

Summary
Based on promising pre-clinical data demonstrating the eradication of systemic B cell malignancies by CD19-targeted T lymphocytes in vivo in SCID beige mouse models, we are launching Phase 1 clinical trials in patients with chronic lymphocytic leukemia (CLL) and acute lymphoblastic leukemia (ALL). We present here the validation of the bioprocess we developed for the production and expansion of clinical grade autologous T cells derived from patients with CLL. We demonstrate that T cells genetically modified with a replication-defective gammaretroviral vector derived from the Moloney murine leukemia virus encoding a chimeric antigen receptor (CAR) targeted to CD19 (1928z) can be expanded with Dynabeads® CD3/CD28. This bioprocess allows us to generate clinical doses of 1928z+ T cells in approximately 2 to 3 weeks in a large-scale semi-closed culture system using the Wave bioreactor. These 1928z+ T cells remain biologically functional not only in vitro but also in SCID beige mice bearing disseminated tumors. The validation requirements in terms of T cell expansion, T cell transduction with the 1928z CAR, biological activity, quality control testing and release criteria were met for all four validation runs using apheresis products from patients with CLL. Additionally, following expansion of the T cells, the diversity of the skewed Vβ T cell receptor repertoire was significantly restored. This validated process will be used in phase I clinical trials in patients with chemo-refractory CLL and in patients with relapsed ALL. It can also be adapted for other clinical trials involving the expansion and transduction of patient or donor T cells using any chimeric antigen receptor or T cell receptor.
Keywords: Autologous T cells, Chimeric Antigen Receptor, biological activity in vitro and in vivo, Dynabeads® CD3/CD28, gammaretroviral vector, validation, Phase I clinical trial, large scale expansion, bioreactor
Introduction
Chronic Lymphocytic Leukemia (CLL), an indolent cancer of B lymphocytes, is the most common adult leukemia. Despite significant progress in the treatment and management of CLL, most patients will ultimately develop progressive, incurable, chemotherapy resistant disease.1 Patients who suffer a relapse of acute lymphoblastic leukemia (ALL) following an allogeneic marrow transplant also have a poor prognosis. 2 For patients relapsing in the first year post transplant, long-term survival is less than 15%, even with second transplants, due to high transplant-related morbidity and mortality as well as a high rate of subsequent relapse.3, 4 As a consequence, alternative treatment approaches are still urgently needed for both patient populations. The adoptive transfer of autologous antigen specific-T cells is one promising approach for therapy in patients with melanoma and certain hematological malignancies.5-10
CD19, a molecule present on the surface of most B cell malignancies including B cell lymphomas, ALL, CLL, hairy cell leukemias, and a subset of acute myelogenous leukemias, is a target of choice for targeted immunotherapy. It is present on normal B cells from early development until differentiation into plasma cells, but it is absent on other normal tissues including pluripotent hematopoietic stem cells. Consequently, we have developed a novel adoptive cell therapy approach based on the genetic modification of patient T cells to recognize the B cell-specific cellular marker CD19 for the treatment of CLL and ALL.11, 12 Our approach uses a replication defective SFG-based gammaretroviral vector13 to deliver a CD19-specific chimeric antigen receptor to T cells.
We have previously shown that a first generation chimeric antigen receptor targeted to human CD19, 19z1, can effectively lyse CD19+ tumor cells in vitro and eradicate systemic tumors in SCID-Beige mice in vivo.11 To promote effective T cell responses against tumors lacking costimulatory molecules, we developed a second generation CAR termed 1928z. The 1928z CAR expresses a single chain fragment-length antibody derived from a murine CD19 specific monoclonal antibody fused to the extracellular, transmembrane and intracellular signaling domains of the co-stimulatory receptor CD28 and the cytoplasmic signaling domain of the T cell receptor associated CD3 ζ chain14. We have demonstrated that this 1928z CAR confers to T cells the ability to eradicate pre-established tumors in vivo that do not express costimulatory molecules in SCID-Beige mice. 12, 15
The method used for expanding T cells prior to infusion is an essential determinant of their in vivo efficacy. It has been previously demonstrated that T cells derived from patients with various lymphoma and leukemias16-20, myeloma21, HIV22-24 or viral antigen-specific T cells25 can be expanded ex vivo with anti-CD3 and anti-CD28 monoclonal antibodies covalently linked to magnetic beads and that these cells exhibit anti-tumor activity in vitro and in vivo. We have also demonstrated that 1928z transduced T cells expanded with Dynabeads® CD3/CD2826 efficiently lyse CD19+ tumor cells both in vitro and SCID-Beige mice 27 similarly to T cells activated with PHA and subsequently restimulated on artificial antigen presenting cells.11
To evaluate the safety and efficacy of autologous T cells genetically modified to express the 1928z CAR in human Phase I clinical trials in patients with CLL and ALL, we developed a manufacturing process based on T cell expansion with Dynabeads® CD3/CD28 for the activation, transduction and expansion of clinical relevant numbers of autologous 1928z+CD3+ T cells. This process allows us to generate clinical doses of biologically functional 1928z+ T cells in approximately 2 to 3 weeks in a large-scale semi-closed culture system using the Wave bioreactor.
Materials and Methods
Selection of a PG13-SFG-1928z clone
A clinical grade high-titer PG13 clone expressing the 1928z chimeric antigen receptor (CAR) was generated by transiently transfecting Phoenix-eco cells with the plasmid encoding the gammaretroviral vector SFG-1928z12 and subsequently infecting PG13 cells with cell-free vector stocks from the transfected Phoenix-eco cells. The PG13-1928z cell population was subsequently subcloned by limiting dilution. Clones were isolated and titers were determined by infecting HeLa cells under standardized conditions. High titer clones were identified by fluorescence activated cell sorting (FACS) using the anti-1928z CAR hamster monoclonal antibody 19E3 that was generated in-house by the MSKCC monoclonal antibody core facility. The high titer PG13-1928z clone 34 was subjected to a second round of subcloning by limiting dilution. The subclone PG13-1928z cl.3 was demonstrated to express the 19-28zCAR and was selected for its ability to efficiently transduce peripheral blood mononuclear cells (PBMCs). Integrity of the retroviral vector construct was demonstrated and a single copy of the integrated proviral vector was detected by Southern blot analysis in the genomic DNA extracted from PG13-1928z clone 3 (data not shown). The PG13-1928z clone 3 was expanded to generate a seed bank (SB) that was tested for absence of mycoplasm, replication competent retrovirus (RCR), and for sterility. The SB passed all required tests.
Generation of a PG13-1928z Master Cell Bank
A master cell bank (MCB) of 100 vials of the resulting PG13-1928z clone 3 was produced and tested according to FDA and NIH recommendations and guidelines (see Results section). The biosafety tests for the MCB were performed by Charles River Laboratories (CRL, Malvern, PA) and the National Gene Vector Laboratory (NGVL, Indianapolis, IN).
