

Abstract
Initiation of warfarin therapy using trial-and-error dosing is problematic. our goal was to develop and validate a pharmacogenetic algorithm. in the derivation cohort of 1,015 participants, the independent predictors of therapeutic dose were: VKORC1 polymorphism −1639/3673 g>a (−28% per allele), body surface area (Bsa) (+11% per 0.25 m2), CYP2C9*3 (−33% per allele), CYP2C9*2 (−19% per allele), age (−7% per decade), target international normalized ratio (inr) (+11% per 0.5 unit increase), amiodarone use (−22%), smoker status (+10%), race (−9%), and current thrombosis (+7%). This pharmacogenetic equation explained 53−54% of the variability in the warfarin dose in the derivation and validation (N = 292) cohorts. For comparison, a clinical equation explained only 17−22% of the dose variability (P < 0.001). in the validation cohort, we prospectively used the pharmacogenetic-dosing algorithm in patients initiating warfarin therapy, two of whom had a major hemorrhage. To facilitate use of these pharmacogenetic and clinical algorithms, we developed a nonprofit website, http://www.WarfarinDosing.org.
Initiation of therapy with the anticoagulant warfarin sodium (Coumadin and others) protects against thrombosis, stroke, and myocardial infarction, but is accompanied by a high incidence of adverse events.1–3 During the first weeks of therapy, the international normalized ratio (INR) is often out of range and risk of bleeding is increased. To reduce the risk of hemorrhage, experts advocate prescribing the anticipated therapeutic dose to patients who are beginning warfarin,4–7 but do not provide a strategy for estimating that dose. The purpose of this study was to develop such a strategy.
Warfarin metabolism varies with single-nucleotide polymorphisms (SNPs) in the cytochrome P450 (CYP) 2C9 gene.1,8–10 Two common CYP2C9 SNPs are associated with impaired metabolism of S-warfarin, the more active enantiomer of warfarin. The SNP in exon 3 (CGT→TGT; Arg144Cys) is denoted as CYP2C9*2 while the SNP in exon 7 (ATT→CTT; Ile359Leu) is called CYP2C9*3. Patients with one or two of these SNPs have reduced warfarin metabolism and a two- to threefold elevated risk of an adverse event when beginning warfarin.1,10,11 Compared to CYP2C9 SNPs, SNPs in VKORC1 may be more important because the latter gene codes for the enzyme inhibited by warfarin, vitamin K epoxide reductase (VKORC1). Recently, we and others retrospectively identified novel VKORC1 SNPs that correlated with warfarin sensitivity.12–16 Other factors thought to affect warfarin dose include age, body size, smoking status, prior venous thromboembolism, race, and certain medications such as amiodarone.7,8
In this report, we develop a warfarin-dosing algorithm by combining CYP2C9 and VKORC1 genotype status with relevant clinical factors8 and prospectively validate the dosing algorithm in patients initiating warfarin therapy.
RESULTS
In the derivation cohort (N = 1,015), the daily therapeutic warfarin dose ranged from 1 to 18 mg/day. The mean age was 65 (range of 18−93); 83% were Caucasian, and 64% were male. The (geometric) mean daily warfarin dose was 4.8 mg (Table 1). The most common indications for warfarin therapy were atrial fibrillation (N = 392) and prior venous thromboembolism (N = 376; 13 of whom also had atrial fibrillation). Patients in the validation cohort (N = 292) were younger, more often female, and had more often (77%) undergone joint replacement as their indication for warfarin therapy (Table 1).
Table 1.
