1) and receptor 2 (flk-1) protein levels were measured using a soluble VEGF-R1/flt-1 ELISA kit (RnD Systems, MVR100) and VEGF-R2/flk-1 ELISA kit (RnD Systems, MVR200B).
Skeletal and cardiac muscle histochemistry
Under anaesthesia (60 mg kg−1 sodium pentobarbital i.p.) the hindlimb muscles (i.e. gastrocnemius, soleus and plantaris muscles) were surgically removed, sliced in half at the muscle mid-belly, and quickly flash frozen with freezing medium gel (TBS Tissue Freezing Medium™, Triangle Biomedical Sciences, Durham, NC, USA) in a liquid N2-cooled isopentane bath. The heart was cut in half perpendicular to the crano-caudal axis and flash frozen in the same manner as skeletal muscle. Muscle samples were stored at −80°C. Both cardiac and skeletal muscles were cut into serial 9 μm tranverse sections and prepared for assessment of muscle capillarity using the alkaline phosphatase capillary staining method (Mrazkova et al. 1986). Skeletal muscle section were also stained for fibre-type expression using the metachromatic dye-ATPase technique (Ogilvie & Feeback, 1990).
Stained muscle sections were viewed by light microscopy and digitally imaged (40× magnification). In the gastrocnemius muscle, capillarity corresponding to the medial superficial, medial deep, lateral superficial, and lateral deep regions of the muscle were separately recorded. All regions together accounted for >90% of the total gastrocnemius cross-sectional area. The acquired images were viewed using computer software (MATLAB version 7.0, The MathWorks, Inc., Natick, MA, USA) for assessment of the number of capillaries and muscle fibre size and type. Within each image, mean fibre area was calculated from the total number of fibres divided by the total muscle area (excluding spaces between muscle fibres and any non-muscle structures). Non-muscle areas were distinguished using an algorithm designed to differentiate colour differences (RGB spectral analysis) between muscle and non-muscle tissue. Stained cardiac muscle sections were viewed by light microscopy and the number of capillaries observed within a 100-point grid with a fix area (62.5 μm2) at 40× magnification were recorded from eight randomly selected sites within the subendocardium region. The average capillary density from all eight sites within each heart was used to compare capillary densities between mVEGF−/− and WT mice.
Skeletal muscle enzyme activities
All enzyme measurements were performed at room temperature with a Beckman model DU 640B spectrophotometer. To assay citrate synthase (CS) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) activities, whole muscle (6–10 mg) was homogenized in 100 volumes (w:v) of buffer (175 mm KCl, 2 mm EDTA; pH 7.4) using a Polythron (PT1200E) homogenizer and subjected to three freeze–thaw cycles using liquid N2, then centrifuged at 7200g for 1 min and the supernatant collected. CS activity was assayed as per Srere (1969). β-HAD activity was assayed as per Bergmeyer (1974) by following the oxidation of NADH spectrophotometrically. Phosphofructokinase (PFK) activity was measured in muscle homogenates by the method of Lowry et al. (1978).
Aerobic exercise performance
All mice were familiarized to a small animal treadmill (model CL-4, Omnitech, Columbus, OH, USA) using a low treadmill speed (5 m min−1, 0 deg incline) for 10 min prior to performing the exercise test. Exercise testing consisted of two separate tests, (1) a submaximal run to exhaustion and (2) determination of maximal running speed, each of which was performed on a separate day.
The submaximal endurance run consisted of treadmill running at 10 m min−1 for the first 10 min (10 deg incline) followed by 15 m min−1 until exhaustion. Exhaustion was defined when the mouse was no longer able to maintain their normal running position and/or frequent contact with the shock grid (≤ 0.2 mA) at the rear of the treadmill.
The maximal running speed test consisted of the following protocol: 8 m min−1 (no incline) for 1 min, after which running speed was increased by 2 m min−1 every 30 s, until exhaustion.
Transthoracic echocardiography
Mice were anaesthetized with isoflurane (5% in 100% oxygen) with a flow rate of 1 l min−1 for 1 min and then maintained at 1% isoflurane throughout transthoracic echocardiography. The anterior chest wall was shaved and small needle electrodes for simultaneous electrocardiogram were inserted into one upper limb and two lower limbs. Transthoracic echocardiography was performed using a Philips, Sonos 5500 instrument with a L15–6 MHz transducer. Measurements of heart rate (HR), left ventricle end-diastolic dimensions (LVEDD) and LV end-systolic dimensions (LVESD), end-diastolic interventricular septal thickness (IVSd) and LV posterior wall thickness (LVPWd) were determined from the LV M-mode tracing. Percentage fractional shortening (%FS) was used as an indicator of systolic cardiac function and was calculated as follows %FS = (LVEDD – LVESD)/LVEDD × 100 (Giordano et al. 2001; Bale et al. 2004).
Cardiac catheterization
Haemodynamic evaluation was performed while mice were under general anaesthesia (ketamine (100 mg kg−1)/xylazine (10 mg kg−1)) and connected to a ventilator. After bilateral vagotomy, 1.4 French (0.46 mm) micromanometer catheters (Millar Instruments, Houston, TX, USA) were inserted (via the common carotid artery) into the ascending aorta, and then across the aortic valve into the LV; phasic and mean pressures were continuously monitored using a WINDAQ (Dataq Instruments, Akron, OH, USA) system. In order to assess inotropic reserve, and to assess potential ischaemic myocardial dysfunction due to reduced capillarity, the responses of micromanometer left ventricular pressure to graded doses of the β-adrenergic agonist dobutamine (0.75, 2, and 4 μg kg−1 min−1) were also determined. Digital post-processing of micromanometer pressure signals, for calculation of maximum and minimum dP/dt, τE and τL (the monoexponential and logistic rate constants of isovolumic pressure fall, respectively) was accomplished as previously described (Bale et al. 2004).
