

Abstract
Fatty liver has been linked to low aerobic fitness, but the mechanisms are unknown. We previously reported a novel model in which rats were artificially selected to be high capacity runners (HCR) and low capacity runners (LCR) that in a sedentary condition have robustly different intrinsic aerobic capacities. We utilized sedentary HCR/LCR rats (generation 17; max running distance equalled 1514 ± 91 vs. 200 ± 12 m for HCR and LCR, respectively) to investigate if low aerobic capacity is associated with reduced hepatic mitochondrial oxidative capacity and increased susceptibility to hepatic steatosis. At 25 weeks of age, LCR livers displayed reduced mitochondrial content (reduced citrate synthase activity and cytochrome c protein) and reduced oxidative capacity (complete palmitate oxidation in hepatic mitochondria (1.15 ± 0.13 vs. 2.48 ± 1.1 nm g−1 h, P < 0.0001) and increased peroxisomal activity (acyl CoA oxidase and catalase activity) compared to the HCR. The LCR livers also displayed a lipogenic phenotype with higher protein content of both sterol regulatory element binding protein-1c and acetyl CoA carboxylase. These differences were associated with hepatic steatosis in the LCR including higher liver triglycerides (6.00 ± 0.71 vs. 4.20 ± 0.39 nmol g−1, P= 0.020 value), >2-fold higher percentage of hepatocytes associated with lipid droplets (54.0 ± 9.2 vs. 22.0 ± 3.5%, P= 0.006), and increased hepatic lipid peroxidation compared to the HCR. Additionally, in rats aged to natural death, LCR livers had significantly greater hepatic injury (fibrosis and apoptosis). We provide novel evidence that selection for low intrinsic aerobic capacity causes reduced hepatic mitochondrial oxidative capacity that increases susceptibility to both hepatic steatosis and liver injury.
Non-alcoholic fatty liver disease (NAFLD) encompasses a gamut of liver maladaptations and is a primary cause of chronic liver disease and liver-related morbidity and mortality. Fatty liver occurs at a higher rate in individuals with obesity and type 2 diabetes and is considered the hepatic component of the metabolic syndrome (Schindhelm et al. 2006). Currently, 34% of the general population (Browning et al. 2004) and 75–100% of obese and extremely obese individuals are estimated to have fatty liver (Bellentani et al. 2000).
Low aerobic capacity, or low cardiorespiratory fitness, is the strongest predictor of early mortality (Myers et al. 2002). Aerobic capacity is dependent upon the ability of the cardiorespiratory system to deliver oxygen and the capacity of mitochondria to utilize oxygen for energy production. In the absence of exercise training, it is believed that genetic inheritance accounts for up to 70% of the variation in intrinsic aerobic capacity (Bouchard et al. 1986). In most cases, exercise training significantly improves aerobic capacity; however, exercise-induced responses are grossly heterogeneous (Kohrt et al. 1991; Bouchard & Rankinen, 2001). Recent evidence also links low aerobic capacity (Nguyen-Duy et al. 2003; Church et al. 2006; McMillan et al. 2007) to an increased deposition of fat in the liver; however, there is no mechanistic information detailing these connections.
Over several generations of selective breeding for high and low treadmill endurance running (performance on 3 graded exercise tests), we have developed two strains of rats with grossly different intrinsic endurance exercise capacities (max run distance during a graded exercise test to exhaustion is ∼7-fold different) and aerobic capacities (30% different at generation 11) (Wisloff et al. 2005). This novel animal model is valuable to study the interaction between aerobic capacities and chronic disease for two main reasons: (1) the contrasting aerobic capacities occur in a sedentary, caged-activity-only condition (Hoydal et al. 2007) removing the conflicting effects of daily exercise training (Wisloff et al. 2005; Bernal-Mizrachi & Semenkovich, 2006); and (2) the strains are a polygenic model of disease that more accurately mimics the pathology of human chronic metabolic disease(s) than single gene mutations. At generation 11, the sedentary low capacity runners (LCR) displayed a higher incidence of both cardiovascular and metabolic syndrome risk factors than sedentary high capacity runners (HCR) (Wisloff et al. 2005). In addition, at generation 13, LCR rats were susceptible to high-fat diet induced obesity and insulin resistance while the HCR rats were largely unaffected (Noland et al. 2007a). In both reports, skeletal muscle mitochondrial oxidative capacity, which is high in the muscle of HCR, and low in the muscle of LCR, was hypothesized to be the primary cause for protection or susceptibility to metabolic dysfunction, respectively.
In the current study, we investigate the potential links between intrinsic aerobic capacity, hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis in generation 17 HCR and LCR rats. Importantly, these studies occurred in sedentary rats which are genetically predisposed to high and low aerobic capacities allowing for the determination of hepatic metabolism independently of the effects of exercise training. We hypothesize that the phenotype of low aerobic capacity is associated with both reduced hepatic mitochondrial content and oxidative capacity which increases both susceptibility to early life hepatic steatosis and significant hepatic injury at an older age.
Methods
Animal strain and protocols
The development of LCR and HCR rats has previously been described (Wisloff et al. 2005; Noland et al. 2007a). Briefly, two-way artificial selective breeding was used to create low capacity runner (LCR) and high capacity runner (HCR) strains that were divergent for treadmill running capacity (run time to exhaustion on a graded exercise test). The founder population (N:NIH stock) and generation of the HCR and LCR strains has been previously reported (Wisloff et al. 2005). In brief, the 13 lowest and 13 highest running capacity rats of each sex were selected from the founder population and randomly paired for mating. At each subsequent generation, within-family selection from 13 mating pairs was practiced for each line, a number of families that maintains a relatively low coefficient of inbreeding (<0.01/generation) and maximizes the retention of genetic variation. After the rats were phenotyped for running capacity they were exposed to no further exercise training or testing and only underwent normal cage activity.