Demographic and clinical factors of participants
| Variable | Derivation cohort N = 1,015 | Validation cohort N = 292 |
|---|---|---|
| Demographic variables | ||
| Age, mean (SD), year | 65 (14) | 57 (14) |
| Gender | ||
| Women, N (%) | 362 (36%) | 152 (52%) |
| Men, N (%) | 653 (64%) | 140 (48%)a |
| Race | ||
| Caucasian, N (%) | 838 (83%) | 242 (83%) |
| African American, N (%) | 153 (15%) | 45 (15%) |
| Other or mixed race, N (%) | 24 (2%) | 5 (2%) |
| Hispanic ethnicity, N (%) | 24 (2%) | 2 (1%) |
| Clinical variables | ||
| Therapeutic warfarin dose, geometric mean (SD), mg per day | 4.8 (1.6) | 4.8 (1.7) |
| Therapeutic INR, mean (SD), units | 2.2 (0.5) | 2.2 (0.4) |
| Target INR, mean (SD), units | 2.4 (0.4) | 2.3 (0.2) |
| Prior DVT or pulmonary embolism | 376 (37%) | 29 (10%)a |
| Body surface area, mean (SD), m2 | 2.0 (0.26) | 2.0 (0.3) |
| Prescribed aspirin, N (%) | 224 (22%) | 78 (27%) |
| Current smokers, N (%) | 131 (13%) | 50 (17%) |
| Takes simvastatin or fluvastatin, N (%) | 192 (19%) | 33 (11%)a |
| Takes amiodarone, N (%) | 36 (4%) | 14 (5%) |
DVT, deep venous thrombosis; INR, international normalized ratio; SD, standard deviation.
Denotes P ≤ 0.01 when comparing values from the two cohorts.
VKORC1 alleles were highly heterogeneous (Table 2), reflecting their original selection as common (>5% allele frequency), informative tagging SNPs (Table 2).12 VKORC1 3673G>A was in high linkage disequilibrium with VKORC1 6853G>C (D’ = 0.97). In both cohorts, all alleles were in Hardy–Weinberg equilibrium. Genotype data from all participants at Washington University and University of Florida have been submitted to the PharmGKB (accession numbers: PS207479 and PS207480 pending).
Table 2.
Allele frequencies in derivation cohort (N = 1,015) and relationship to race and therapeutic warfarin dose
| SNP alleles | Gene location | White (N = 838) (%) | African American (N = 153) (%) | Other or mixed race (N = 24) (%) | Univariate association with dose in total population, per allele (95% CI) |
|---|---|---|---|---|---|
| CYP2C9*2 (C>T) | 3608C>T | 13.1 | 5.2 | 10.4 | −17% (−22 to −12%) |
| CYP2C9*3 (A>C) | 42614A>C | 6.0 | 1.0 | 4.2 | −30% (−36 to −24%) |
| CYP2C9*5 (C>G) | 42619C>G | 0 | 1.3 | 2.1 | NS |
| VKORC1 861 C>A | −4451C>A | 36.0 | 8.8 | 26.1 | 14% (9−18%) |
| VKORC1 3673 G>A | −1639G>A | 36.6 | 9.5 | 41.7 | −29% (−31 to −26%) |
| VKORC1 5808 T>G | IVS1+324T>G | 25.1 | 4.6 | 8.3 | 25% (29−22%) |
| VKORC1 6853 G>C | IVS2+124G>C | 37.2 | 24.3 | 41.7 | −37% (−42 to −32%) |
| VKORC1 9041 G>A | 626G>A | 39.4 | 51.3 | 41.7 | 18% (14−23%) |
| F2 Thr165Met | 494C>T | 13.3 | 1.3 | 30.0 | −5.8% (−11.4−0.0%) |
CI, confidence interval; NS, not significant; SNP, single-nucleotide polymorphism.
Frequency values are reported for the variant allele at each SNP. CYP2C9 gene locations taken from http://www.cypalleles.ki.se/cyp2c9.htm.
Pharmacogenetic model development
The VKORC1 3673G>A SNP was the first variable to enter the stepwise regression model (Table 3); each VKORC1 3673A allele was associated with a 28% reduction (95% confidence interval 25−30%) in the therapeutic warfarin dose. Once VKORC1 3673G>A entered the model, none of the other VKORC1 SNPs was an independent predictor of warfarin dose. Body surface area (BSA) was the second variable to enter the model, and each 0.25 m2 increase in BSA was associated with an 11% (95% confidence interval 9−14%) increase in the therapeutic warfarin dose. CYP2C9*3 was associated with a 33% (95% confidence interval 29−37%) decrement, and CYP2C9*2 with a −19% (−22 to −15%) decrement in the warfarin dose per allele. Other factors that entered the regression model were: target INR, amiodarone usage, smoking status, race, and prior venous thromboembolism as the indication for warfarin therapy. Overall, the pharmacogenetic model contained 10 significant variables and explained more than half of the variance in the therapeutic warfarin dose (R2 = 53.1%). Interaction terms between the genotype and key medications were not statistically significant. Once VKORC1 3673G>A entered the model, VKORC1 haplotype12 was not an independent predictor of dose.