Statistics
Data shown are means ±s.e.m., unless otherwise indicated. Comparisons between mVEGF−/− and control groups were made using Student's unpaired t-test. In the case of serial haemodynamic measurements, a repeated measures ANOVA was applied, following by post hoc testing of individual variables at discrete time points. In all cases, significance was accepted at a P < 0.05 level.
Results
Body mass and gastrocnemius muscle mass were both lower in mVEGF−/− than control (WT) mice by an average of 11% (P < 0.01) and 14% (P < 0.02), respectively (Table 1), such that muscle/body mass ratio was unchanged. Heart mass was not significantly different, and therefore a 9% rise in heart/body mass ratio in mVEGF−/− compared to controls was due to lowered body mass and not cardiac muscle hypertrophy. Blood haemoglobin and haematocrit levels were not different between mVEGF−/− and control mice (Table 1).
Table 1.
Body and organ mass and haematological variables
| WT (n= 25) | mVEGF−/− (n= 29) | P | |
|---|---|---|---|
| Age (days) | 153 ± 6 | 147 ± 7 | 0.54 |
| Body mass (g) | 25.6 ± 0.7 | 22.9 ± 0.7* | 0.01 |
| Gastrocnemius m. (mg) | 212 ± 7 | 186 ± 5* | 0.02 |
| Gastrocnemius/body mass (%) | 0.82 ± 0.01 | 0.83 ± 0.01 | 0.77 |
| Heart (mg) | 119 ± 5 | 116 ± 5 | 0.64 |
| Heart/body mass (%) | 0.46 ± 0.01 | 0.50 ± 0.01* | 0.02 |
| Lung (mg) | 158 ± 3 | 152 ± 4 | 0.56 |
| Kidney (mg) | 332 ± 9 | 297 ± 11 | 0.13 |
| Haemoglobin (g dl−1) | 12.8 ± 0.2 | 13.4 ± 0.3 | 0.08 |
| Haematocrit (%) | 40 ± 1 | 41 ± 1 | 0.48 |
Data are means ±s.e.m.
Significantly different compare to WT (P < 0.05).
VEGF and VEGF receptor protein levels
VEGF protein levels in the gastrocnemius of mVEGF−/− mice were reduced by 90% (P < 0.01) compared to control mice (Fig. 1). An 80% reduction (P < 0.05) of VEGF protein in mVEGF−/− was also observed in cardiac muscle compared to control (Fig. 1). Muscle specificity of the VEGF deletion was evident by the fact that no differences in VEGF protein levels were observed between mVEGF−/− and control mice when examining non-muscle tissue, i.e. lung and kidney (Fig. 1).
Figure 1. Protein expression of VEGF in skeletal muscle, heart and other body organs in 5-month old muscle VEGF-deficient (mVEGF−/−) and wild-type (WT) control mice.

Data are means ±s.e.m.n= 5–7/group. *Significantly different compared to WT (P < 0.05).
VEGF receptor 1 (VEGF-R1) and VEGF receptor 2 (VEGF-R2) expression were also significantly decreased by 63% and 69%, respectively, in gastrocnemius muscle of mVEGF−/− mice compared to controls (Fig. 2). In the heart, there was significant 51% decline in VEGF-R2 (P < 0.05) compared to a 26% non-significant decline in VEGF-R1. No differences in either VEGF-R1 or-R2 were observed in the non-muscle organs, i.e. lung and kidney (Fig. 2).
Figure 2. Protein expression of VEGF receptor 1 & 2.

Data are means ±s.e.m. A, protein expression of VEGF receptor 1, i.e. flt-1, measured from the samples in Fig. 1A. B, protein expression of VEGF receptor 2, i.e. flk-1, measured from the samples in Fig. 1A. *Significantly different compared to WT (P < 0.05).
Skeletal muscle capillarity and muscle fibre type composition
Histological assessment of muscle capillarity in the gastrocnemius muscle revealed a 48% reduction in the capillary-to-fibre ratio (C:F, P < 0.05) and 39% reduction in capillary density (CD, P < 0.05) in mVEGF−/− mice. No significant differences in muscle fibre cross-sectional area were detected (Fig. 3). The distribution of gastrocnemius muscle fibre types was also not different between mVEGF−/− and WT mice (Table 2).
Figure 3. Alkaline phosphatase staining of muscle sections obtained from deep and superficial areas within the gastrocnemius muscle of wild-type (WT) control and muscle VEGF-deficient (mVEGF−/−) mice.

A, representative images showing alkaline phosphatase stained muscle sections obtained from deep and superficial areas within the gastrocnemius muscle of wild-type (WT) control and muscle VEGF deficient (mVEGF−/−) mice. Images depicted are transverse sections magnified at 40× (bar = 50 μm). Capillaries appear dark purple (or black) and muscle fibres are yellow stained. B, morphometric analysis of pooled (deep and superficial) data from gastrocnemius muscle of mVEGF−/− and WT mice. Data show differences in capillary-to-fibre ratio and capillary density, but no difference in the average muscle fibre cross-sectional area between mVEGF−/− and WT mice. Data are means ±s.e.m. *Significantly different compared to WT (P < 0.05).
Table 2.