The animal protocols were approved by the Institutional Animal Care and Use Committees at the University of Missouri and the University of Michigan and the Subcommittee for Animal Safety at the Harry S. Truman Memorial VA Hospital.
Two separate sets of ∼25-week-old male LCR (n= 7) and HCR (n= 8) rats from generation 17 were used for the investigation of hepatic steatosis and hepatic mitochondrial oxidative capacity. The rats arrived at ∼14 weeks of age, were provided standard rat chow and water ad libitum, and were kept on a 12 h light–12 h dark time schedule until killed at ∼25 weeks of age. On the morning of tissue collection, rats were intraperitoneally injected with sodium Nembutal (100 mg kg−1), blood was collected, the animals were exsanguinated, and tissues were frozen with liquid nitrogen and stored at –80°C or fixed for immunohistochemistry. Tissue was collected in the same manner in a second group of 25-week-old, generation 17, HCR (n= 8) and LCR (n= 8) rats and ∼500 mg of liver was quickly removed and placed in ice-cold buffer for the isolation of hepatic mitochondria or frozen for later measurement of peroxisomal enzyme activity. Additional generation 17 rats (HCR, n= 4; LCR, n= 6) were aged until natural death or until they were killed because of undue suffering at approximately 24–35 months of age at the University of Michigan where the rats were generated. After death animals underwent necropsy within 4 h and livers were fixed for immunohistochemistry only. Weekly food consumption and changes in body weight were measured in an additional set of HCR/LCR rats (n= 10 in both groups) from 5 to 25 weeks of age. In addition, determinations of physical activity and oxygen consumption by indirect calorimetry were taken in the rats over a 2 day period at 25 weeks of age.
Intrahepatic lipid content
Hepatic triglycerides (TAG) were measured as referenced previously using a commercially available kit (Sigma, F6428) (Rector et al. 2008).
Histology analysis
Liver and epididymal adipose tissues were collected and fixed in 10% formalin for histological analysis. Routine hematoxylin–eosin (H&E) staining was performed in both liver and adipose and sirius red staining for collagen deposition was performed in paraffin liver sections as previously referenced (Wei et al. 2008). Hepatic steatosis and the percentage of hepatocytes associated with lipid droplets were determined as referenced previously (Ibdah et al. 2005) and morphometric determinations of adipocyte cell volume were carried out as previously referenced (Laye et al. 2006).
Immunohistochemistry
Liver paraffin sections were de-paraffinized and non-specific antibody binding was blocked with 5% normal goat or rabbit serum and 5% BSA in phosphate-buffered saline (PBS). These sections were incubated with anti-carbamoylphosphate synthetase 1 (CPS1), transforming growth factor β (TGF-β) or 4-hydroxynonenal (4-HNE) antibodies at 1:200 dilutions for overnight at 4°C. After washing, the sections were incubated with a second antibody conjugated with alexa-586 for 1 h at room temperature and mounted with DAPI (Vector) or biotin conjugated second antibody, following by 0.3% H2O2, ABC (Vector), NovaRed (Vector), and counterstained with hematoxylin. The primary antibody was omitted from this procedure for additional sections to ensure that results were not due to non-specific binding. Images were acquired with a light and fluorescence microscope (Nikon, Eclipse 50i).
Apoptosis by TUNEL staining
To evaluate apoptotic cell death, terminal desoxynucleotide transferase-mediated dUTP nick end labelling (TUNEL) was performed in liver sections using an in situ Apoptosis Detection Kit (Roche Applied Science, Indianapolis, IN, USA) as per the manufacturer's instructions. TUNEL-positive and-negative nuclei were counted at five random fields for each section. Results are expressed as number of TUNEL-positive cell/total cells × 100%.
Western blotting
Peroxisome proliferator-activated receptor (PPAR) γ, PPARα, and sterol regulatory binding protein-1c (SREBP-1c) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against Acetyl CoA carboxylase (ACC) and cytochrome c were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA) and 4-HNE was purchased from Oxis International Inc. (Beverly Hills, CA, USA). Liver samples were homogenized in ice cold buffer, separated by SDS-PAGE gels, transferred to PVDF membranes and probed with primary antibodies. Protein bands were quantified using a densitometer and band densities were corrected for total protein loaded by staining with 1% amido-black (Sigma) as described previously (Rector et al. 2008).
Hepatic mitochondrial enzyme activities, isolation, and palmitate oxidation
Citrate synthase and β-hydroxyacyl-CoA dehydrogenase (β-HAD) activity were both measured as previously described in whole liver homogenates (Rector et al. 2008). Mitochondrial suspensions were prepared according to modified methods of Koves et al. (2005). A portion of the liver was quickly excised from unconscious rats and placed in 5 ml of fresh Buffer A (100 mm KCl, 50 mm Mops, 5 mm MgSO4, 1 mm EGTA, 1 mm ATP, pH 7.4). The liver was then minced and suspended 7-fold (w/v) in Buffer A, and homogenized for 15 s. The homogenate was then centrifuged at 800 g for 10 min at 4°C and the supernatant was filtered through gauze and set aside. The remaining pellet was resuspended in Buffer A, homogenized, and the homogenate was then centrifuged at 800 g for 10 min at 4°C. The remaining supernatant was added to the previously set aside supernatant after filtering through gauze. The pooled supernatants were centrifuged at 9000 g to pellet the mitochondria. The pellet was resuspended in Buffer B (Buffer A, 0.2% BSA, pH 7.4), and centrifuged at 8000 g before the pellet was washed and resuspended in Buffer C (100 mm KCL, 50 mm Mops, 0.5 mm EGTA, pH 7.4) and centrifuged for 10 min at 7000 g. The final pellet was resuspended in 0.5 ml of sucrose EDTA (SET) buffer and protein content was determined. Palmitate oxidation was measured with radiolabelled [1-14C]palmitate (American Radiolabeled Chemicals, Inc., St Louis, MO, USA) in the freshly isolated liver mitochondria preparation using methods previously reported (Laye et al. 2008; Rector et al. 2008). Both 14CO2, representing complete fatty acid oxidation, and 14C labelled acid soluble metabolites, representing incomplete fatty acid oxidation, were collected in the previously described trapping device and then counted on a liquid scintillation counter. In addition to [14C]palmitate the reaction buffer contained a final concentration of 50 μm cold palmitate and 0.5% BSA.