Table 3.
Warfarin-dosing equation in derivation cohort (N = 1,015)
| Entry into model | Variable | Effect on warfarin dose | R2 after entry | P value |
|---|---|---|---|---|
| 1 | VKOR 3673G>A | −28% (−30 to −25%) | 25% | <0.0001 |
| 2 | BSA, per 0.25 m2 | 11% (9−14%) | 34% | <0.0001 |
| 3 | CYP2C9*3 | −33% (−37 to −29%) | 40% | <0.0001 |
| 4 | Age, per decade | −7% (−9 to −6%) | 45% | <0.0001 |
| 5 | CYP2C9*2 | −19% (−22 to −15%) | 50% | <0.0001 |
| 6 | Target INR, per 0.5 increase | 11% (7−14%) | 51% | <0.0001 |
| 7 | Amiodarone | −22% (−30% to −14%) | 52% | <0.0001 |
| 8 | Current smoker | 10% (3−16%) | 52.4% | 0.002 |
| 9 | African-American race | −9% (−14 to −3%) | 52.8% | 0.002 |
| 10 | Venous thromboembolism | 7% (1−13%) | 53.1% | 0.013 |
BSA, body surface area in meters2; DVT, deep venous thrombosis; INR, international normalized ratio; PE, pulmonary embolism; SNP, single-nucleotide polymorphism. The optimal pharmacogenetics algorithm that estimated the daily warfarin dose (mg/day) was:
To quantify the benefit of incorporating genotype when estimating the therapeutic warfarin dose, we excluded genetic factors and repeated the stepwise regression in the derivation cohort. This clinical algorithm without genetic factors explained 21.5% of the variance (R2) in the derivation cohort. The optimal clinical warfarin dose (mg/day) was:
where exp is the exponential function; BSA is in m2; race is 1 if African American (0 otherwise); target INR is the desired INR (e.g., 2.5); amiodarone is 1 if the patient is taking that drug (and 0 otherwise); and DVT/PE is 1 if the indication for warfarin is deep venous thrombosis (DVT) or pulmonary embolism (PE). The accuracy of the derived pharmacogenetics model depended on race: R2 was 57% in 838 Caucasian participants and 31% in 153 African-American patients.
Validation cohort
In the validation cohort (Figure 1), the therapeutic dose correlated significantly (P < 0.001) more with the pharmacogenetic predicted dose (R2 = 54%) than with the clinical predicted dose (R2 = 17%; P < 0.001). The pharmacogenetic predicted dose also had a significantly lower prediction error (Table 4). In the validation cohort, the R2 was 40% in 45 African-American patients and 55% in 242 Caucasian participants.
Figure 1.
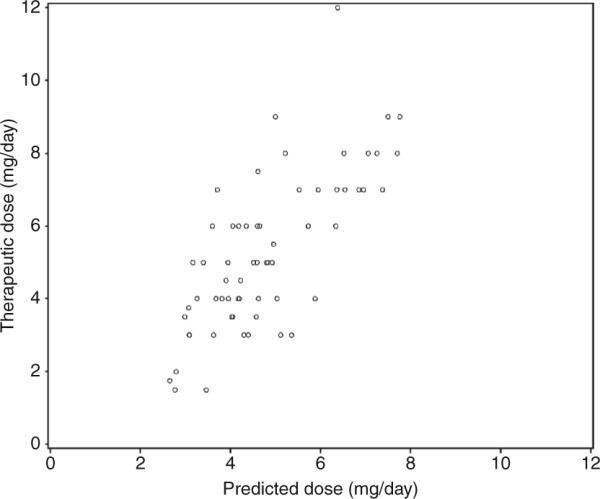
Scatter plot showing the relationship between the pharmacogenetics-predicted dose (horizontal axis) and therapeutic dose (vertical axis) in the validation cohort.
Table 4.
Dosing accuracy in validation cohort
| Accuracy metric | Pharmacogenetics | Clinical | P value |
|---|---|---|---|
| R2 (SD) | 54% (5%) | 17% (4%) | <0.0001 |
| Median (mean) absolute prediction error, mg/day | 1.0 (1.3) | 1.5 (1.8) | <0.001 |
SD, standard deviation.