Muscle fibre type, enzyme activities, and exercise capacity
| WT | mVEGF−/− | |
|---|---|---|
| Gastrocnemius muscle fibre type composition (%) | ||
| Type-I | 4 ± 1 | 6 ± 1 |
| Type-IIA | 83 ± 5 | 81 ± 4 |
| Type-IIB | 13 ± 5 | 12 ± 4 |
| Exercise capacity | ||
| Maximal running speed (m min–1) | 33.5 ± 1.2 | 23.0 ± 1.2 ** |
| Run time to exhaustion (min) | 95 ± 8 | 18 ± 1 ** |
Data are means ±s.e.m. Symbols indicate significantly different than WT: *P < 0.05
P < 0.001.
Exercise performance
Shown in Table 2, time to exhaustion during submaximal endurance treadmill running (at 15 m min−1, 10 deg incline) was decreased by 81% in mVEGF−/− mice compared to controls (P < 0.001). Assessment of maximal running speed was also significantly lower (by 34%) in mVEGF−/− compared to control mice (P < 0.001).
Muscle metabolic adaptation to reduced VEGF levels
In the gastrocnemius muscle, the enzymatic activities of citrate synthase (CS), phosphofructokinase (PFK), and β-hydroxyacyl CoA dehydrogenase (β-HAD) were increased by 21%, 18%, and 32%, respectively (P < 0.05 each) in mVEGF−/− mice compared to the control group (Table 3).
Table 3.
Skeletal muscle enzyme activity
| WT (n= 6) | mVEGF−/− (n= 6) | |
|---|---|---|
| Gastrocnemius | ||
| Phosphofructokinase | 67.0 ± 3.3 | 79.3 ± 3.9* |
| β-Hydroxyacyl CoA dehydrogenase | 7.0 ± 0.2 | 9.3 ± 0.6* |
| Citrate synthase | 27.6 ± 0.05 | 33.4 ± 1.3* |
| Diaphragm | ||
| Phosphofructokinase | 16.2 ± 1.1 | 18.7 ± 1.4 |
| β-Hydroxyacyl CoA dehydrogenase | 8.7 ± 1.3 | 12.9 ± 1.4* |
| Citrate synthase | 29.3 ± 3.1 | 40.1 ± 2.0* |
Cardiac muscle capillarity and function
Similar to the skeletal muscle, there was also a significant 61% reduction in cardiac muscle capillarity (Fig. 4). 2D-echocardiography revealed that cardiac dimensions (left ventricular and septal wall thickness at end diastole) were not significantly different in mVEGF−/− mice compared to WT, except for a 15% reduction in fractional shortening (FS) in mVEGF−/− mice compared to WT (P < 0.01, Table 4). Nevertheless the average FS in the mVEGF−/− mice was within the limits of normality. Contractile function assessed by LV micromanometry during increasing β-adrenergic (dobutamine) stimulation revealed a small, but significant, overall drug effect of dobutamine (P < 0.05) between mVEGF−/− and WT in several of the cardiac variables, i.e. maximum dP/dt, minimum dP/dt, monoexponential decay of LV pressure decay during isovolumic diastole (τE), and end-diastolic pressure, and heart rate (Fig. 5). However, post hoc testing at basal or specific dobutamine doses did not reveal significant differences between mVEGF−/− and WT controls.
Figure 4. Alkaline phosphatase staining of cardiac muscle sections and average capillary density for wild-type (WT) control and muscle VEGF-deficient (mVEGF−/−) mice.

A, representative images showing alkaline phosphatase stained cardiac muscle sections from WT and mVEGF−/− mice. Images shown at 12.5× magnification (Bar = 5 μm). Capillaries appear as dark purple stained. B, average capillary density (number of capillary per mm2) in WT and VEGF deficient (mVEGF−/−) mice (n= 3/group). Data are means ±s.e.m. *Significantly different compared to WT (P < 0.05).
Table 4.
Transthoracic echocardiography under basal conditions
| WT (n= 16) | mVEGF−/− (n= 11) | P | |
|---|---|---|---|
| Heart Rate (bpm) | 529 ± 75 | 559 ± 44 | ns |
| IVSd (mm) | 0.61 ± 0.01 | 0.59 ± 0.01 | ns |
| LVPWd (mm) | 0.60 ± 0.01 | 0.61 ± 0.01 | ns |
| LVEDD | 48.3 ± 11.2 | 46.5 ± 8.5 | ns |
| LVESD | 12.7 ± 4.3 | 15.8 ± 4.8 | ns |
| FS (%) | 42.3 ± 4.4 | 36.0 ± 3.3* | 0.008 |
| Mean Vcf (circ s–1) | 9.01 ± 1.46 | 8.27 ± 1.07 | ns |
| LVEDD/BW | 0.16 ± 0.02 | 0.18 ± 0.02 | ns |
Data are means ±s.d. IVSd, intraventricular septal thickness diastole; LVPWd, left ventricular posterior wall thickness diastole; LVEDD, left ventricle end-disystolic dimension; LVESD, left ventricle end-systolic dimension; FS, fractional shortening ([LVEDD-LVESD/LVEDD]× 100); Vcf, mean velocity of circumferential fibre shortening in circumference per second; BW, body weight.
Figure 5. Haemodynamic data obtained from left ventricular micromanometer catheterization of muscle VEGF-deficient (mVEGF−/−) and wild-type (WT) control mice under general anaesthesia.

Data are means ±s.e.m. mVEGF−/−, n= 7; WT, n= 5. ANOVA results identify P-valve for main effect of genotype obtained from two-way ANOVA (comparing genotype and drug dose).