Hepatic peroxisomal enzyme activity
Acyl-CoA oxidase activity was determined using a modification of methods previously described (Small et al. 1985). Briefly, H2O2 production was measured spectrophotometrically (520 nm) through oxidative coupling with 3,5-dichloro-2-hydroxybenzenesulfonic acid to 4-aminoantipyrine in the presence of horseradish peroxidase. Reaction wells contained 20 μl of liver homogenate and 200 μl of reaction buffer consisting of 40 mm aminotriazole, 0.02% Triton X-100, 0.5 mm 4-aminoantipyrine, 4 mm 3,5-dichloro-2-hydroxybenzenesulfonic acid, 8 U horseradish peroxidase, 11 mm potassium phosphate; pH 7.4. They were incubated for 5 min at 37°C. The reaction was initiated by the addition of acyl-CoA substrate (50 μm) and absorbance was monitored continuously over 30 min at 37°C. Catalase activity was determined by the spectrophotometric (520 nm) oxidative coupling of 3,5-dichlorobenzene-sulfonic acid to 4-aminoantipyrine in the presence of HRP and H2O2 (Sigma) as previously described (Noland et al. 2007b).
Serum assays and blood pressure
Serum TAG (Sigma) and insulin (Linco Research, St Charles, MO, USA) were measured using commercially available kits. Serum alanine aminotransferase (ALT) concentrations were determined utilizing an Olympus AU400e Chemistry Immuno Analyzer (Olympus America, Inc., Center Valley, PA, USA). Systolic blood pressure was measured in triplicate using the tail-cuff method a week before sacrifice.
Food consumption, ambulatory physical activity, and oxygen consumption
Both ad libitum food consumption and total body weights were measured every week from the post-weaning period (∼5 week) until 25 weeks of age. In addition, oxygen consumption  , carbon dioxide production
, carbon dioxide production 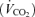 , and spontaneous motor activity were measured using a Comprehensive Laboratory Monitoring System (CLAMS, Columbus Instruments, Columbus, OH, USA) over a 2 day period at 25 weeks of age at the University of Michigan Animal Phenotyping Core. The measurements were carried out continuously for 48 h in an experimentation room set at 20–23°C with 12–12 h (18.00 h∼06.00 h) dark–light cycles.
, and spontaneous motor activity were measured using a Comprehensive Laboratory Monitoring System (CLAMS, Columbus Instruments, Columbus, OH, USA) over a 2 day period at 25 weeks of age at the University of Michigan Animal Phenotyping Core. The measurements were carried out continuously for 48 h in an experimentation room set at 20–23°C with 12–12 h (18.00 h∼06.00 h) dark–light cycles. 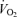 and
and 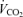 in each chamber were sampled sequentially for 5 s in a 5 min interval and the motor activity was simultaneously recorded every second in X and Z dimensions. The air flow rate through the chambers was adjusted at the level to keep the oxygen differential around 0.3% at resting conditions.
in each chamber were sampled sequentially for 5 s in a 5 min interval and the motor activity was simultaneously recorded every second in X and Z dimensions. The air flow rate through the chambers was adjusted at the level to keep the oxygen differential around 0.3% at resting conditions.
Statistics
For each outcome measure, a one-way analysis of variance was performed using SPSS 15.0 (Chicago, IL, USA). Pearson correlational analysis was also used on select data. Values are reported as means ±s.e.m., and a P value < 0.05 was considered statistically significant.
Results
Animal characteristics
The HCR rats ran ∼9-fold further distance (P < 0.001) than LCR rats during the graded exercise test conducted at 11 weeks of age (Table 1). In the rats killed at 25 weeks, the LCR rats displayed overt characteristics of the metabolic syndrome compared to the HCR including heavier body weights, elevated serum TAG (P= 0.045) and insulin (P= 0.005) and higher systolic blood pressures (P= 0.039) (Table 1). Serum ALT levels, a marker of liver injury, were not significantly different between groups (Table 1).
Table 1.
Characteristics LCR and HCR rats at 25 weeks of age
| Variable | LCR | HCR |
|---|---|---|
| Body weight (g) | 497 ± 48* | 350 ± 28 |
| Max run distance at 11 weeks (m) | 188 ± 15* | 1486 ± 86 |
| Serum insulin (ng ml−1) | 3.03 ± 0.44* | 34 ± 0.24 |
| Serum TAG (mg dl−1) | 61.1 ± 19.5* | 16.7 ± 1.8 |
| Serum ALT (U l−1) | 61.1 ± 14.1 | 57.4 ± 38.4 |
| Systolic blood pressure (mmHg) | 160.2 ± 2.3* | 151.1 ± 1.3 |
Means ±s.e.m.
Significantly different from HCR, P < 0.05; m, metres.
Ambulatory physical activity, oxygen consumption and food consumption
At 25 weeks of age, the LCR rats had 40% lower ambulatory physical activity in the 12 h dark phase (P < 0.001) and a 22% lower activity in the light phase (P= 0.008) compared to the HCR (Table 3). This corresponded with a 16% and 11% lower total oxygen consumption in both the light (P= 0.005) and dark phase (P < 0.001), respectively (Table 3). Average weekly food consumption (averaged from ∼4 to 25 weeks of age) was also significantly lower in the LCR rats (P= 0.03) (Table 3). Collectively, these data indicate that the contrasting levels of ambulatory activity and oxygen consumption measured between the two strains corresponded with different rates of food consumption. Feeding efficiency (weight gain divided by food intake) was not different between the groups between 9 weeks and the time of kill at 25 weeks suggesting that overall energy balance was similar for 16 weeks prior to kill (data not shown).