For prospective patients who were dosed pharmacogenetically, we prescribed the pharmacogenetic dose using the dosing equation from the derivation cohort (Table 3) or from a preliminary version of this equation that was nearly identical (absolute mean difference = 0.1 mg/day). We did not decrease the very first warfarin dose to account for CYP2C9*2 or CYP2C9*3 alleles for logistic and pharmacologic reasons. Logistically, in 62 (21%) patients, CYP2C9 genotype was not available when the first dose was prescribed. Pharmacologically, we wished to avoid a delay until reaching a therapeutic INR that can be caused by initially prescribing the small predicted dose in slow metabolizers.17 Following this initial dose, we prescribed the pharmacogenetically estimated dose (rounded to the nearest 0.5 mg) for 2 additional days with regular INR monitoring. If the INR was rising gradually, patients in the prospective cohort continued on the pharmacogenetic dose, but otherwise clinicians adjusted that dose on day 4. During the 30-day follow-up, two patients dosed prospectively had a major hemorrhage or PE and three patients had a symptomatic DVT. None of the 43 individuals dosed prospectively in the validation cohort who stopped warfarin prior to becoming therapeutic suffered an adverse event.
DISCUSSION
For several reasons, warfarin is the ideal drug to test the hypothesis that pharmacogenetics can reduce drug toxicity: it is commonly prescribed, has a narrow therapeutic/toxic ratio, and is affected by common genetic polymorphisms. Using clinical factors alone we explained 17−21% of the variability in the therapeutic warfarin dose, similar to other clinical algorithms.18 However, by including CYP2C9 and VKORC1 genotypes, we could explain 53−54% of this variability. These encouraging results are similar to conclusions of smaller, retrospective studies of homogenous populations that found that the addition of genetic information substantially reduces the unknown intersubject variability in warfarin dose.13,19,20 These previous studies did not provide prospectively validated algorithms for individualized warfarin dosing, but made it clear that this issue had to be addressed.
In the present study, the most important factor in predicting the warfarin dose was VKORC1 3673G>A (located 1,639 bases upstream of the translation start site of VKORC1). This polymorphism alters a VKORC1 transcription factor binding site15 and haplotypes associated with the A allele result in lower VKORC1 mRNA expression in human liver.12 This change in gene expression likely lowers the steady-state concentration of tissue vitamin K epoxide reductase, the rate-limiting enzyme in the vitamin K cycle.21 The clinical effect was that individuals with this allele were more susceptible to inhibition by warfarin, resulting in a 28% decrease in the therapeutic warfarin dose per allele. Because of this decrease and the prevalence of the VKORC1 3673G>A (Table 2), this SNP was the most important predictor of dose at the time of warfarin initiation. We confirmed that this SNP is present at a low frequency in African-American patients and intermediate frequency in Caucasian patients, consistent with previous work.12,16,22 Genotyping of this single informative VKORC1 SNP should simplify clinical genetic testing and implementation of pharmacogenetics-based therapy.
Each CYP2C9*2 and CYP2C9*3 allele predicted 19 and 33% reductions (respectively) in the therapeutic dose of warfarin. These alleles correlate with slower clearance of the S-warfarin enantiomer,19,23 a longer drug half-life, and a delay until stable dosing.1,22 To ameliorate this delay, we gave a slightly greater first dose in poor warfarin metabolizers; we did not reduce their initial dose despite the presence of CYP2C9*2 and/or CYP2C9*3.
In contrast to previous single-center studies of Caucasian patients,13,19,20 we genotyped a diverse, multicenter cohort. To further enhance generalizability, we included patients from multiple anticoagulation centers regardless of their comorbid conditions, target INR, and concomitant therapy. To improve clinical usefulness, we focused on the effects of SNPs that were suspected to be clinically relevant. One interesting result was the inclusion of the VKORC1 3673 SNP as the only significant VKORC1 SNP in predicting warfarin dose. An important strength of the current study is that we prospectively validated pharmacogenetics-based warfarin therapy, thereby demonstrating its feasibility and safety.
All patients in the validation cohort received pharmacogenetic therapy without comparison to patient management vianongenetic algorithms. In the future, other genetic variants may strengthen the predictive power of pharmacogenetic warfarin therapy. For example, one study showed significant associations between SNPs in factor VII and in γ−glutamyl carboxylase and warfarin dose,24 but this association has not been replicated in a subset of our cohort (B.F. Gage and C. Eby unpublished data) or in other cohorts.20 The factor II Thr165Met SNP, previously associated with lower warfarin dose in studies of 147 Italian24 and 45 Japanese patients,25 was of borderline significance in our univariate analysis and not an independent predictor of warfarin dose. In keeping with our clinical focus, we did not consider additional SNPs26,27 that are too rare to significantly improve dosing accuracy in the general population.