Manufacture of cGMP-like clinical grade vector stocks
cGMP-like grade PG13-1928z vector stocks were prepared as previously described28. Briefly, cells were initially seeded using one certified MCB cryovial containing 107 cells and ultimately expanded into four 10-tray Cell Factories. Viral stocks were harvested from 4 Cell Factories in a 5 L sterile bioprocessing bag using a peristaltic pump on each of 3 consecutive days. Viral stocks were filtered, temporary stored at 4°C, pooled on the third day and frozen at −80°C in cryobags. Cell expansion and viral stocks production was performed in DMEM, 10% FBS. In order to release the vector stocks, biosafety testing was performed according to FDA and NIH guidelines and recommendations. The biosafety tests were performed by CRL and the NGVL.
Apheresis products from patients with B-CLL
Frozen CLL patient-derived apheresis products were obtained under MSKCC Institutional Review Board approval. Apheresis products were washed using a Cytomate™ cell washer (Baxter, Deerfield, IL) to remove gross platelet and red blood cell contamination29, followed by freezing in a Cryomed controlled rate freezer according to Standard Operating Procedures (SOPs). Frozen products were stored in liquid nitrogen vapor phase until use.
Large clinical scale T cell expansion
All processes were validated within a class 10,000 cleanroom environment. At Day 0, frozen apheresis products obtained from patients with B-CLL were thawed and washed with PBS using a Cytomate™ cell washer 29 (Baxter, Deerfield IL) and filtered using a 40/150 Micron Dual Screen Blood filter (Baxter). Cells were incubated for 1 hour with agitation at room temperature with Xcyte™ Dynabeads® kindly provided by M. Bonyhadi (VR1 to VR3) or Dynabeads® ClinExVivo™ CD3/CD28 beads (VR4) (Invitrogen, Carlsbad, CA) at a ratio of 3:1 (beads:cells). Following incubation cells were bound and enriched using a Dynal ClinExVIVO™ MPC magnet (Invitrogen). CD3+ enriched cells were resuspended at approximately 1×106/ml in XVIVO-15 medium (Cambrex, East Rutherford, NJ) + 5% AB serum (Gemini, West Sacramento, CA) and 100U/ml Interleukin-2 (Proleukin, Chiron) and transferred to a 37°C incubator. On Day 3, Origen Permalife PL240 bags (Origen Biomedical, Austin, TX) were injected with 50ml of Retronectin (Takara, Japan) at a final concentration of 15 ug/mL in water for injection (WFI). Bags were incubated at ambient temperature for two hours. After discarding the Retronectin solution, bags were injected with 60ml of 0.5% Human Serum Albumin (HSA) (Talecris, Research Triangle Park, NC) in D-PBS, followed by a 30 minutes incubation at ambient temperature. Finally, bags were washed twice with 50ml of D-PBS.
Activated T cells were resuspended at 2×106/ml, mixed 1:1 (v/v) with clinical grade 1928z-vector stocks and transferred to the Retronectin coated Origen bags. Bags were spinoculated for 1 hour at ambient temperature at 186g using an HL-2B rotor in a Sorval RC-3BP centrifuge and subsequently transferred to a 37°C incubator. The spinoculation was repeated at a ratio of 1:1 (v/v) on Day 4 and cells received a media feed on Day 5. After two rounds of transduction, the cells were transferred to a 2L WAVE bag on a WAVE EHT Bioreactor (GE Healthcare, Somerset, NJ) and expanded using a constant perfusion regime as previously described29 in XVIVO-15 medium supplemented with 5% AB serum and increasing levels of Interleukin-2 (100U/ml, 200U/ml and 500U/ml respectively). Once the target cell dose of 3 × 107 1928z+ T cells/ kg was reached (using a reference weight of 70kg), cells were pumped out of the WAVE, debeaded using a Dynal ClinEXVIVO™ MPC magnet (Invitrogen), washed, volume reduced and resuspended in Plasmalyte A (Baxter) + 1% HSA using the Cytomate™ cell washer.
Lactate levels within the WAVE bioreactor were measured using Lactate Pro test strips and test meter (ARKRAY, Inc, Kyoto, Japan). Glucose levels were monitored using an Accu-Chek blood glucose testing system (Roche Diagnostics, Indianapolis, IN).
Cell counts and immunophenotyping by FACS analysis
Viable cell counts were performed using standard Trypan blue exclusion method. Expanded T cells were analyzed for the expression of 1928z CAR following staining with a PE-conjugated 1928z specific hamster monoclonal antibody (in-house 19E3 monoclonal antibody) and mouse anti-human CD3 FITC (Caltag/Invitrogen, Carlsbad, CA, USA). CD4 and CD8 were detected using mouse anti-human CD4-PerCP Cy5.5 (BD Biosciences, San Jose, CA, USA) or CD4-TRI (Caltag/Invitrogen) and mouse anti-human CD8-APC (Caltag/Invitrogen), respectively. Untransduced cells and relevant isotype antibodies were used as controls. The B-CLL tumor cells were detected in apheresis and end products by co-staining with CD45 APC H7, CD19 FITC, CD5 PE (BD Biosciences, San Jose, CA, USA). Staining with 7AAD (Beckman Coulter, Fullerton, CA) was used to gate out dead cells. Stained cells were acquired on a FACSCanto (BD Biosciences, San Jose, CA, USA). For phenotyping of end of production T cells, frozen T cell products were thawed, washed in PBS and stained at room temperature. Antibodies used were 19E3 PE, CD62L FITC, CD28 FITC, CD45RA (Caltag/Invitrogen), CCR7 PE-Cy7, CD27 APC, CD8 PE-Cy7 (BD Biosciences), CD8 APC-AF750 and CD127 Pacific Blue (eBiosciences, San Diego, CA). 7AAD (Beckman Coulter) was added prior to acquisition on an LSRII flow cytometer (BD Biosciences, San Jose, California). All frozen products contained >98% CD3+ and were gated on 7AAD-1928z+ chimeric antigen positive events. Analysis was performed using FloJo software (TreeStar, Ashland, OR).
Quantitative Real-Time PCR
DNA was extracted from expanded T cells when the number of 1928z+ T cells reached 30% to 100% of the target dose using a Gentra Puregene kit (QIAGEN, Valencia, CA) as per manufacturers' directions). Quantification of the SFG-1928z vector copy number was performed using the Applied Biosystems 7500 real-time PCR system. The PCR mix contained 1X Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA), 800nM each of the primers (Fisheroligo.com), 200nM each of labeled probes (Applied Biosystems) and sterile nuclease free water. The primer sets SFG forward and reverse, Human Albumin forward and reverse and the labeled probes for SFG-based vector and human albumin were previously described30,. Reactions were set up in MicroAmp Optical 96-well reaction plates (Applied Biosystems), with 24ul PCR mix per well. 1ul containing 100ng DNA was added per well, plates sealed with optical adhesive cover (Applied Biosystems) and centrifuged at 100×g 2mins. Unknown samples, no template controls and standards were run in triplicate. 1ul of serially diluted SFG-based plasmid and human albumin plasmid standards contained 107 to 102 plasmid copies in 100ng salmon sperm DNA. Q-PCR running conditions were 50°C for 2mins, 95°C for 10mins, then 40 cycles of 95°C for 15 secs, 60°C for 1 min. Analysis was performed using the 7500 System SDS software v.1.2.3. Average SFG-1928z vector copy number per cell was calculated by normalizing to the endogenous number of diploid albumin copies.