Discussion
The major purpose of this study was to determine whether lifelong postnatal depletion of muscle VEGF affected (1) muscle capillarity, and/or (2) maximal and endurance exercise capacity. In addition, we also sought to uncover any compensatory mechanisms in skeletal muscle that might mitigate an associated fall-off in exercise performance in the face of VEGF reduction. To accomplish these goals we engineered a mouse model in which skeletal muscle VEGF expression is reduced by >90% and found that skeletal muscle capillarity is reduced by nearly 50% (Fig. 3). Similarly cardiac VEGF levels were reduced by 80% and subsequent cardiac microvessel density decreased by 61%. The consequence of reducing VEGF-dependent muscle capillarity was major impairment in overall exercise capacity. These data clearly indicate that while muscle VEGF deficient mice survive to adulthood, VEGF is required to maintain muscle microvasculature and support exercise capacity. This study highlights the functional importance of this growth factor for muscle capillarity, oxygen availability and exercise performance throughout life.
VEGF-dependent capillary survival in postnatal mice
The fact that the mVEGF−/− mice survive embryogenesis and postnatal development can likely be attributed to the timing of muscle creatine kinase (MCK) expression in skeletal muscle. In skeletal muscle, MCK levels are first detectable in the fetus around day 16 (after the heart has formed) and rapidly increase >5-fold after 28 days post-birth (Trask & Billadello, 1990). To ensure that in our mice VEGF was indeed reduced throughout their postnatal life (and not just around the time we studied them at 5 months of age) we also measured muscle VEGF levels in separate groups of mice at 1 and 3 months and found the same >90% loss in VEGF as that seen 5-month-old adult mice (data not shown). Likewise, skeletal and cardiac muscle capillarity were also significantly reduced (by 34% and 45%, respectively; P < 0.05) in the 1-month-old mice. This confirms an early loss of VEGF in our mice and demonstrates that VEGF is necessary to regulate muscle capillary number throughout postnatal life. In support of this, previous data from our laboratory has shown that localized VEGF inactivation within skeletal muscle (via AAV/Cre injection in VEGFloxP floxed adult mice) results in a 60–70% loss in capillaries in the muscle region surrounding the injection site (Tang et al. 2004). Together, these data suggest that the myocyte is a critical source for paracrine VEGF production and that other cellular sources (such as endothelial cells, fibroblasts, pericytes and macrophages) are unable to compensate for the loss of myocyte-expressed VEGF.
In contrast, endothelial cell-specific deletion of VEGF (i.e. VEGFECKO) leads to sudden and early death in more than half of VEGFECKO mice, despite the fact that VEGFECKO mice have normal tissue (i.e. organ) and circulating VEGF mRNA and protein levels (Lee et al. 2007). This clearly also suggests a vital autocrine role for VEGF (at least within endothelial cells), which may seem to argue against a paracrine role suggested by our study. However, we would note, Lee et al. (2007) found normal VEGF expression in the heart and also showed that myocardial vessel density was unaltered in VEGFECKO mice. Thus, while it is evident that autocrine expression of VEGF is required for endothelial cell survival and integrity of vessel homeostasis per se, the development or architectural design of microvessel networks within muscle appears to be dependent on paracrine expression of VEGF. However, irrespective of where VEGF production occurs, these data unequivocally demonstrate that other putative angiogenic programmes and factors cannot compensate for the loss of VEGF at any stage in life. Moreover, given the profound reductions in exercise capacity we observed (Table 2) – without compensatory alterations in oxygen carrying capacity (i.e. haemoglobin, Table 1), muscle fibre type composition (Table 2) and/or reductions in metabolic enzyme activities (Table 3) – these data highlight the importance of VEGF in the physiological regulation of skeletal muscle microvasculature.
Contribution of cardiac VEGF deficiency and capillary regression to exercise intolerance
In the present study a 61% reduction in cardiac capillary density was found that could contribute to cardiac dysfunction, a potential factor in the limitation of exercise capacity. However, non-invasive transthoracic echocardiography used to measure cardiac dimensions and systolic function under basal conditions found no significant structural abnormalities (Table 4). There was evidence of reduced cardiac contractility (measured by a 15% reduction in fractional shortening, %FS) in mVEGF−/− compared to WT mice (P < 0.05). Yet, despite this reduction in %FS, it should be noted the absolute value of %FS in mVEGF−/− mice still remained within the normal physiological range (i.e. 35–50%) (Tanaka et al. 1996). Thus there appears to only be mild cardiac dysfunction in mVEGF−/− compared to WT mice.
We also evaluated the effect of cardiac muscle VEGF gene deletion by left ventricular micromanometry using retrograde arterial catheterization to measure inotropic and lusitropic reserve in response to β-adrenergic agonist (dobutamine) stimulation. This approach affords the opportunity to step-wise escalate myocardial oxygen demand by pharmacologically increasing contractility and intracavitary LV pressure and showed that end-diastolic pressure and τE (the time constant of LV pressure decay during isovolumic diastole) were only slightly increased, while maximal dP/dt was an average of 20% lower in mVEGF−/− mice compared to controls (P < 0.05). Taken together our data argue against a significant cardiac contribution to the exercise intolerance observed. Nevertheless, we cannot exclude the possibility that even a small diminution in cardiac function, such as we observed during dobutamine challenge, may exist and contribute to impaired exercise capacity. While we used a drug intervention to increase myocardial oxygen demand, the actual cardiac responses during treadmill exercise could have been greater compared to the pharmacologically induced responses we report here. Indeed, the effect of anaesthesia (which was required in order to make these cardiac measurements) could have itself masked or altered cardiac function.