Table 3.
Physical activity, oxygen consumption and weekly food consumption
| LCR | HCR | |||||
|---|---|---|---|---|---|---|
| Variable | Dark | Light | 24 h | Dark | Light | 24 h |
| Ambulatory activity (counts h−1) | 345.5 ± 16.0* | 78.7 ± 6.8* | 216.7 ± 0.7* | 573.4 ± 26.2 | 102.0 ± 4.7 | 345.8 ± 13.7 |
| O2 consumption (ml kg–1 h–1) | 1282.2 ± 36.4* | 1042.1 ± 30.1* | 1167.2 ± 34.8* | 1532.7 ± 43.5 | 1176.0 ± 28.0 | 1362.7 ± 34.0 |
| LCR | HCR | |||||
| Food consumption (g wk−1 per 100 g BW) | 52.05 ± 2.09* | 58.79 ± 2.01 | ||||
Mean ±s.e.m.
Significantly lower than HCR, P < 0.05; BW = body weight; ambulatory activity and O2 consumption were measured over a 2 day period at 25 weeks of age. Average weekly food consumption was measured from 5 weeks of age (post-weaned) until 25 weeks of age.
Hepatic mitochondrial enzyme activity, palmitate oxidation and peroxisomal enzyme activity
β-HAD activity, the rate limiting enzyme for β-oxidation, was 65% higher (P < 0.001) in the HCR livers compared to LCR livers (Fig. 2A). Citrate synthase activity and cytochrome c protein content, markers of mitochondrial content, were 32–42% higher (P < 0.001) in the HCR livers compared to LCR (Fig. 2B and C). Immunoflourescence staining for CPS-1, a hepatic mitochondrial marker, qualitatively revealed a greater hepatic mitochondrial content in the HCR vs. the LCR (Fig. 2D). The HCR mitochondria also had higher rates of fatty acid oxidation than the LCR. Isolated hepatic mitochondria from the HCR rats had a 63% (Fig. 3A; P < 0.0001) higher rate of complete palmitate oxidation to CO2 and a 22% higher (Fig. 3B; P < 0.001) rate of total palmitate oxidation (CO2+ acid soluble metabolite production) than the LCR. The LCR rats also displayed significantly higher peroxisomal enzyme activity for both acyl CoA oxidase (Fig. 4A; P= 0.039) and catalase (Fig. 4B; P= 0.02) compared to the HCR rats, suggestive of a compensatory increase in peroxisomal activity due to reduced mitochondrial oxidative capacity. Collectively these data show that LCR livers have both a lower mitochondrial content and lower mitochondrial oxidative capacity (decreased fatty acid oxidation per mitochondria).
Figure 2. Hepatic mitochondrial content and enzyme activity.

The activity of mitochondrial enzymes β-HAD (A) and citrate synthase (B) and the protein content of cytochrome c (C) were significantly lower in the livers of LCR rats than HCR rats at 25 weeks of age. D, representative immunofluorescence photomicrographs (magnification = 400×) for mitochondrial marker CPS1 (red) showed granular staining patterns in liver sections. The LCR livers visibly display decreased CPS-1 staining compared to the HCR at 25 weeks of age.
Figure 3. Hepatic mitochondrial fat oxidation.

Both complete palmitate oxidation to CO2 (A) and total palmitate oxidation (CO2+ ASM) (B) were significantly lower in isolated mitochondria from the LCR rats than the HCR at 25 weeks of age. *Significant difference (P < 0.05); values are reported as means ±s.e.m.; n= 7.
Figure 4. Hepatic peroxisomal activity.

Hepatic peroxisomal enzyme activities of both acyl CoA oxidase (A) and catalase (B) were significantly greater in LCR than HCR rats at 25 weeks of age. *Significant difference (P < 0.05); values are reported as means ±s.e.m.; n= 7.
Hepatic lipogenesis
The content of SREBP-1c, a transcriptional regulator of fatty acid synthesis genes (Shimano et al. 1996; Horton et al. 2002), was significantly higher in the livers of LCR rats than HCR rats (Fig. 5A). Specifically, SREBP-1c is an upstream transcriptional regulator of both acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS). ACC protein content, the enzyme responsible for malonyl CoA production (a potent inhibitor of fatty acid oxidation and the initial substrate for FAS), was 3-fold higher (P= 0.049) in the LCR liver than the HCR liver (Fig. 5B). There was no difference in serine79 phosphorylation of ACC protein, a marker of ACC inhibition (data not shown), suggesting that ACC differences were due to total content of the enzyme and not due to differences in covalent modification. We found no difference in FAS content (Fig. 5C). Protein content of PPARγ and PPARα, vital transcriptional regulators of lipid synthesis and oxidation also were not different in the livers of LCR and HCR rats (Fig. 5C).
Figure 5. Hepatic lipid synthesis proteins.

The protein content of SREBP-1c (A) and ACC (B) were significantly higher in the livers of LCR than HCR rats. C, no differences were observed for PPARα, PPARγ, or FAS protein contents between groups. Representative western blots are shown. *Significant difference (P < 0.05); values are reported as means ±s.e.m.; n= 7–8.
Hepatic and extrahepatic lipid storage
Total hepatic TAG concentrations were significantly higher in the LCR than the HCR rats (P= 0.02) (Fig. 1C). Within the LCR rats, hepatic TAG levels inversely correlated with maximum running distance (r=−0.724; P= 0.034), demonstrating an influence of endurance capacity within the LCR rat strain. The LCR livers also had a higher steatosis score (P= 0.004) and >2 fold higher number of hepatocytes associated with lipid droplets than the HCR (Table 2 and Fig. 1). The adipocyte cell volumes of both epidydimal and omental fat pads were 35% (P= 0.003) and 50% (P= 0.025) larger in the LCR than the HCR rats (Table 2). Although not measured herein, a previous report has shown that total epididymal fat pad weight is significantly higher in the LCR than HCR rats (Noland et al. 2007a). Taken together, the LCR showed both increased ectopic lipid storage in liver and expansion of fat pads over the HCR.