Detection of GaLV envelope by PCR for testing of Replication Competent Retrovirus (RCR)
The presence of DNA sequences encoding the GaLV viral envelope was investigated in the genomic DNA extracted from 5 × 106 untransduced and transduced patient's T cells using QIAamp DNA Blood Midi Kit (QIAGEN). The PCR method uses a set of primers designed to detect GaLV envelope sequences: 5′-ACCACAGGCGACAGACTTTT, 5′-TGAGACAGCCTCTCTTTTAGTCCT and Actin primers utilized as amplification control: 5′-ACACTGTGCCCATCTACGAGG, 5′-AGGGGCCGGACTCGTCATACT. The sensitivity of the assay was determined to be as low as 6 copies of the DNA sequences in samples spiked with the positive control plasmid (pGEM-Gal-Seato). The primers and the pGEM-Gal-Seato plasmid were kindly provided by K. Cornetta, NGVL, Indianapolis, IN.
Points to Consider (PTC) Replication Competent Retrovirus testing (RCR) and Mycoplasma Testing
PTC testing of cells, supernatant or a mix of cell and supernatant, as required, was performed by CRLand NGVL according to FDA guidelines as previously described 28 31 32
Endotoxin Detection
Endotoxin level in final products was determined using the FDA-approved Endosafe® -PTS Portable Test System (Charles River Laboratories) as per the manufacturer's recommendations.33 The PTS mimics LAL kinetic chromogenic methodology by measuring color intensity directly related to the endotoxin concentration in a sample and was performed according to manufacturer's guidelines. The level of sensitivity is between 0.10-10 EU/ml. Results were converted to EU/kg based on patient weight and product volume.
Rapid Mycoplasma Testing
Mycoplasma detection was performed on final products using the MycoAlert® Mycoplasma Detection Kit (Cambrex). Assays were performed according to manufacturer's guidelines.
Residual Dynabeads ClinExVivo CD3/CD28
Residual bead count was determined on samples containing 107 cells. Cells were lysed using chlorine bleach, centrifuged, resuspended in 10 ul and loaded onto a chamber on a hemacytometer. Counts were determined by counting beads contained in the nine large squares of the grid that contain 0.1 μl/square that was determined to contain an equivalent of 105 cells/square.
In vitro cytotoxic assay
Cytotoxic activity of 1928z transduced T cells on autologous B-CLL cells and Raji cells, a Burkitt's lymphoma cell line expressing CD19, was determined using a standard chromium release assay. 1 × 104 51Cr-labeled target cells were incubated with the effector cells at various E:T ratios (25:1 to 3.125:1) in 96-well microplates14. Following 4 h incubation at 37°C, supernatants were harvested and radioactivity was counted in a microplate scintillation counter (Packard Instruments Co., Meriden, CT).
In vivo activity of 1928z+ T cells in SCID beige mice
FOX CHASE C.B-17 (SCID-Beige) mice (Taconic, Germantown NY) inoculated intravenously by tail vein injection with 5 ×105 Raji cells develop hind-limb paralysis in 3 to 5 weeks after tumor cell injection, secondary to spinal cord compression from vertebral bone marrow tumor involvement.11 Mice bearing established Raji tumors, six days after intravenous injection, were treated with 107 1928zCD3+ transduced T cells from VR4 by tail-vein injections. Untransduced T cells from the same donor served as a control.
Spectratype Analysis
RNA was isolated from 1× 107 T cells derived from either the apheresis product (starting material) or from the 1928z transduced T cells collected at the end of the validation run (end of production T cells). RNA was isolated using TRIzol reagent following manufacturer's instructions (Invitrogen). Synthesis of cDNA was performed using 3 ug of RNA as template, random hexamers as primers and the Superscript III synthesis kit following manufacturer's instructions (Invitrogen). Aliquots of cDNA were used as templates for Vβ PCR amplification. A Cβ primer labeled with FAM34 and variable region primers for β families 1-2435 were used for PCR. The PCR was performed under the following conditions: 94 °C for 2 min, followed by 35 cycles at 94° C for 1 min, 55 °C for 1 min and 72 °C for 1 min, and completed at 72 °C for 10 min hold. Aliquots of the PCR were run on an ABI 3100 and the resulting fragments analyzed using Peakscanner software (Applied Biosystems, Foster City, CA).
Results
PG13-1928z Master Cell Bank Qualification
We produced a PG13-1928z Master Cell Bank of 100 vials. The PG13-1928z MCB was certified after a broad range of safety and identity testing was performed (Table 1). 21CFR610 compliant sterility testing was negative, PTC mycoplasm testing on combined end of production (EOP) cells and cell supernatant was negative. Analysis of genomic DNA by Southern blot demonstrated expected band sizes (data not shown). The MCB biosafety testing included in vitro and in vivo adventitious virus testing, replication competent retrovirus (RCR) testing and screening for a range of human pathogenic viruses. No virus was detected in any of these assays. In summary, the PG13-1928z MCB passed all required testing (Table 1). We also performed small-scale vector productions and evaluated the suitability of the vector titer on HeLa cells for clinical use. Titration of these small-scale vector stocks on T cells derived from healthy donors (n=2) gave an average titer of 6× 105 TU/ml corresponding to transduction efficiency in the range of 22 to 34%.
TABLE 1. Biosafety testing performed on PG13-1928z cl.3 Master Cell Bank (MCB) and PG13-1928z cl.3 Vector Stocks.
| 1928z cl. 3 MCB | 1928z cl.3 Vector Stocks | |||
|---|---|---|---|---|
| Test | Method | Specification | Result | Result |
| Identity | Southern Blot | Band of predicted size | As expected | N/A |
| Vector Titration | Transduction of healthy donor T cells | For information | 6×105 TU/ml | 9×105 TU/ml |
| Species Determination | Isozyme analysis | Murine | Murine | N/A |
| Final Product Sterility | 21CFR610.12 Direct Inoculation | Sterile | Sterile | N/A |
| Final Product Sterility | 21CFR610.12 Membrane Filtration | Sterile | N/A | Sterile |
| Bulk Sterility | 21CFR610.12 Membrane Filtration | Sterile | N/A | Sterile |
| Mycoplasma (EOP cells & supernatant) | Cell lines PTC | Negative | Negative | N/A |
| Mycoplasma (EOP cells) | Cell lines PTC | Negative | N/A | Negative |
| Mycoplasma (EOP supernatant) | Cell lines PTC | Negative | N/A | Negative |
| In vitro Adventitious Virus | Cell Culture Assay | ND | ND | N/A |
| In vivo Adventitious Virus | Live Animal Assay | ND | ND | N/A |
| Ecotropic Retrovirus Testing (EOP cells) | Marker-Rescue Cell Culture Assay | ND | ND | N/A |
| GalV RCR Testing (EOP cells) | Marker-Rescue Cell Culture Assay | ND | ND | ND |
| GalV RCR Testing (supernatant) | Marker-Rescue Cell Culture Assay | ND | ND | ND |
| Transmission Electron Microscopy | Thin Section EM | Type-C Retrovirus Particles only | Type-C Retrovirus Particles only | Type-C Retrovirus Particles only |
| General Safety | USP 21CFR610.11 | Pass | N/A | Pass |
| MAP Testing with LCMV Challenge | Cell lines PTC | Negative | Negative | N/A |
EM: Electron Microscopy; GaLV: Gibbon Ape Leukemia Virus; EOP, end of production; MAP: Mouse Antibody Production;
N/A: Not applicable; ND, none detected; LAL: Limulus Amoebocyte Lysate; PCR: Polymerase Chain Reaction;
PTC, Points to consider; RCR, Replication Competent Retrovirus; USP: United States Pharmacopeia;
RCR: Replication Competent Retrovirus; CFR: Code of Federal Regulations
PG13-1928z Vector Stocks Qualification
Seven liters of cell-free clinical grade 1928z vector stocks were prepared as previously described28 and frozen at -80°C in 250ml aliquots. The vector stocks were certified after a broad range of safety and identity testing was performed. Membrane-filtration 21CFR610.12 compliant sterility testing on both the bulk and final products was negative. PTC mycoplasma testing performed on EOP cell and EOP supernatant was negative. GaLV RCR envelope was undetectable on both the EOP cells and EOP supernatant and the supernatant passed USP 21CFR610.11 general safety testing. The vector stocks passed all tests (Table 1). Titration of the large-scale vector stocks on HeLa indicator cells was less than 105 TU/ml. Titration on T cells derived from healthy donors (n=2) gave an average titer of 9× 105 IU/ml corresponding to an average transduction efficiency of 46%.