The aetiology of this ventricular dysfunction is not readily apparent. One explanation could be ischaemic myocardial dysfunction based on hypoperfusion (resulting from capillary rarefaction). In contrast to our model (producing an 80% reduction in VEGF protein), Giordano et al. (2001) have reported that cardiac-specific deletion of VEGF (through cre recombinase expressed under the control of the myosin light chain 2v (MLC2v) promoter) results in an even greater loss (to undetectable levels) of cardiac VEGF protein and significant cardiac dysfunction, i.e. reduced resting cardiac output, reduced ejection fraction and decreased dP/dt in response to dobutamine-induced stress. In that study, erythropoietin (EPO) levels and other hypoxia-inducible genes (i.e. Glut-1 and transforming growth factor-β) were significantly elevated in the myocardium of cardiac-specific VEGF KO mice, implicating a hypoxic/ischaemic mechanism in this more severe cardiac-specific VEGF gene ablation mouse model.
While we did not measure EPO levels in this study, we would point out that neither haemoglobin nor haematocrit levels were different between mVEGF−/− and controls (Table 1). Also, the VEGF receptors are hypoxia-inducible, but neither were elevated. To the contrary, both VEGF-R1 (flt-1) and VEGF-R2 (flk-1) were reduced (Fig. 2). These findings question whether hypoxia/ischaemia is the mechanism at play under these circumstances.
Irrespective of the mechanism involved, both studies clearly establish that VEGF is vital to microvascular development (Fig. 4) and function of the adult myocardium. While the level of cardiac dysfunction between Giordano's cardiac-specific VEGF KO model and our present muscle VEGF-deficient model is different, it is possible that the 20% VEGF protein found in cardiac muscle of our mVEGF−/− mice might account for the relatively small changes in cardiac function we observed. We also cannot exclude the possibility that metabolic adaptations (similar to that seen in skeletal muscle) might have played a role in protecting the myocardium and potentially aided cardiac systolic function.
Respiratory muscle function
The respiratory skeletal muscles (i.e. diaphragm, intercostals muscle, etc.) would also be expected to have had reduced VEGF expression in this muscle-specific VEGF gene deletion mouse model. Indeed, we observed a 95% and 40% reduction in VEGF protein and VEGF receptors, respectively (P < 0.05), and a similar reduction in diaphragm capillarity to that seen in the locomotor skeletal muscle. Accordingly, it is possible that impaired respiratory muscle function could also have contributed to reduced exercise capacity in mVEGF−/− mice.
Unfortunately, it is not currently possible to assess respiratory muscle function as a separate contributor to exercise limitation in an intact mouse. Moreover, it is not feasible to obtain arterial blood during exercise to measure 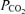 as an indication of adequate (or inadequate) ventilation. However, in a separate group of mice, we did measure resting minute ventilation using established plethysmographic methods (Drorbaugh & Fenn, 1955; Jacky, 1978). To our surprise, we found ventilation was elevated (by 19%, P < 0.05) under normoxic conditions in mVEGF−/− compared to the control group. The ventilatory response to acute hypoxia (
as an indication of adequate (or inadequate) ventilation. However, in a separate group of mice, we did measure resting minute ventilation using established plethysmographic methods (Drorbaugh & Fenn, 1955; Jacky, 1978). To our surprise, we found ventilation was elevated (by 19%, P < 0.05) under normoxic conditions in mVEGF−/− compared to the control group. The ventilatory response to acute hypoxia (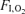 = 0.10) resulted in similar increases (∼50%) in resting ventilation in both groups of mice (data not shown). While these limited measurements cannot allow us to conclude that ventilation was normal during treadmill exercise, these data do suggest that an impairment of ventilation during exercise is unlikely.
= 0.10) resulted in similar increases (∼50%) in resting ventilation in both groups of mice (data not shown). While these limited measurements cannot allow us to conclude that ventilation was normal during treadmill exercise, these data do suggest that an impairment of ventilation during exercise is unlikely.
Physiological compensation for VEGF-dependent capillary loss
The reduction in exercise capacity is remarkable in its severity, especially in the presence of minimal cardiac dysfunction, and further supports the notions that muscle perfusion and/or the ability of the muscle to convey O2 by diffusion from capillaries to mitochondria are dependent on the size of the capillary–fibre interface (Gayeski & Honig, 1986; Groebe & Thews, 1990; Hepple et al. 2000). Potential compensatory responses to reduced O2 transport, such as increases in haemoglobin/haematocrit or alterations in skeletal muscle fibre types, which have been observed in other mouse models involving reduced O2 transport (i.e. myoglobin knockout mice) (Garry et al. 1998; Gödecke et al. 1999; Meeson et al. 2001), were looked for but did not occur in mVEGF−/− mice. Moreover, that litter sizes were normal (6–8 pups), with no unexplained premature deaths, suggests that other potential genetic/physiological compensations did not selectively provide a survival phenotype. It should also be noted that no evidence of increased myocyte apoptosis (assessed via in situ caspase-3 staining) was seen in mVEGF−/− mice (data not shown). There were, however, significant increases in skeletal muscle metabolic enzyme activities for both glycolytic (phosphofructokinase) and oxidative systems (β-hydroxyacyl CoA dehydrogenase and citrate synthase, Table 3), which are likely to reflect an attempt to offset lower availability of oxygen by facilitating ATP production through glycolysis or fatty acid oxidation. This response is perhaps not surprising since similar observations have been made in patients with muscle dysfunction due to peripheral arterial disease (Bylund et al. 1976; Jansson et al. 1988; Lundgren et al. 1989) and chronic obstructive pulmonary disease (Sanchez et al. 1988; Gea et al. 2001). Nevertheless, it is clear that increasing enzymatic activities cannot overcome the limitation in oxygen transport due to fewer capillaries. Thus, despite this compensatory response to the loss of VEGF (and the subsequent loss of muscle capillarity), mice with VEGF-deficient muscles display a significant intolerance to exercise. Combined with evidence implicating the importance of skeletal muscle capillarity in determining O2 conductance across the muscle (Hepple et al. 2000; Howlett et al. 2003), these data support the notion that peripheral limitations (e.g. reduced locomoter skeletal muscle capillarity) play a principal role in limiting maximal exercise capacity in an untrained (sedentary) state. Given the reduced maximal running capacity observed in mVEGF−/− mice (Table 2), the marked decline in submaximal endurance capacity (i.e. 81% difference in time to exhaustion, Table 2) likely implies that greater relative work was performed by mVEGF−/− compared to WT. Some of this, however, is also mitigated by the lower body mass of mVEGF−/− compared to WT (Table 1).