Figure 1. Hepatic lipids.

A, representative liver sections with H&E staining reveal greater steatosis in the LCR livers than HCR. B, representative liver sections of 4-HNE staining also show greater lipid peroxidation in LCR livers. C, hepatic triglyceride content is significantly higher in the LCR than the HCR rats. D, lipid peroxidation quantified by western blotting with 4-HNE antibody is significantly higher in the LCR livers than HCR. Samples were obtained from 25-week-old rats. *Significant difference (P < 0.05); values are reported as means ±s.e.m.; n= 7–8.
Table 2.
Hepatic lipids and adipocyte cell volume
| Variable | LCR | HCR |
|---|---|---|
| Steatosis score | 2.20 ± 0.28* | 1.17 ± 0.07 |
| Hepatocytes with lipids (%) | 54.0 ± 9.2* | 22.0 ± 3.5 |
| Epidydimal fat pad cell | 160.0 ± 9.8* | 118.4 ± 3.4 |
| volume (pl) | ||
| Omental fat pad cell volume (pl) | 122.3 ± 13.3* | 85.7 ± 3.9 |
Means ±s.e.m.
Significantly different than HCR, P < 0.05; pl, picolitres
Hepatic lipid peroxidation
LCR livers also displayed higher levels of lipid peroxidation (4-HNE), a marker of oxidative stress, than HCR livers (Fig. 1D (P= 0.04) (Western blot) and Fig. 1B (immunohistochemistry)) indicating that LCR livers possess both increased lipids and oxidative stress early in life.
Hepatic injury
Staining for apoptotic hepatocytes revealed that the LCR rats had a significantly higher percentage of apoptotic nuclei than their HCR counterparts both at 25 weeks of age (P < 0.001; data not shown) and after aging to natural death (age at death ranged between 24 and 35 months) (Fig. 6B; P= 0.007). Collagen deposition and fibrosis were assessed by sirius red staining in formalin-fixed liver sections. No visual fibrosis was detected from either group at 25 weeks of age (data not shown); however, the LCR livers display a greater percentage of fibrotic area than the HCR rats after aging to natural death (Fig. 6A; P= 0.005). These results suggest that early hepatic mitochondrial defects in animals with low intrinsic aerobic capacity are associated with advanced disease development and hepatic injury later in life.
Figure 6. Hepatic injury.

Hepatic injury was greater in the LCR than the HCR rats when measured at the time of natural death. A, sirius red staining for hepatic fibrosis (quantified by % of fibrotic area); B, apoptosis measured by TUNEL staining of apoptotic hepatocytes nuclei. C, TGF-1β protein, a strong fibrogenic signal, was visually greater in livers from LCR than HCR rats at the time of natural death. *Significant difference (P < 0.05); values are reported as means ±s.e.m.; n= 4–6.
Discussion
The major findings of this study are that intrinsically low aerobic capacity in the LCR rats was associated with reduced hepatic mitochondrial content, decreased mitochondrial capacity to oxidize fatty acids, and an increased hepatic de novo lipogenic profile. Furthermore, the LCR rats developed hepatic steatosis by 25 weeks of age and greater hepatic fibrosis and apoptosis at death after natural aging compared to the HCR rats.
Low aerobic capacity, independent of physical activity levels, is the best predictor of early mortality (Blair et al. 1989; Myers et al. 2002) and is linked to type 2 diabetes (Mootha et al. 2003) and cardiovascular disease (Myers et al. 2002). Recently, Church et al. (2006) reported an inverse association between aerobic capacity assessed by a graded exercise test and the prevalence of NAFLD in a large cohort of male subjects, a finding that also has been replicated in other studies (Nguyen-Duy et al. 2003; McMillan et al. 2007). These studies suggest that low aerobic capacity either directly impacts hepatic metabolism or peripheral factors (insulin sensitivity or visceral adiposity) that lead to the increased storage of fat in the liver. To our knowledge, there are no data describing the hepatic biochemical/molecular adaptations that occur in the livers of animals or humans with intrinsically low or high aerobic capacity. Early reports and editorials on the HCR/LCR model (Wisloff et al. 2005; Bernal-Mizrachi & Semenkovich, 2006; Hawley & Spargo, 2006; Noland et al. 2007a) postulated that LCR rats possessed low rates of mitochondrial oxidative capacity in skeletal muscle predisposing them to insulin resistance, obesity, and metabolic complications, a common hypothesis supported by studies in the skeletal muscle of humans (Mootha et al. 2003; Petersen et al. 2004). We report herein that intrinsically low aerobic capacity in sedentary animals is also linked to decreased mitochondrial content and mitochondrial oxidative capacity in liver, a phenotype that we postulate leads to an increased susceptibility to early life hepatic steatosis.
Mobilized fatty acids produced from the lipolysis of visceral adipose are directly trafficked through the liver via portal circulation making it no surprise that increased visceral adiposity is strongly correlated to fatty liver (Nguyen-Duy et al. 2003; Church et al. 2006; McMillan et al. 2007). The LCR rats displayed increased expansion of visceral fat (increased adipocyte cell volume of omental fat pad) along with a greater storage of hepatic fat compared to the HCR, suggesting a connection between increased visceral adiposity and fatty liver in this model. Excessive daily food consumption and postprandial increases in circulating lipids also contribute to excessive fat storage in the liver and the expansion of visceral fat stores. However, it is unlikely that increased food consumption is the primary cause of excessive fat storage in the LCR rats as the results presented here and in previous reports (Noland et al. 2007a) show that the LCR actually have a significantly lower (∼12–25%) daily food consumption per body weight than the HCR. In summary, we postulate that reduced hepatic mitochondrial oxidation is the primary event mediating the increased fat deposition in the liver of the LCR although it is impossible to completely discount the contributing effects of increased adiposity or circulating insulin.