Large scale bioprocessing of 1928z+ transduced T cells
We undertook validation studies of the large scale transduction and expansion process to demonstrate that i) suitable levels of transduction could be achieved in tissue culture bags using the certified PG13-1928z vector stocks, ii) sufficient numbers of 1928z+ T cells could be manufactured in a reasonable time frame and iii) our clinical process was free of fungal, bacterial and mycoplasma contamination or replication competent retrovirus. The validation study was designed to represent the process that would be used for the upcoming Phase 1 clinical trials. This process is outlined in Fig 1.
FIGURE 1.

Scheme of the manufacturing process. The semi-closed system relies on the use of the Cytomate™ to wash the apheresis product prior to freezing and after thawing, the capture of CD3+CD28+ T cells with Dynabeads® and subsequent selection on the ClinExVivo magnetic particle concentrator (MPC), expansion in the Wave™ bioreactor, debeading on the ClinExVivo MPC and formulation using the Cytomate™.
Apheresis products were used as starting materials for the validation runs and were obtained from patients with CLL (termed VR1, VR2, VR3 and VR4). The patients had received a range of prior treatment regimens including previous gene therapy, Rituximab, fludarabine and other chemotherapy regimens (Table 2). On day 0, the apheresis products were evaluated for various cell markers by fluorescence activated cell sorting (FACS), viability and total number of cells (TNC) after thawing and washing using the Cytomate™. Characteristics of the initial products are shown in Table 2. Starting CD3+ ranged from 0.5% to 8.4% and TNC ranged from 0.66 to 3.24 × 1010. All four products had high levels of CD5+CD19+ CLL tumor cells ranging from 45% to 96%. Washed apheresis products were then activated using Dynabeads® CD3/CD28 and CD3+CD28+ T cells were enriched using a ClinExVivo MPC magnet. A volume of apheresis product, containing 3×108 CD3+ cells (VR1-VR3) or 2.5×108 CD3+ cells (VR4) was diluted to 100ml and incubated with 9×108 beads. After incubation, CD3+CD28+ T cells were enriched on the ClinExVivo MPC magnet and cells were resuspended at 1×106/ml. Using this process, sufficient cells were obtained and activated. On day 3 and 4, T cells were transduced with the 1928z vector stocks as described in Materials and Methods.
TABLE 2. Characteristics of the apheresis and final products after expansion of T cells with Dynabeads® in the Wave bioreactor.
| Apheresis | Final Product | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Previous treatment | Total WBC | CD3+ | CD5+ CD19+ | CD4: CD8 | Total WBC | CD3+ | CD5+ CD19+ | CD3+ 1928z+ | CD4:CD8 |
| VR1 | Rituximab | 0.66 × 1010 | 8.4% | 82% | 2.9:1 | 2.27 ×1010 | 100% | 0.22% | 43% | 12:1 |
| VR2 | Fluadarabine | 1.08×1010 | 2.9% | 45% | 2.8:1 | 0.8×1010 | 98% | <BLD | 40% | 4.6:1 |
| VR3 | Adenoviral GT | 2.24×1010 | 0.5% | 96% | 1.95:1 | 2.39 ×1010 | 99% | 0.28% | 26% | 2.7:1 |
| VR4 | FCR, PCR | 3.24×1010 | 5% | 94% | 2.2:1 | 2.5 ×1010 | 100% | 0.04%s | 14.6% | 1.7:1 |
GT: Gene Therapy; FCR: Fludarabine, Cyclophosphamide, Rituximab; PCR: Pentostatin, Cyclophosphamide, Rituximab; WBC: White Blood Cells; BLD: below level of detection
During the early stages of our pre-clinical development, we investigated various options to optimize the expansion of patient T cells in the WAVE bioreactor. A continuous perfusion regime was selected. In this system, fresh media is pumped continuously into the WAVE bioreactor, while an equal volume of media is pumped out. During the expansion of T cells in the WAVE bioreactor, we adjust rocking of the WAVE platform to provide increase oxygen exchange (6-15rpm), we increase perfusion volumes over 24hr periods (200ml-1600ml) and we increase Interleukin-2 levels (100-500U/ml). T cells expanded a total of 87 to 668-fold over a period of 13 to 18 days (Fig 2A). Cell densities ranged from 13.2×106/ml to 31×106/ml (Fig 2B) with final viabilities of 91.7% to 98.9% (Table 5). Total T cell counts ranged from 0.8 to 2.5 ×1010 (Table 2). In addition to cell counts and viability, we also monitored glucose (Fig. 2C) and lactate levels (Fig. 2D) in the WAVE bioreactor cultures. We consistently observed that as the cell concentrations increased, the levels of lactate decreased while the levels of glucose increased.
FIGURE 2.

T cell expansion, T cell concentration, glucose and lactate levels in the Wave Bioreactor during the course of VR1, VR2, VR3 and VR4. A, Total cumulative fold expansion of patient T cells. Data is shown from initial activation until completion of the expansion runs. B, Viable cell concentrations of patient T cells during activation and within the WAVE bioreactor. C, Glucose and D, Lactate levels were measured daily during the course of the validation runs.