Muscle-type specificity of VEGF dependence
The present findings of no resting ventilatory deficiency and minimal cardiac dysfunction, despite significant reductions in capillary density in both the diaphragm and the heart raises interesting questions about the muscle specific differences with respect to structure–function consequences in the face of reduced capillarity. For example, despite a slightly smaller loss of VEGF protein in cardiac compared to skeletal muscle (80%vs. 95% reduction, respectively), the heart experienced a relatively greater reduction in capillary density compared to the gastrocnemius (61%vs. 39%, respectively), and yet minimal dysfunction. To our knowledge these are first data to show this potential muscle type differences between cardiac and skeletal muscle vascularity. One potential explanation for this difference may involve the expression of VEGF receptors levels. For example, in adult mice treated pharmacologically to either bind VEGF (using VEGF-Trap) or block VEGF receptor activation (and thereby inhibit VEGF signalling), organ specific differences in capillarity have been observed between the heart and other organs (such as the pancreas, thyroid, adrenal cortex, pituitary, adipose, choroids plexus) (Baffert et al. 2004; Kamba et al. 2006). These studies reveal that organs susceptible to VEGF receptor blockade contained higher levels of VEGF-R2 and VEGF-R3. In our study, a significant reduction in VEGF-R2 was detected in both the VEGF-deficient heart and skeletal muscle (Fig. 2). Since VEGF-R2 levels are higher in the cardiac compared to skeletal muscle (as seen in Fig. 2), it could be that the heart is more effected from a loss of VEGF paracrine signalling. Yet, despite a greater loss in cardiac muscle capillarity, myocardial function was only minimally affected. One possibility for this functional difference is the fact that basal capillarity is nearly three times greater in the heart compared to skeletal muscle. Thus, it may be that coronary vessels have a greater reserve designed to protect the heart from ischaemia-related insults.
In summary, we have shown that myocyte VEGF deficiency initiated during late gestation and present throughout life leads to permanent diminution of muscle capillarity and reduced capacity to perform endurance exercise. While effects of cardiac and/or ventilatory impairment cannot be completely ruled out in the intolerance to exercise observed, we suggest that reduced muscle capillarity impairs locomotor muscle O2 availability by both reducing muscle perfusion and diminished diffusive transport of O2 from red cells to mitochondria. The inability of other factors and/or mechanisms to compensate for the loss of VEGF by maintaining the number of muscle capillaries in adult mice clearly demonstrates that VEGF (which is essential during growth and development) is also essential and required to develop and maintain the normal complement of muscle capillaries.
Acknowledgments
We are grateful to Neoploena Ferrara, MD (Genentech, Inc., South San Franciso, CA) for generously providing VEGF/LoxP mice, Ronald Kahn, MD (Joslin Diabetes Center, Boston, MA) for permission to use MCK/Cre mice they created, Randall Johnson (UCSD, Dept. of Biological Sciences) for supplying the MCK/Cre mice, and to Odile Mathieu-Costello, PhD and Li Cui, PhD (UCSD, Dept. of Medicine) for their assistance and consultation on muscle histology and morphology assessment. We thank Dr. Frank Powell for his assistance and advice in setting up and analyzing in vivo ventilation studies using the body plethysmography method, and Joe Merlone in helping to perform the ventilation measurements. This research was supported by funds from the Tobacco-Related Disease Research Program of the University of California, Grant number 14KT-0091 (I.M.O.) and by the National Institutes of Health HL091830 (P.D.W.). I.M.O. was a Parker B. Francis pulmonary fellow. Cardiovascular studies were supported by the Edith and William Perlman Fund for Cardiovascular Research (K.L.P.).
Glossary
Abbreviations
- CD
capillary density
- C:F
capillary-to-muscle fibre ratio
- Cre
Cre recombinase
- CS
citrate synthase
- FS
fractional shortening
- β-HAD
β-hydroxyacyl-CoA dehydrogenase
- IVSd
intraventricular septal thickness diastole
- LVEDD
left ventricle end-disystolic dimension
- LVESD
left ventricle end-systolic dimension
- LVPWd
left ventricular posterior wall thickness diastole
- MCK
muscle creatine kinase
- mVEGF−/−
muscle specific VEGF knockout mouse
- PFK
phosphofructokinase
- τE
monoexponential decay of LV pressure during isovolumic diastole
- VCF
mean velocity of circumferential fibre shortening in circumference per second
- VEGF
vascular endothelial growth factor
- VEGF-R1
vascular endothelial growth factor receptor-1
- VEGF-R2
vascular endothelial growth factor receptor-2
- VEGFloxP
mice whose VEGF gene is floxed with LoxP
- WT
wild-type mouse
Author contributions
I.M.O., K.T., P.D.W. and E.C.B. developed the concept and the mouse model described. I.M.O. and R.A.H. performed skeletal muscle measurements. N.D.D. and Y.G. performed the cardiac assessments, and together with K.L.P. and I.M.O. assisted in the interpretation and analysis of the cardiac data. All authors contributed to the study design, analysis, interpretation and writing (and/or revising) the manuscript.