This study demonstrates the feasibility of pharmacogenetic warfarin therapy and supports the need for a multicenter, randomized control trial quantifying its effect on laboratory and clinical outcomes. Several companies have developed genotyping platforms to facilitate pharmacogenetic dosing, and at least two have received Food and Drug Administration approval. The Food and Drug Administration has also changed the Coumadin/warfarin labeling to encourage lower initial doses in patients who have the VKORC1 3673G>A, CYP2C9*2, or CYP2C9*3 allele.
Two recent trials have randomized patients to pharmacogenetic vs. clinically-based warfarin initiation but had conflicting findings. Caraco and colleagues at Hadassah University found that CYP2C9 genotyping improved INR control and reduced minor (but not major) bleeding compared to dosing by a computerized clinical algorithm.28 In the other randomized trial, Anderson and colleagues in Utah found no significant benefit of pharmacogenetic vs. clinically dosed warfarin.29 The studies were similarly sized—each having ∼100 patients per treatment arm. The Anderson-dosing algorithm had an R2 of 47%; Caraco did not report the accuracy of their algorithm, but a similar CYP2C9-dosing algorithm had an R2 of 39%.8 Given these findings, a multicenter randomized and controlled trial is needed to quantify how pharmacogenetic therapy affects INR control and clinical adverse events.
To facilitate a trial and the use of the dosing algorithms in this report, we have developed a nonprofit website, http://www.WarfarinDosing.org. While the genotyping is pending, WarfarinDosing.org estimates the warfarin dose using the clinical algorithm in this report. Once genotype is available, WarfarinDosing.org uses the more accurate pharmacogenetic algorithm (Table 3). If an INR is available after three doses, WarfarinDosing.org uses a pharmacogenetic algorithm and the INR response to estimate the therapeutic dose (R2 = 79%).30 A randomized controlled trial should test the hypothesis that pharmacogenetic dosing improves laboratory and clinical outcomes in patients beginning warfarin for a variety of indications. The National Institutes of Health has announced their intention to fund such a trial via RFP-NHLBI-HV-08−03.
METHODS
From individuals in the derivation cohort (1,015 participants who had therapeutic warfarin doses), we collected DNA and clinical factors to develop a warfarin-dosing algorithm. In the validation cohort, we validated this algorithm prospectively, by prescribing pharmacogenetics-based warfarin therapy in 292 patients. The research protocol was approved by the Human Subjects’ Committee at the participating medical centers. All subjects gave written, informed consent.
Derivation cohort
We genotyped adult patients who had their warfarin therapy managed at one of the anticoagulation clinics affiliated with Barnes–Jewish Hospital at Washington University Medical Center (St. Louis), the University of Florida (Gainesville), or the Anticoagulation Clinics of North America (San Antonio), including some patients in prior studies.11,12,16 The derivation cohort also included 203 additional participants from the PREVENT (PREvention of VENous Thromboembolism) study who were randomized to long-term, low-intensity warfarin therapy (target INR 1.5−2.0) for prevention of recurrent venous thromboembolism.31 To control for the lower target INR in these patients (and higher target INR in participants with mechanical heart valves), we included target INR in the regression equation. To prevent referral bias, we excluded four participants who had been referred for extraordinary warfarin doses (<1 or >20 mg/day). As in our prior work,30,32 therapeutic dose was defined as the warfarin dose (unchanged for at least 6 days), which resulted in an INR that was between 70 and 130% of the target INR (i.e., in the range of 1.8−3.2 for someone with a target INR of 2.5).
Validation cohort
To validate the dosing algorithm, we recruited 335 patients who were scheduled to begin warfarin therapy for a variety of medical and surgical indications at Barnes–Jewish Hospital, 295 of whom we dosed prospectively. We excluded patients who had contraindications to 1 month of warfarin therapy and patients whose therapeutic warfarin dose was known from prior therapy. For our calculation of dosing accuracy, we excluded patients who stopped their warfarin therapy before achieving a therapeutic dose (N = 292). To improve generalizability, we included patients regardless of comorbid conditions and concomitant medications. The Barnes–Jewish Hospital Anticoagulation Service prospectively followed all patients in the validation cohort for symptomatic adverse events; we did not screen for asymptomatic thromboses.