TABLE 5. Release testing criteria and results on the EOP 1928z transduced T cells.
| VR1 | VR2 | VR3 | VR4 | |||
|---|---|---|---|---|---|---|
| Test | Method | Specification | Results | |||
| Transduction efficiency (EOP cells) | Flow cytometry | >10% | 43% | 40% | 26% | 14.6% |
| CD3+ purity (EOP cells) | Flow cytometry | >95% | 100% | 98% | 99% | 100% |
| Residual CD5+ CD19+ tumor (EOP cells) | Flow cytometry | <total CD5+ CD19+ in apheresis | 0.22% | <BLD | 0.28% | 0.04% |
| Average Vector Copy number (In process) | Taqman | ≤ 4 copies/cell | 0.97 | 0.58 | 0.58 | 0.20 |
| Sterility, In process | Current USP | Sterile | Sterile | Sterile | Sterile | Sterile |
| Sterility, In process | Gram Stain | Sterile | Sterile | Sterile | Sterile | Sterile |
| Sterility, Final Product | Current USP | Sterile | Sterile | Sterile | Sterile | Sterile |
| Sterility, Final Product | Gram Stain | Sterile | Sterile | Sterile | Sterile | Sterile |
| Sterilty, Bulk Supernatant | Current USP | Sterile | Sterile | Sterile | Sterile | Sterile |
| Mycoplasma (EOP cells + supernatant) | Cell lines PTC | Negative | Negative | Negative | Negative | Negative |
| Mycoplasma (in process) | MycoAlert Rapid Detection Kit | Negative | Negative | Negative | Negative | Negative |
| Final Product Bacterial Endotoxin | Kinetic Chromogenic LAL Assay USP <85> | <5 EU/kg | <0.01 EU/ml=0.1 EU/kg | <0.052 EU/ml=0.17EU/kg | <0.01 EU/ml=0.1 EU/kg | 0.029 EU/ml |
| GalV RCR Testing (EOP cells) | Marker-Rescue Cell Culture Assay | No RCR detected | No RCR detected | No RCR detected | No RCR detected | No RCR detected |
| GalV RCR Testing (in process) | PCR for GalV Envelope | No RCR detected | No RCR detected | No RCR detected | No RCR detected | No RCR detected |
| Residual Dynabeads (EOP cells) | Microscopy | <100 beads/3×106 cells | N/D | 0 | 0 | 12 |
| Viability (EOP cells) | Trypan blue exclusion | >80% | 92.4% | 94.9% | 98.9% | 91.7% |
To set the validation specifications, patient weight was considered to be 70kg. EOP: end of production cells; GaLV: Gibbon Ape Leukemia Virus;
N/D: not done; PCR: Polymerase Chain Reaction; RCR: Replication Competent Retrovirus; USP: United States Pharmacopeia;
Transduction efficiency, as measured by FACS analysis using the 19E3 monoclonal antibody specific for the 1928z CAR, ranged from 14.6% to 43% (Table 2). CD4:CD8 ratios changed during the course of Dynabeads® CD3/CD28 activation and subsequent expansion in the WAVE bioreactor. An increase in the CD4 content of cultures was observed in three of the four validation runs (Table 2).
The downstream T cell processing involves the use of the ClinExVivo MPC magnet and Cytomate™ cell washer. The ClinExVivo MPC magnet consists of a large primary magnet that binds the majority of beads plus a smaller secondary magnet that allows final clarification before the de-beaded cells enter the collection transfer pack. 200ml fractions of the bulk culture are pumped onto the primary magnet using a Watson Marlow pump (Watson Marlowm, Wilmington, MA), incubated to allow bead capture and drained over the smaller secondary magnet into the collection transfer pack. This process is repeated until the entire bulk culture (1 to 1.2 liters) is passed over both magnets. The residual bead contamination was very low and even undetectable in two out of the 3 tested validation runs (Table 5). The final stage of our bioprocess is a volume reduction and wash using the Cytomate™. The de-beaded bulk culture is washed and volume reduced into a solution of Plasmalyte A and 1% HSA. The final product can be further diluted with Plasmalyte A and 1% HSA if required.
At each stage of cell processing, particularly during selection and volume reduction, loss of cells can occur. We calculated the downstream processing efficiency during the course of the validation runs (Table 3). In all cases, the TNC recovery after the downstream ClinExVivo MPC magnet de-beading and Cytomate™ washing/volume reduction steps was ≥ 66%. The downstream processing had no impact on the viability of cells, as the viability at all stages and in all validation runs was > 91%.
TABLE 3. Downstream bioprocessing efficiency.
| VR1 | VR2 | VR3 | VR4 | |||||
|---|---|---|---|---|---|---|---|---|
| Process Step | Viable WBC | Viability | Viable WBC | Viability | Viable WBC | Viability | ViableWBC | Viability |
| Filtered WAVE T cells | 2.92×1010 | 93% | 1.16×1010 | 97.2% | 2.16×1010 | 99% | 2.26×1010 | 96% |
| Debeaded ClinExVivo | 2.08×1010 | 97.9% | 1.04×1010 | 97.6% | 2.81×1010 | 99% | 2.1×1010 | 97% |
| Cytomate™ Volume reduced | 3.29×1010 | 93.8% | 0.86×1010 | 93% | 2.92×1010 | 98.9% | 1.5×1010 | 91.7% |
| % Recovery | 113% | 74% | 135% | 66% | ||||
The downstream processing efficiency was determined from the time the expanded 1928z+ T cells were removed from the WAVE bioreactor until after volume reduction. Briefly, T cells were removed from the WAVE bioreactor and the residual Dynabeads CD3/CD28 were removed using a ClinExVivo magnet. The debeaded 1928z+ T cell products were then volume reduced using a Cytomate cell washer; WBC: White Blood Cells;
Depletion of B-CLL tumors in EOP 1928z T cells
It is important that carry over of B-CLL tumor cells present in the initial apheresis product be monitored during the bioprocessing of 1928z T cells. CD3+ purity was greater than 95% at completion of all four runs, ranging from 98% to 100%. Final CD5+CD19+ tumor contamination in the EOP 1928z+ T cells was very low in VR1, VR3 and VR4 (0.04% to 0.28%) and undetectable in VR2, indicating that the cultures were highly enriched in CD3+ T cells. We also observed that the clearance of contaminating tumor cells was accelerated in cultures expressing the 1928z CAR indicating that the CD5+CD19+ cells which remain post-selection and activation with Dynabeads® are lost during expansion, likely due to lysis upon coculture with 1928z+ CD3+ cells (data not shown).
In vitro cytotoxic studies and in vivo biological activity of 1928z+ T cells
We have previously shown that human T cells expressing 19z1 and 1928z CARs can efficiently lyse CD19+ Raji lymphoma cells both in vitro and eradicate tumors in vivo11, 12. As part of the validation process, we sought to demonstrate that the 1928z transduced T cells, when manufactured at large scale and expanded with Dynabeads®, possessed in vitro cytotoxic activity and in vivo biological activity. To this end, we tested cells from each of the four validation runs in vitro in chromium release assays. We choose both Raji lymphoma cells and patient autologous B-CLL cells as targets (Fig. 3A). On autologous patient B cells, VR1 – VR3 T cell final products demonstrated greater than 50% lysis activity at 25:1 effector to target ratio and VR4 gave 37% lysis. Control untransduced T cells lacking the 1928z CAR showed no significant lysis. In order to determine the biological activity of the transduced T cells in vivo, 1928z+ T cells from VR4 were injected in mice bearing pre-established Raji tumors (Fig. 3B). The 1928z+ T cells were able to eradicate the tumors in 90% of the mice followed for more than 120 days while all the mice treated with untransduced T cells had to be sacrificed around 25 to 30 days following tumor cell infusion due to hind limb paralysis.
FIGURE 3.