Aerobic capacity, hepatic mitochondrial oxidative capacity and steatosis
Our research group and other labs have shown that impaired hepatic fatty acid oxidation can be a primary cause or strongly associated with fatty liver (Ibdah et al. 2005; Rector et al. 2008); in contrast, others have shown increased rates of hepatic fatty acid oxidation in obese models of fatty liver disease (Brady et al. 1985; Sanyal et al. 2001; Miele et al. 2003). To explain these divergent findings, it is hypothesized that increased hepatic fatty acid oxidation is a secondary but insufficient adaptation to a chronic oversupply of lipids to the liver. Because of these conflicting results utilizing varied methods, it is difficult to determine if hepatic fatty acid oxidation differences are related to alterations in hepatic mitochondrial fatty acid oxidation or if they involve the extra-mitochondrial organelle, peroxisomes, that boost fatty acid catabolism (Noland et al. 2007a). PPARα is a transcription regulator of genes involved in hepatic fatty acid oxidation, including mitochondrial β-oxidation proteins (Memon et al. 2000). However, hepatic PPARα also increases peroxisomal proliferation and the capacity of peroxisomes to oxidize fatty acids (Reddy & Rao, 2006; Schoonjans et al. 1995; Schoonjans et al. 1996). Hepatic PPARα protein content did not differ between groups in our study. However, complete hepatic mitochondrial fatty acid oxidation to CO2 and total fatty acid oxidation (CO2+ ASM) was suppressed in the LCR rats compared with the HCR, suggesting that oxidative capacity of hepatic mitochondria is reduced in LCR livers. In addition, the suppressed enzyme activity or protein content at all three critical steps of mitochondrial fat oxidation (β-oxidation, TCA cycle, and electron transport) in the whole liver homogenate combined with the reduced staining for CPS-1 strongly suggest that the LCR livers also have lower mitochondrial content. In agreement with (Noland et al. 2007a), we also found no differences in rates of hepatic fatty acid oxidation between HCR and LCR rats when measurements were made in whole liver homogenate (data not shown), a technique that prevents the distinction between mitochondrial and peroxisomal contributions. The lack of a significant difference between groups using a liver whole homogenate suggested that LCR rats have a compensatory increase in peroxisomal activity. Indeed, we found that the LCR livers displayed higher peroxisomal activity (higher acyl-CoA oxidase and catalase activity) than their HCR counterparts. Increased peroxisomal activation can contribute to significant lipid peroxidation or oxidative stress, as acyl CoA oxidase produces hydrogen peroxide (Reddy & Hashimoto, 2001). Taken together these results clearly suggest that intrinsically low aerobic capacity is associated with a reduced hepatic mitochondrial quality and content and a compensatory increase in peroxisomal activity. Importantly, a pathogenesis of reduced mitochondrial oxidative capacity is documented in patients with NAFLD making these findings clinically relevant (Perez-Carreras et al. 2003).
It is impossible to completely eliminate the influence of skeletal muscle mitochondrial function on the hepatic phenotypes observed in the HCR and LCR rats, as the model was created separating low and high running capacity, a selective pressure largely dependent on the function of skeletal muscle and the cardiorespiratory system. It is possible that the lower levels of ambulatory physical activity witnessed in the LCR rats may have led to reductions in hepatic mitochondrial content compared to the HCR rats. This is unlikely as previous results from our group studying far larger contrasting levels of physical activity (daily voluntary wheel running at a level of ∼6 km a day for 6 weeks vs. sedentary cage only activity) caused no significant difference in hepatic mitochondrial content in healthy rats (Laye et al. 2008). Our results clearly show that selective breeding for low aerobic capacity resulted in lower hepatic mitochondrial content and function in rats at 25 weeks of age, which we speculate causes the increased susceptibility for hepatic steatosis in the LCR rats. In support of this concept, preliminary results from our laboratory show that the phenotype of reduced lipid oxidation in the LCR livers is retained in primary cell culture (data not shown). Just as the selective breeding for low running capacity reduced skeletal muscle mitochondrial ATP production needed to fuel contractions, it also likely lowered the need for hepatic mitochondrial ATP production, which is needed to fuel the energy-costly process of gluconeogenesis needed to maintain glucose homeostasis during exercise.
Increased lipid peroxidation can lead to the production of reactive aldehydes that damage mitochondrial DNA (Demeilliers et al. 2002) or inactivate respiratory chain proteins (Chen et al. 2000), resulting in reduced mitochondrial content or function and hepatic injury. Despite the strong link between obesity or aging and reduced mitochondrial DNA in skeletal muscle, we were unable to find studies linking hepatic lipid peroxidation or oxidative stress to reduced liver mitochondrial DNA that do not involve liver damage induced by alcohol or pharmacological agents. Here we demonstrate that the aging LCR rats have increased hepatic levels of lipid peroxidation which is subsequent to reduced hepatic mitochondrial function compared with the HCR rats. This could be partially attributed to increased adiposity (Noland et al. 2007a) and potentially greater hepatic free fatty acid flux in the LCR animals. However, the LCR rats had lower relative food consumption and similar feed efficiencies compared to the HCR rats, indicating similar energy balance between the two strains. Collectively, these findings support our contention that hepatic mitochondrial dysfunction plays a primary role in non-alcoholic fatty liver disease development and progression in animals with low intrinsic aerobic capacity. Future studies are warranted to pair feed the two strains and/or attempt to match ambulatory activity levels across the lifespan.