Genotyping
We obtained 15-ml mouthwash samples from University of Florida participants (N = 327) and 10-ml whole blood in EDTA or sodium cit-rate tubes from other participants (N = 980). After centrifugation, we isolated genomic DNA from buccal cells or buffy coat. We genotyped for CYP2C9*2[rs1799853] using PCR/restriction fragment length polymorphism and Pyrosequencing.8,12,33–35 We also Pyrosequenced all other SNPs in the validation cohort (listed with dbSNP reference SNP identifiers): CYP2C9*3[rs1057910], CYP2C9*5 [rs1057910], VKORC1 C861A [rs17880887], VKORC1 G3673A [rs9923231; more recently designated as –1639 G>A], VKORC1 A5808C [rs2884737], VKORC1 G6853C [rs8050894], VKORC1 G9041A [rs7294], and one factor II variant (C/T Thr/Met; rs5896)24 that was associated with warfarin sensitivity. We performed and interpreted genotyping while blinded to clinical and demographic variables.
In the validation cohort, we genotyped for those SNPs that were significant predictors of the warfarin dose in the derivation cohort using three different methods: (i) melting curve analysis on a SmartCycler (Cepheid, Sunnyvale, CA),30 (ii) Invader assay (Third Wave Technologies, Madison, WI) with the TECAN GENios FL fluorescence plate reader (Zurich, Switzerland), or (iii) INFINITI analyzer (Autogenomics, Carlsbad, CA). In blinded comparisons, genotyping was 99−100% accurate for all methods.36
Clinical variables
Using a paper-based survey, we collected demographic variables, laboratory data, smoking status, and key interacting medications8 (including aspirin, amiodarone, simvastatin, or fluvastatin) (Table 1). We used height and weight to calculate BSA,37 which correlated more strongly with warfarin dose than either variable alone. We used continuous variables for warfarin dose, age, and target INR (which was 1.75 for PREVENT participants and 2.5 for most others).
Statistical analyses
In the derivation cohort, we used stepwise regression to derive the pharmacogenetic model that predicted the therapeutic warfarin dose. To use an additive genetic model, we coded the number of variant alleles at each locus as 0, 1, or 2. We tested for the effect of several clinical and demographic variables: African-American race, Hispanic ethnicity, age, gender, BSA, PVT, target INR, and use of aspirin, amiodarone, and simvastatin, or fluvastatin. The genetic factors were CYP2C9*2, CYP2C9*3, CYP2C9*5, VKORC1 861C>A, 3673G>A, 5808T>G, 6853G>C, 9041G>A, and F2 Thr165Met. We retained variables in the regression model if they were significant (two-tailed P < 0.05) independent predictors of warfarin dose in the derivation cohort. To assess model fit, we examined residuals, including partial residuals. To improve model fit and limit heteroscedascity we used a logarithmic transformation of warfarin dose.
We tested the regression model in an independent validation cohort (N = 335), where we calculated the prediction error from the formula: |pharmacogenetics predicted dose—therapeutic dose|. We also captured incident hemorrhages or thromboses in the 30 days after warfarin initiation in patients (N = 295) who were prospectively dosed. We used bootstrapping38 to quantify the standard deviations of the prediction error and the R2. We performed statistical analyses in SAS (Version 9; SAS Institute, Cary, NC).
ACkNOWLEDGMENTS
This study was funded by National Institutes of Health grants R01 HL074724 and R01 HL71083. We thank Rhonda Porche-Sorbet, Christi King, and Lynda Stauffer for assistance in genotyping, Susan Gatchel for entering data and recruiting patients, and Ann Barnett for help with statistical analyses. The study sponsor had no role in gathering or interpreting data or right to approve/disapprove of the manuscript. BG and ED had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design were made by BG, CE, and HM. Acquisition of data was done by BG, CE, JJ, MR, CA, PM, LG, LF, and DV. Analysis and interpretation were done by BG, CE, JJ, PR, and HM. Drafting of the manuscript was done by BG. Critical revision of the manuscript for important intellectual content was done by BG, CE, JJ, MR, PR, AR, and HM. Statistical analysis was done by BG and ED. Administrative, technical, or material support was given by CA, LG, SM, and TL. Study supervision was done by BG.