In vitro and in vivo cytotoxic activity of the 1928z+ T cells. A, In vitro chromium release assays performed on 1928z+ EOP T cells derived from VR1, VR2, VR3 and VR4. Both autologous patient tumor B-CLL cells and Raji lymphoma cell line were used as target cells. B, Survival of mice bearing established disseminated Raji tumors after infusion of EOP 1928z+ T cells. Six days after intravenous injection of 5 × 105 Raji tumor cells, mice received 107 fresh 1928z CD3+ transduced T cells from VR4 run. Untransduced T cells from the same donor grown in parallel served as control. Note: we previously demonstrated that untransduced T cells and T cells transduced with an irrelevant CAR are equally unable to eradicate Raji tumors in vivo (data not shown).
Spectratype analysis
Spectratype analysis was performed on samples from the initial apheresis products as well as on the final 1928z transduced T cell products expanded with the Dynabeads® derived from VR1 and VR2. Skewed receptor repertoire was observed in the apheresis products in many of the Vβ families characterized by oligoclonal patterns showing one or two predominant peaks (Fig. 4). Following expansion, repertoire skewing was reduced and replaced with a Gaussian distribution of peaks representing a more diverse repertoire.
FIGURE 4.
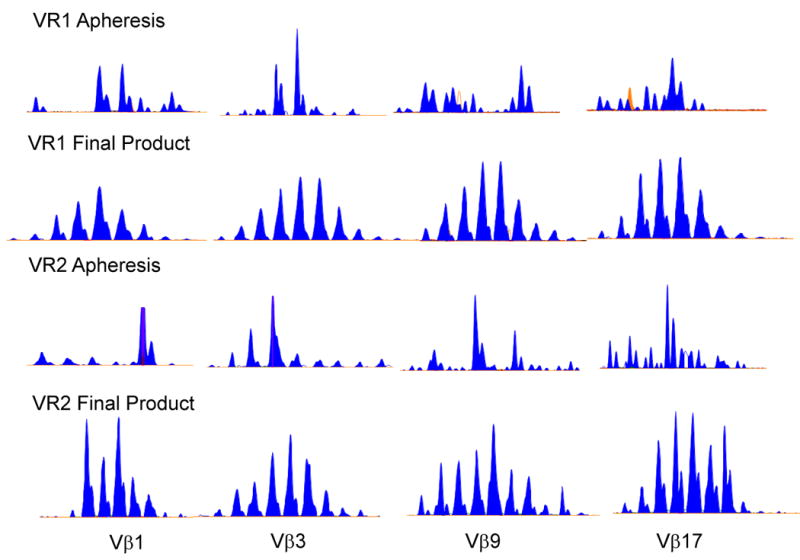
Spectratyping analysis in T cells from the apheresis product and in the EOP 1928z T cells from VR1 and VR2. Representative spectratype analysis of Vβ1, Vβ3, Vβ9 and Vβ17 shows a skewed TCR repertoire in the T cells from the apheresis product and restored TCR repertoire in the EOP 1928z T cells after expansion with Dynabeads® (final product).
Immunophenotype of the EOP 1928z+ T cells
The immunophenotype of the 1928z+ EOP T cells is shown in Table 4. In VR2, VR3 and VR4 1928z+ transduced T cells were CD62L-, CCR7-, and CD127-. In VR1, 1928z+ transduced T cells were also CD62L- and CCR7-, but did show CD127 expression (56.6%). In all four validation runs, the 1928z+ T cells remained CD28+ (≥ 85%) following expansion with the Dynabeads® CD3/CD28 and a significant proportion of transduced T cells (23% to 44%) retained CD27 expression. 36.7% of 1928z+ T cells in VR4 also remained CD45RA+.
TABLE 4. Immunophenotyping of the EOP 1928z transduced T cells.
| Marker | VR1 | VR2 | VR3 | VR4 |
|---|---|---|---|---|
| CD27 | 37.3 | 24.4 | 23 | 44 |
| CD28 | 98.6 | 90.2 | 87.6 | 85.7 |
| CD62L | 2.1 | 7.3 | 0.9 | 5.2 |
| CCR7 | 0.8 | 0.2 | 0.8 | 1.0 |
| CD127 | 56.6 | 6.3 | 1.2 | 0.2 |
| CD45RA | 5.9 | 5.2 | 8.5 | 36.7 |
Results are shown as percentages of 7AAD- 1928z+ events.
Release and biosafety testing
During the development of this T cell process, we sought to use a semi-closed system that would allow us to culture cells in the WAVE bioreactor without the addition of antibiotics. Extensive testing was performed during the validation runs to ensure that our process was free of bacterial, fungal, yeast and mycoplasma contamination (Table 4). 14 day USP sterility testing and Gram staining were performed on T cell cultures during expansion, on bulk supernatant during final washes on the Cytomate™ and on the EOP cells. All tests performed in process and at the end of the production were negative in all validation runs.
Levels of endotoxin determined on EOP cells to rapidly detect bacterial contamination were below specifications in all validation runs as well. We also undertook rapid mycoplasma testing during expansion of T cells in combination with PTC Mycoplasma testing on EOP cells. All tests were negative for the presence of mycoplasma.
As we are using an SFG-based gammaretroviral vector13 to deliver the 1928z CAR, several important biosafety tests were performed including the determination of the average number of integrated SFG-1928z vector copies in transduced T cells. Real time PCR analysis was performed on genomic DNA extracted from EOP cells using a plasmid DNA standard curve. The average vector copy numbers ranged from 0.2 to 0.97 and fell within the specified range of ≤ 4 copies/ cell as per FDA recommendation (Table 5). We also tested the presence of replication competent retrovirus (RCR) by PCR using the GaLV envelope as the target sequence as well as a marker-rescue cell culture assay. RCR testing by PCR and marker-rescue assays were performed on in-process and EOP 1928z T cells respectively for each of the four validation runs. All tests were negative for the presence of RCR (Table 5).
Stability of the formulated 1928z+ T cells
The design of our Phase I clinical trials requires that the EOP 1928z T cells be infused fresh, without cryopreservation. To determine the stability of the T cells at 4°C and provide an expiration time/date for the product, we undertook a stability study. Formulated EOP cells from VR1 and VR2 were placed at 4°C and the viability was tested at 24hr, 48hr, 72hr and 96hr. EOP cells from both VR1 and VR2 were >80% viable after 48hrs at 4°C (data not shown). The viability at 72 hrs was 74% and 79% and 70% and 78% at 96 hrs in VR1 and VR2 respectively. Our specification for T cell infusion requires a minimum of 80% viability. We have therefore set our expiration date for 1928z transduced T cells at 48hrs post-final formulation.
Discussion
Several recent studies describe the promising use of autologous adoptive T cell therapies for cancer immunotherapy.5-10 Here, we demonstrate that T cells genetically modified to express a CAR targeted to CD19 can be effectively expanded using Dynabeads® CD3/CD28. The semi-closed system that we developed for the activation, transduction, expansion and formulation of 1928z+ T cells for phase I clinical trials in patients with CLL and ALL using Dynabeads® CD3/CD28 and the Wave bioreactor is not only efficient, but rapid, cost effective and convenient to use. We were able to generate sufficient number of 1928z+ T cells to achieve the cell dose of 3 × 107 1928z+ CD3+ T cells in a one liter culture volume as required in the phase I clinical trial that we initiated in patients with purine analog-refractory CLL (http://clinicaltrials.gov/ct2/show/NCT00466531).