Aerobic capacity, hepatic lipogenesis and steatosis
Both biochemical and histological measures showed increased hepatic fat deposition in the LCR compared to the HCR rats at 25 weeks of age. These findings contradict an earlier report (Noland et al. 2007a) which studied the effect of high fat diet on the metabolic phenotypes of the HCR and LCR strains. That study did not find a significant difference in hepatic fat storage between the LCR and HCR strains on a normal chow or high-fat diet; however, that study used the less sensitive and qualitative Oil Red O technique to stain neutral lipids in a notably small subset (n= 3) of liver samples, while our report used the standard biochemical measure of hepatic triglycerides in a larger sample size. In addition, rats were studied from a different generation between the two studies (generation 13 in former report vs. generation 17 in this study) and it is difficult to discern what impact further selective breeding had upon metabolic or hepatic phenotypes. However, a comparison of reports from previous generations shows comparable values for both strains between generation 11 (Wisloff et al. 2005), 13 (Noland et al. 2007a), and 17 (this report) for measures of body weight, and serum insulin and triglycerides suggesting that methodological differences, and not generational differences, were the main reason that hepatic triglycerides were not previously reported to be higher in the LCR rats.
Insulin increases hepatic fatty acid synthesis, in part through increases in SREBP-1c transcriptional regulation of FAS and ACC (Shimomura et al. 1999). In the LCR rats, it could be speculated that the elevated SREBP-1c protein content was due to elevated serum insulin levels resulting from peripheral insulin resistance (Noland et al. 2007a). The elevated SREBP-1c content in liver of LCR rats was matched with a 2-fold higher protein content of ACC over the HCR rat, with no observed differences in FAS protein content or PPARγ. ACC synthesizes malonyl-CoA, a metabolite that is both a substrate for de novo synthesis of fatty acids through FAS and a potent inhibitor of fatty acid oxidation (McGarry et al. 1977). Although malonyl CoA was not measured herein, the elevated hepatic ACC content also suggests that hepatic CPT-1 activity in the LCR could allosterically be reduced due to a presumed increase in hepatic malonyl-CoA levels.
In conclusion, these results show that the hepatic phenotype associated with reduced aerobic capacity is characterized by both reduced mitochondrial content and oxidative capacity, leading to early hepatic steatosis and lipid peroxidation and the later development of elevated markers of liver injury. These findings provide insight into the pathogenesis of hepatic steatosis and may have important clinical implications. Lower intrinsic aerobic capacity due to physical inactivity and/or genetic heritability may lead to reduced hepatic mitochondrial oxidative capacity and increased susceptibility to hepatic steatosis and injury. Therefore, the clinical measurement of aerobic capacity may serve as a valuable prognostic tool. More importantly, because exercise training and/or increased physical activity is the proven method to increase aerobic capacity, it should remain the cornerstone therapy for fatty liver disease.
Glossary
Abbreviations
- ACC
acetyl CoA carboxylase
- ALT
serum alanine aminotransferase
- CPS1
anti-carbamoylphosphate synthetase 1
- FAS
fatty acid synthase
- LCR
low capacity runner
- β-HAD
β-hydroxyacyl-CoA dehydrogenase
- HCR
high capacity runner
- TAG
triglyceride
- H&E
haematoxylin–eosin
- 4-HNE
4-hydroxynonenal
- NAFLD
non-alcoholic fatty liver disease
- PPARα
peroxisome proliferator-activated receptor α
- SREBP-1c
sterol regulatory binding protein-1c
- TGF-β
transforming growth factor β
- TUNEL
terminal desoxynucleotide transferase-mediated dUTP nick end labelling
- PPARγ
peroxisome proliferator-activated receptor γ
References
- Bellentani S, Saccoccio G, Masutti F, Croce LS, Brandi G, Sasso F, Cristanini G, Tiribelli C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med. 2000;132:112–117. doi: 10.7326/0003-4819-132-2-200001180-00004. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi C, Semenkovich CF. Fast predators or fast food, the fit still survive. Nat Med. 2006;12:46–47. doi: 10.1038/nm0106-46. discussion 47. [DOI] [PubMed] [Google Scholar]
- Blair SN, Kohl HW, 3rd, Paffenbarger RS, Jr, Clark DG, Cooper KH, Gibbons LW. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA. 1989;262:2395–2401. doi: 10.1001/jama.262.17.2395. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Lesage R, Lortie G, Simoneau JA, Hamel P, Boulay MR, Perusse L, Theriault G, Leblanc C. Aerobic performance in brothers, dizygotic and monozygotic twins. Med Sci Sports Exerc. 1986;18:639–646. [PubMed] [Google Scholar]
- Bouchard C, Rankinen T. Individual differences in response to regular physical activity. Med Sci Sports Exerc. 2001;33:S446–451. doi: 10.1097/00005768-200106001-00013. discussion S452–453. [DOI] [PubMed] [Google Scholar]
- Brady LJ, Brady PS, Romsos DR, Hoppel CL. Elevated hepatic mitochondrial and peroxisomal oxidative capacities in fed and starved adult obese (ob/ob) mice. Biochem J. 1985;231:439–444. doi: 10.1042/bj2310439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- Chen J, Petersen DR, Schenker S, Henderson GI. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcoholism Clin Exp Res. 2000;24:544–552. [PubMed] [Google Scholar]
- Church TS, Kuk JL, Ross R, Priest EL, Biltoft E, Blair SN. Association of cardiorespiratory fitness, body mass index, and waist circumference to nonalcoholic fatty liver disease. Gastroenterology. 2006;130:2023–2030. doi: 10.1053/j.gastro.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Demeilliers C, Maisonneuve C, Grodet A, Mansouri A, Nguyen R, Tinel M, Letteron P, Degott C, Feldmann G, Pessayre D, Fromenty B. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology. 2002;123:1278–1290. doi: 10.1053/gast.2002.35952. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Spargo FJ. It's all in the genes, so pick your parents wisely. J Appl Physiol. 2006;100:1751–1752. doi: 10.1152/japplphysiol.00150.2006. [DOI] [PubMed] [Google Scholar]
- Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoydal MA, Wisloff U, Kemi OJ, Britton SL, Koch LG, Smith GL, Ellingsen O. Nitric oxide synthase type-1 modulates cardiomyocyte contractility and calcium handling: association with low intrinsic aerobic capacity. Eur J Cardiovasc Prev Rehabil. 2007;14:319–325. doi: 10.1097/hjr.0b013e3280128bef. [DOI] [PubMed] [Google Scholar]
- Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, Matern D, Rinaldo P, Cline JM. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology. 2005;128:1381–1390. doi: 10.1053/j.gastro.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Kohrt WM, Malley MT, Coggan AR, Spina RJ, Ogawa T, Ehsani AA, Bourey RE, Martin WH, 3rd, Holloszy JO. Effects of gender, age, and fitness level on response of VO2max to training in 60–71 yr olds. J Appl Physiol. 1991;71:2004–2011. doi: 10.1152/jappl.1991.71.5.2004. [DOI] [PubMed] [Google Scholar]
- Koves TR, Noland RC, Bates AL, Henes ST, Muoio DM, Cortright RN. Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am J Physiol Cell Physiol. 2005;288:C1074–1082. doi: 10.1152/ajpcell.00391.2004. [DOI] [PubMed] [Google Scholar]
- Laye MJ, Rector RS, Borengasser SJ, Naples SP, Uptergrove GM, Ibdah JA, Booth FW, Thyfault JP. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J Appl Physiol. 2008;106:161–168. doi: 10.1152/japplphysiol.91186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laye MJ, Thyfault JP, Stump CS, Booth FW. Inactivity induces increases in abdominal fat. J Appl Physiol. 2006;102:1341–1347. doi: 10.1152/japplphysiol.01018.2006. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. 1977;60:265–270. doi: 10.1172/JCI108764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan KP, Kuk JL, Church TS, Blair SN, Ross R. Independent associations between liver fat, visceral adipose tissue, and metabolic risk factors in men. Appl Physiol Nutr Metab. 2007;32:265–272. doi: 10.1139/h06-112. [DOI] [PubMed] [Google Scholar]
- Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. Up-regulation of peroxisome proliferator-activated receptors (PPAR-α) and PPAR-γ messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-γ-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology. 2000;141:4021–4031. doi: 10.1210/endo.141.11.7771. [DOI] [PubMed] [Google Scholar]
- Miele L, Grieco A, Armuzzi A, Candelli M, Forgione A, Gasbarrini A, Gasbarrini G. Hepatic mitochondrial β-oxidation in patients with nonalcoholic steatohepatitis assessed by 13C-octanoate breath test. Am J Gastroenterol. 2003;98:2335–2336. doi: 10.1111/j.1572-0241.2003.07725.x. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, Houstis N, Daly MJ, Patterson N, Mesirov JP, Golub TR, Tamayo P, Spiegelman B, Lander ES, Hirschhorn JN, Altshuler D, Groop LC. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346:793–801. doi: 10.1056/NEJMoa011858. [DOI] [PubMed] [Google Scholar]
- Nguyen-Duy TB, Nichaman MZ, Church TS, Blair SN, Ross R. Visceral fat and liver fat are independent predictors of metabolic risk factors in men. Am J Physiol Endocrinol Metab. 2003;284:E1065–1071. doi: 10.1152/ajpendo.00442.2002. [DOI] [PubMed] [Google Scholar]
- Noland RC, Thyfault JP, Henes ST, Whitfield BR, Woodlief TL, Evans JR, Lust JA, Britton SL, Koch LG, Dudek RW, Dohm GL, Cortright RN, Lust RM. Artificial selection for high capacity endurance running is protective against high fat diet-induced insulin resistance. Am J Physiol Endocrinol Metab. 2007a;293:E31–41. doi: 10.1152/ajpendo.00500.2006. [DOI] [PubMed] [Google Scholar]
- Noland RC, Woodlief TL, Whitfield BR, Manning SM, Evans JR, Dudek RW, Lust RM, Cortright RN. Peroxisomal-mitochondrial oxidation in a rodent model of obesity-associated insulin resistance. Am J Physiol Endocrinol Metab. 2007b;293:E986–E1001. doi: 10.1152/ajpendo.00399.2006. [DOI] [PubMed] [Google Scholar]
- Perez-Carreras M, Del Hoyo P, Martin MA, Rubio JC, Martin A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rector RS, Thyfault JP, Morris RT, Laye MJ, Borengasser SJ, Booth FW, Ibdah JA. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G619–626. doi: 10.1152/ajpgi.00428.2007. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Hashimoto T. Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Rao MS. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am J Physiol Gastrointest Liver Physiol. 2006;290:G852–858. doi: 10.1152/ajpgi.00521.2005. [DOI] [PubMed] [Google Scholar]
- Sanyal AJ, Campbell-Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120:1183–1192. doi: 10.1053/gast.2001.23256. [DOI] [PubMed] [Google Scholar]
- Schindhelm RK, Diamant M, Dekker JM, Tushuizen ME, Teerlink T, Heine RJ. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab Res Rev. 2006;22:437–443. doi: 10.1002/dmrr.666. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARS) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Watanabe M, Suzuki H, Mahfoudi A, Krey G, Wahli W, Grimaldi P, Staels B, Yamamoto T, Auwerx J. Induction of the acyl-coenzyme A synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J Biol Chem. 1995;270:19269–19276. doi: 10.1074/jbc.270.33.19269. [DOI] [PubMed] [Google Scholar]
- Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura I, Bashmakov Y, Horton JD. Increased levels of nuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–30032. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- Small GM, Burdett K, Connock MJ. A sensitive spectrophotometric assay for peroxisomal acyl-CoA oxidase. Biochem J. 1985;227:205–210. doi: 10.1042/bj2270205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Clark SE, Morris EM, Thyfault JP, Uptergrove GM, Whaley-Connell AT, Ferrario CM, Sowers JR, Ibdah JA. Angiotensin II-induced non-alcoholic fatty liver disease is mediated by oxidative stress in transgenic TG(mRen2)27(Ren2) rats. J Hepatol. 2008;49:417–428. doi: 10.1016/j.jhep.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernstrom M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307:418–420. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]