References
- Baffert F, Thurston G, Rochon-Duck M, Le T, Brekken R, McDonald DM. Age-related changes in vascular endothelial growth factor dependency and angiopoietin-1-induced plasticity of adult blood vessels. Circ Res. 2004;94:984–992. doi: 10.1161/01.RES.0000125295.43813.1F. [DOI] [PubMed] [Google Scholar]
- Bale TL, Hoshijima M, Gu Y, Dalton N, Anderson KR, Lee KF, Rivier J, Chien KR, Vale WW, Peterson KL. The cardiovascular physiologic actions of urocortin II: acute effects in murine heart failure. Proc Natl Acad Sci U S A. 2004;101:3697–3702. doi: 10.1073/pnas.0307324101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro E, Schols AM, Polkey MI, Galdiz JB, Gosker HR, Swallow EB, Coronell C, Gea J. Cytokine profile in quadriceps muscles of patients with severe chronic obstructive pulmonary disease. Thorax. 2008;63:100–107. doi: 10.1136/thx.2007.078030. [DOI] [PubMed] [Google Scholar]
- Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–165. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeyer HU. Methods in Enzymatic Analysis. New York: Academic Press; 1974. [Google Scholar]
- Breen EC, Johnson EC, Wagner H, Tseng H-M, Sung LA, Wagner PD. Angiogenic growth factor mRNA responses in muscle to a single bout of exercise. J Appl Physiol. 1996;81:355–361. doi: 10.1152/jappl.1996.81.1.355. [DOI] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- Bylund AC, Hammarsten J, Holm J, Schersten T. Enzyme activities in skeletal muscles from patients with peripheral arterial insufficiency. Eur J Clin Invest. 1976;6:425–429. doi: 10.1111/j.1365-2362.1976.tb00538.x. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeety S, Kieckens L, Geertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Riasu W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics. 1955;16:81–87. [PubMed] [Google Scholar]
- Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56:794–814. doi: 10.1046/j.1523-1755.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Folkman J. The vascularization of tumors. Sci Am. 1976;234:58–73. doi: 10.1038/scientificamerican0576-58. [DOI] [PubMed] [Google Scholar]
- Garry DJ, Ordway GA, Lorenz JN, Radford NB, Chin ER, Grange RW, Bassel-Duby R, Williams RS. Mice without myoglobin. Nature. 1998;395:905–908. doi: 10.1038/27681. [DOI] [PubMed] [Google Scholar]
- Gayeski TEJ, Honig CR. O2 gradients from sarcolemma to cell interior in red muscle at maximal VO2. Am J Physiol Heart Circ Physiol. 1986;251:H789–H799. doi: 10.1152/ajpheart.1986.251.4.H789. [DOI] [PubMed] [Google Scholar]
- Gea JG, Pasto M, Carmona MA, Orozco-Levi M, Palomeque J, Broquetas J. Metabolic characteristics of the deltoid muscle in patients with chronic obstructive pulmonary disease. Eur Respir J. 2001;17:939–945. doi: 10.1183/09031936.01.17509390. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- Giordano FJ, Gerber HP, Williams SP, VanBruggen N, Bunting S, Ruiz-Lozano P, Gu Y, Nath AK, Huang Y, Hickey R, Dalton N, Peterson KL, Ross J, Jr, Chien KR, Ferrara N. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci U S A. 2001;98:5780–5785. doi: 10.1073/pnas.091415198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gödecke A, Flögel U, Zanger K, Ding Z, Hirchenhain J, Decking UK, Schrader J. Disruption of myoglobin in mice induces multiple compensatory mechanisms. Proc Natl Acad Sci U S A. 1999;96:10495–10500. doi: 10.1073/pnas.96.18.10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groebe K, Thews G. Calculated intra-and extracellular PO2 gradients in heavily working red muscle. Am J Physiol Heart Circ Physiol. 1990;259:H84–92. doi: 10.1152/ajpheart.1990.259.1.H84. [DOI] [PubMed] [Google Scholar]
- Gustafsson T, Puntschart A, Kaijser L, Jansson E, Sundberg CJ. Exercise-induced expression of angiogenesis-related transcription and growth factors in human skeletal muscle. Am J Physiol Heart Circ Physiol. 1999;276:H679–685. doi: 10.1152/ajpheart.1999.276.2.H679. [DOI] [PubMed] [Google Scholar]
- Hepple RT, Hogan MC, Stary C, Bebout DE, Mathieu-Costello O, Wagner PD. Structural basis of muscle O2 diffusing capacity: evidence from muscle function in situ. J Appl Physiol. 2000;88:560–566. doi: 10.1152/jappl.2000.88.2.560. [DOI] [PubMed] [Google Scholar]
- Holloszy J. Adaptations of muscular tissue to training. Prog Cardiovasc Dis. 1976;18:445–458. doi: 10.1016/0033-0620(76)90011-6. [DOI] [PubMed] [Google Scholar]
- Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol. 1984;56:831–838. doi: 10.1152/jappl.1984.56.4.831. [DOI] [PubMed] [Google Scholar]
- Howlett RA, Gonzalez NC, Wagner HE, Fu Z, Britton SL, Koch LG, Wagner PD. Genetic models in applied physiology: skeletal muscle capillarity and enzyme activity in rats selectively bred for running endurance. J Appl Physiol. 2003;94:1682–1688. doi: 10.1152/japplphysiol.00556.2002. [DOI] [PubMed] [Google Scholar]