We performed four successful validation runs in which we demonstrated that T cells expressing the 1928z CAR could be expanded with Dynabeads® CD3/CD28 and retained their cytotoxic activity both in vitro and in vivo in SCID beige mice bearing pre-established systemic Burkitt lymphoma (Raji tumor). We observed greater than 37% lysis activity at 25:1 effector to target ratio demonstrating the in vitro cytotoxic activity of the expanded 1928z+ T cells in all patients. The cytotoxic activity was also demonstrated to be dependent on the presence of the 1928z CAR, as no significant killing was observed with non-genetically modified T cells. In vivo, the biological activity of the 1928z+ T cells expanded in this manner was comparable to that of 1928z+ T cells activated with artificial antigen presenting cells11, 27 (and manuscript in preparation).
This process allowed us to manufacture 0.8 to 2.5 ×1010 T cells in 13 to 17 days at 98 to 100% CD3+ T cell purity. These results are well in line with the results obtained by Hami et al. 18 and Tran et al. 29 and indicate that the stimulation of the T cells through CD3 and CD28 does not interfere with the function of the 1928z CAR. The selection of the CD3+CD28+ T cells with the Dynabeads in conjunction with the expression of the 1928z CAR resulted in the decrease of the B-CLL tumors in the EOP 1928z+ T cells by approximately 3 logs.
T cell expansion (range 87- 668 fold), transduction efficiencies (range 14.6%- 43%) and CD4:CD8 ratios (range 12:1- 1.7:1) varied widely between patients, possibly due to variations in patient age, prior chemotherapy regimens and stage of disease. We also observed similar variations in transduction efficiencies during our pre-clinical investigations.30 CD4:CD8 ratios evolved during the course of Dynabeads® CD3/CD28 activation and subsequent expansion in the WAVE bioreactor. An increase in the CD4 content of cultures was observed in three out of four validations runs without any bias in the transduction efficiency in either subset. The increase in CD4 content did not affect the cytotoxicity of the cells in vitro nor in vivo in SCID beige mice bearing pre-established Raji tumors (manuscript in preparation). Indeed, these data are consistent with a recently published study demonstrating that autologous CD4+ T cell clones with specificity for the melanoma-associated antigen NY-ESO-1 could mediate a durable clinical remission in a patient with melanoma and led to endogenous responses against melanoma antigens other than NY-ESO-1.8
Alterations in the Vβ TCR repertoire, specifically decreased diversity, has been observed in the peripheral blood T cells derived from patients with CLL36. It has been previously demonstrated that the diversity of the Vβ TCR repertoire can be restored from a skewed repertoire to a normal repertoire following expansion of the T cells with Xcyte Dynabeads17. Our study demonstrates similar findings when comparing the Vβ TCR repertoire from the T cells present in the apheresis product derived from two patients with CLL to the Vβ TCR repertoire in the EOP 1928z transduced T cells. Following expansion, repertoire skewing was reduced and a Gaussian distribution of peaks representing a more diverse repertoire was observed.
The immunophenotype of the expanded 1928z+ EOP T cells at the end of the runs correlates best with that of effector T cells still expressing significant levels of CD28 and CD27. Importantly, these effector T cells were derived not only from T cells with an effector phenotype but also from T cells in the apheresis product that display a memory phenotype.37-39
We also demonstrated that the 1928z+ T cells from all four validation runs passed all release criteria and biosafety testing. During these studies, we were able to demonstrate that the ClinExVivo MPC magnet consisting of a large primary magnet combined to a smaller secondary magnet reproducibly and efficiently allowed the removal of the beads from the EOP 1928z+ T cells. In all four validation runs, the EOP 1928z+ T cells and cell supernatant were consistently free of fungal, bacterial and mycoplasma contamination and the levels of endotoxin were below 5 EU per kg in in-process samples as well. This aspect of the validations is crucial for the safety of the patients in the phase I clinical trials as the EOP 1928z+ T cells will be infused fresh, pending 14 days USP sterility and PTC Mycoplasma test results. Therefore, final products will be released on the basis of negative Gram stains, negative rapid mycoplasma testing and levels of endotoxin meeting specifications. Additionally, we demonstrated that the in-process testing for RCR using a PCR assay that detects the GaLV env was also negative in all the runs. This is critical for the safety of the patients as well as the results of the marker-rescue cell culture assay will also be pending at the time of the patient infusion.
We consistently observed that as the T cell concentrations increased, the levels of lactate decreased while the levels of glucose increased. The monitoring of these parameters is important as we are using a continuous perfusion culture. These factors together with cell counts provided information regarding the metabolism of the cells in culture. These data suggest that the perfusion regime provides sufficient levels of nutrients and gas exchange to maintain cell expansion despite high cell concentrations (≥ 31 ×106/ml) at the late stages of the cultures.
In summary, we have designed a large scale semi-closed manufacturing process based on the expansion of T cells using Dynabeads® CD3/CD28 and the Wave bioreactor capable of producing clinical doses of genetically engineered T cells in a minimum period of time. This validated process will be used in our upcoming phase I clinical trials in patients with chemo-refractory CLL and relapsed ALL. It can easily be adapted for other clinical trials involving the expansion and transduction of patient or donor T cells using any CAR or TCR.
Acknowledgments
We would like to thank M. Bonyhadi for providing the Xcyte™ Dynabeads®; F. Weis Garcia & Lisa Denzin from the MSKCC monoclonal antibody core facility for the development of the 19E3 antibody; K. Smith from the Clinical Immunology Laboratory for fluorescence activated cell sorting analyses. This work is supported by P30 CA-008748, PO1 CA-059350, PO1 CA-023766, P50 CA086438, Damon Runyon Clinical Investigator Award (RJB), the Annual Terry Fox Run for Cancer Research (New York, NY) organized by the Canada Club of New York, the Geoffrey Beene Cancer Foundation, Golfers Against Cancer and by Mr. William H. Goodwin and Mrs. Alice Goodwin of the Commonwealth Cancer Foundation for Research & the Experimental Therapeutics Center of MSKCC.
Footnotes
Financial disclosure: All authors have declared there are no financial conflicts of interest in regards to this work.
References
- 1.Johnson AJ, Mone AP, Abhyankar V, Byrd JC. Advances in the therapy of chronic lymphocytic leukemia. Curr Opin Hematol. 2003 Jul;10(4):297–305. doi: 10.1097/00062752-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Frassoni F, Barrett AJ, Granena A, et al. Relapse after allogeneic bone marrow transplantation for acute leukaemia: a survey by the E.B.M.T. of 117 cases. Br J Haematol. 1988 Nov;70(3):317–320. doi: 10.1111/j.1365-2141.1988.tb02488.x. [DOI] [PubMed] [Google Scholar]
- 3.Barrett AJ, Locatelli F, Treleaven JG, Gratwohl A, Szydlo R, Zwaan FE. Second transplants for leukaemic relapse after bone marrow transplantation: high early mortality but favourable effect of chronic GVHD on continued remission. A report by the EBMT Leukaemia Working Party. Br J Haematol. 1991 Dec;79(4):567–574. doi: 10.1111/j.1365-2141.1991.tb08083.x. [DOI] [PubMed] [Google Scholar]
- 4.Michallet M, Tanguy ML, Socie G, et al. Second allogeneic haematopoietic stem cell transplantation in relapsed acute and chronic leukaemias for patients who underwent a first allogeneic bone marrow transplantation: a survey of the Societe Francaise de Greffe de moelle (SFGM) Br J Haematol. 2000 Feb;108(2):400–407. doi: 10.1046/j.1365-2141.2000.01851.x. [DOI] [PubMed] [Google Scholar]
- 5.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005 Apr 1;23(10):2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006 Oct 6;314(5796):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008 May 28; doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hunder NN, Wallen H, Cao J, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med. 2008 Jun 19;358(25):2698–2703. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci U S A. 2004 Oct 5;101 2:14639–14645. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002 Dec 10;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brentjens RJ, Latouche JB, Santos E, et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9(3):279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- 12.Brentjens RJ, Santos E, Nikhamin Y, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007 Sep 15;13(18 Pt 1):5426–5435. doi: 10.1158/1078-0432.CCR-07-0674. [DOI] [PubMed] [Google Scholar]
- 13.Riviere I, Brose K, Mulligan RC. Effects of retroviral vector design on expression of human adenosine deaminase in murine bone marrow transplant recipients engrafted with genetically modified cells. Proc Natl Acad Sci U S A. 1995;92(15):6733–6737. doi: 10.1073/pnas.92.15.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 15.Stephan MT, Ponomarev V, Brentjens RJ, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007 Dec;13(12):1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 16.Porter DL, Levine BL, Bunin N, et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006 Feb 15;107(4):1325–1331. doi: 10.1182/blood-2005-08-3373. [DOI] [PubMed] [Google Scholar]
- 17.Bonyhadi M, Frohlich M, Rasmussen A, et al. In vitro engagement of CD3 and CD28 corrects T cell defects in chronic lymphocytic leukemia. J Immunol. 2005 Feb 15;174(4):2366–2375. doi: 10.4049/jimmunol.174.4.2366. [DOI] [PubMed] [Google Scholar]
- 18.Hami LS, Green C, Leshinsky N, Markham E, Miller K, Craig S. GMP production and testing of Xcellerated T Cells for the treatment of patients with CLL. Cytotherapy. 2004;6(6):554–562. doi: 10.1080/14653240410005348. [DOI] [PubMed] [Google Scholar]
- 19.Rapoport AP, Levine BL, Badros A, et al. Molecular remission of CML after autotransplantation followed by adoptive transfer of costimulated autologous T cells. Bone Marrow Transplant. 2004 Jan;33(1):53–60. doi: 10.1038/sj.bmt.1704317. [DOI] [PubMed] [Google Scholar]
- 20.Laport GG, Levine BL, Stadtmauer EA, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003 Sep 15;102(6):2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 21.Rapoport AP, Stadtmauer EA, Aqui N, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005 Nov;11(11):1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 22.Levine BL, Bernstein WB, Aronson NE, et al. Adoptive transfer of costimulated CD4+ T cells induces expansion of peripheral T cells and decreased CCR5 expression in HIV infection. Nat Med. 2002 Jan;8(1):47–53. doi: 10.1038/nm0102-47. [DOI] [PubMed] [Google Scholar]
- 23.Levine BL, Cotte J, Small CC, et al. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. J Hematother. 1998 Oct;7(5):437–448. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- 24.Humeau LM, Binder GK, Lu X, et al. Efficient lentiviral vector-mediated control of HIV-1 replication in CD4 lymphocytes from diverse HIV+ infected patients grouped according to CD4 count and viral load. Mol Ther. 2004 Jun;9(6):902–913. doi: 10.1016/j.ymthe.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Kalamasz D, Long SA, Taniguchi R, Buckner JH, Berenson RJ, Bonyhadi M. Optimization of human T-cell expansion ex vivo using magnetic beads conjugated with anti-CD3 and Anti-CD28 antibodies. J Immunother. 2004 Sep-Oct;27(5):405–418. doi: 10.1097/00002371-200409000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Neurauter AA, Bonyhadi M, Lien E, et al. Cell isolation and expansion using Dynabeads. Adv Biochem Eng Biotechnol. 2007;106:41–73. doi: 10.1007/10_2007_072. [DOI] [PubMed] [Google Scholar]
- 27.Stefanski J, Brentjens RJ, Hollyman D, Bonyhadi M, Sadelain M, Rivière I. CD19-Targeted Normal and CLL Patient T Cells Expanded with Beads Can Eradicate Systemic Tumors In Vivo. Molecular Therapy. 2006;13:S102. [Google Scholar]
- 28.Przybylowski M, Hakakha A, Stefanski J, Hodges J, Sadelain M, Riviere I. Production scale-up and validation of packaging cell clearance of clinical-grade retroviral vector stocks produced in Cell Factories. Gene Ther. 2006 Jan;13(1):95–100. doi: 10.1038/sj.gt.3302648. [DOI] [PubMed] [Google Scholar]
- 29.Tran CA, Burton L, Russom D, et al. Manufacturing of large numbers of patient-specific T cells for adoptive immunotherapy: an approach to improving product safety, composition, and production capacity. J Immunother. 2007 Sep;30(6):644–654. doi: 10.1097/CJI.0b013e318052e1f4. [DOI] [PubMed] [Google Scholar]
- 30.Quintas-Cardama A, Yeh RK, Hollyman D, et al. Multifactorial optimization of gammaretroviral gene transfer into human T lymphocytes for clinical application. Hum Gene Ther. 2007 Dec;18(12):1253–1260. doi: 10.1089/hum.2007.088. [DOI] [PubMed] [Google Scholar]
- 31.FDA. US Food and Drug Administration Center for Biologics Evaluation and Research. Supplemental Guidance on Testing for RCR in Retroviral Vector Based Gene Therapy Products and During Follow-up of Patients in Clinical Trials Using Retroviral Vectors. Rockville, MD: Nov, 2006. [Google Scholar]
- 32.FDA. PTC on Characterization of Cell Lines Used To Produce Biologicals. Jul, 1993. [Google Scholar]
- 33.Gee AP, Sumstad D, Stanson J, et al. A multicenter comparison study between the Endosafe PTS rapid-release testing system and traditional methods for detecting endotoxin in cell-therapy products. Cytotherapy. 2008;10(4):427–435. doi: 10.1080/14653240802075476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi YW, Kotzin B, Herron L, Callahan J, Marrack P, Kappler J. Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc Natl Acad Sci U S A. 1989 Nov;86(22):8941–8945. doi: 10.1073/pnas.86.22.8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genevee C, Diu A, Nierat J, et al. An experimentally validated panel of subfamily-specific oligonucleotide primers (V alpha 1-w29/V beta 1-w24) for the study of human T cell receptor variable V gene segment usage by polymerase chain reaction. Eur J Immunol. 1992 May;22(5):1261–1269. doi: 10.1002/eji.1830220522. [DOI] [PubMed] [Google Scholar]
- 36.Kipps T, Castro E, Wierda WMK. A phase I/II trial of Xcellerated T cells in patients with chronic lymphocytic leukemia. Blood. 2003;102:109a. [Google Scholar]
- 37.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 38.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006 Jun;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008 Jan;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]