- Jacky JP. A plethysmograph for long-term measurements of ventilation in unrestrained animals. J Appl Physiol. 1978;45:644–647. doi: 10.1152/jappl.1978.45.4.644. [DOI] [PubMed] [Google Scholar]
- Jansson E, Johansson J, Sylven C, Kaijser L. Calf muscle adaptation in intermittent claudication. Side-differences in muscle metabolic characteristics in patients with unilateral arterial disease. Clin Physiol. 1988;8:17–29. doi: 10.1111/j.1475-097x.1988.tb00258.x. [DOI] [PubMed] [Google Scholar]
- Kamba T, Tam BY, Hashizume H, Haskell A, Sennino B, Mancuso MR, Norberg SM, O’Brien SM, Davis RB, Gowen LC, Anderson KD, Thurston G, Joho S, Springer ML, Kuo CJ, McDonald DM. VEGF-dependent plasticity of fenestrated capillaries in the normal adult microvasculature. Am J Physiol Heart Circ Physiol. 2006;290:H560–576. doi: 10.1152/ajpheart.00133.2005. [DOI] [PubMed] [Google Scholar]
- Kivela R, Silvennoinen M, Lehti M, Jalava S, Vihko V, Kainulainen H. Exercise-induced expression of angiogenic growth factors in skeletal muscle and in capillaries of healthy and diabetic mice. Cardiovasc Diabetol. 2008;7:13. doi: 10.1186/1475-2840-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, Ferrara N, Nagy A, Roos KP, Iruela-Arispe ML. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691–703. doi: 10.1016/j.cell.2007.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry CV, Kimmey JS, Felder S, Chi MM, Kaiser KK, Passonneau PN, Kirk KA, Lowry OH. Enzyme patterns in single human muscle fibers. J Biol Chem. 1978;253:8269–8277. [PubMed] [Google Scholar]
- Lundgren F, Dahllof AG, Schersten T, Bylund-Fellenius AC. Muscle enzyme adaptation in patients with peripheral arterial insufficiency: spontaneous adaptation, effect of different treatments and consequences on walking performance. Clin Sci (Lond) 1989;77:485–493. doi: 10.1042/cs0770485. [DOI] [PubMed] [Google Scholar]
- Meeson AP, Radford N, Shelton JM, Mammen PP, DiMaio JM, Hutcheson K, Kong Y, Elterman J, Williams RS, Garry DJ. Adaptive mechanisms that preserve cardiac function in mice without myoglobin. Circ Res. 2001;88:713–720. doi: 10.1161/hh0701.089753. [DOI] [PubMed] [Google Scholar]
- Mrazkova O, Grim M, Carlson BM. Enzymatic heterogeneity of the capillary bed of rat skeletal muscles. Am J Anat. 1986;177:141–148. doi: 10.1002/aja.1001770203. [DOI] [PubMed] [Google Scholar]
- Ogilvie RW, Feeback DL. A metachromatic dye-ATPase method for the simultaneous identification of skeletal muscle fiber types I, IIA, IIB and IIC. Stain Technol. 1990;65:231–241. doi: 10.3109/10520299009105613. [DOI] [PubMed] [Google Scholar]
- Olfert IM, Breen EC, Mathieu-Costello O, Wagner PD. Skeletal muscle capillarity and angiogenic mRNA levels after exercise training in normoxia and chronic hypoxia. J Appl Physiol. 2001;91:1176–1184. doi: 10.1152/jappl.2001.91.3.1176. [DOI] [PubMed] [Google Scholar]
- Prior BM, Lloyd PG, Yang HT, Terjung RL. Exercise-induced vascular remodeling. Exerc Sport Sci Rev. 2003;31:26–33. doi: 10.1097/00003677-200301000-00006. [DOI] [PubMed] [Google Scholar]
- Prior BM, Yang HT, Terjung RL. What makes vessels grow with exercise training? J Appl Physiol. 2004;97:1119–1128. doi: 10.1152/japplphysiol.00035.2004. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Wagner H, Mudaliar SR, Henry R, Noyszewski EA, Wagner PD. Human VEGF gene expression in skeletal muscle: effect of acute normoxic and hypoxic exercise. Am J Physiol Heart Circ Physiol. 1999;277:H2247–2252. doi: 10.1152/ajpheart.1999.277.6.H2247. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–865. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- Sanchez J, Brunet A, Medrano G, Debesse B, Derenne JP. Metabolic enzymatic activities in the intercostal and serratus muscles and in the latissimus dorsi of middle-aged normal men and patients with moderate obstructive pulmonary disease. Eur Respir J. 1988;1:376–383. [PubMed] [Google Scholar]
- Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–5. [Google Scholar]
- Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996;94:1109–1117. doi: 10.1161/01.cir.94.5.1109. [DOI] [PubMed] [Google Scholar]
- Tang K, Breen EC, Gerber HP, Ferrara NM, Wagner PD. Capillary regression in vascular endothelial growth factor-deficient skeletal muscle. Physiol Genomics. 2004;18:63–69. doi: 10.1152/physiolgenomics.00023.2004. [DOI] [PubMed] [Google Scholar]
- Trask RV, Billadello JJ. Tissue-specific distribution and developmental regulation of M and B creatine kinase mRNAs. Biochim Biophys Acta. 1990;1049:182–188. doi: 10.1016/0167-4781(90)90039-5. [DOI] [PubMed] [Google Scholar]