Footnotes
CONFLICT OF INTEREST
Dr Gage has consulted for Bristol-Myers Squibb on work unrelated to this article. Drs Rieder and Rettie report having applied for a patent (application serial no. 10/967,879) on the use of VKORC1 haplotypes and SNPs. The other authors declared no conflict of interest.
References
- 1.Higashi MK, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287:1690–1698. doi: 10.1001/jama.287.13.1690. [DOI] [PubMed] [Google Scholar]
- 2.White RH, Beyth RJ, Zhou H, Romano PS. Major bleeding after hospitalization for deep-venous thrombosis. Am. J. Med. 1999;107:414–424. doi: 10.1016/s0002-9343(99)00267-3. [DOI] [PubMed] [Google Scholar]
- 3.Beyth RJ, Quinn L, Landefeld CS. A multicomponent intervention to prevent major bleeding complications in older patients receiving warfarin. A randomized, controlled trial. Ann. Intern. Med. 2000;133:687–695. doi: 10.7326/0003-4819-133-9-200011070-00010. [DOI] [PubMed] [Google Scholar]
- 4.Hirsh J, Fuster V, Ansell J, Halperin JL. American heart association/American college of cardiology foundation guide to warfarin therapy. Circulation. 2003;107:1692–1711. doi: 10.1161/01.CIR.0000063575.17904.4E. [DOI] [PubMed] [Google Scholar]
- 5.Ezekowitz MD, James KE, Radford MJ, Rickles FR, Redmond N. Initiating and maintaining patients on warfarin anticoagulation: the importance of monitoring. J. Cardiovasc. Pharmacol. Ther. 1999;4:3–8. doi: 10.1177/107424849900400102. [DOI] [PubMed] [Google Scholar]
- 6.Ansell J, et al. Managing oral anticoagulant therapy. Chest. 2001;119(suppl 1):22S–38S. doi: 10.1378/chest.119.1_suppl.22s. [DOI] [PubMed] [Google Scholar]
- 7.Ageno W, Squizzato A, Dentali F, Crowther M. Tailoring warfarin induction doses to reflect individual and disease-specific factors. Am. J. Med. 2005;118:143–144. doi: 10.1016/j.amjmed.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Gage BF, Eby C, Milligan PE, Banet GA, Duncan JR, McLeod HL. Use of pharmacogenetics and clinical factors to predict the maintenance dose of warfarin. Thromb. Haemost. 2004;91:87–94. doi: 10.1160/TH03-06-0379. [DOI] [PubMed] [Google Scholar]
- 9.Aithal GP, Day CP, Kesteven PJL, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353:717–719. doi: 10.1016/S0140-6736(98)04474-2. [DOI] [PubMed] [Google Scholar]
- 10.Margaglione M, et al. Genetic modulation of oral anticoagulation with warfarin. Thromb. Haemost. 2000;84:775–778. [PubMed] [Google Scholar]
- 11.Voora D, et al. Prospective dosing of warfarin based on cytochrome P-450 2C9 genotype. Thromb. Haemost. 2005;93:700–705. doi: 10.1160/TH04-08-0542. [DOI] [PubMed] [Google Scholar]
- 12.Rieder MJ, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005;352:2285–2293. doi: 10.1056/NEJMoa044503. [DOI] [PubMed] [Google Scholar]
- 13.Wadelius M, et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenomics J. 2005;5:262–270. doi: 10.1038/sj.tpj.6500313. [DOI] [PubMed] [Google Scholar]
- 14.D'Andrea G, et al. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005;105:645–649. doi: 10.1182/blood-2004-06-2111. [DOI] [PubMed] [Google Scholar]
- 15.Yuan HY, et al. A novel functional VKORC1 promoter polymorphism is associated with inter-individual and inter-ethnic differences in warfarin sensitivity. Hum. Mol. Genet. 2005;14:1745–1751. doi: 10.1093/hmg/ddi180. [DOI] [PubMed] [Google Scholar]
- 16.Aquilante CL, et al. Influence of coagulation factor, vitamin K epoxide reductase complex subunit 1, and cytochrome P450 2C9 gene polymorphisms on warfarin dose requirements. Clin. Pharmacol. Ther. 2006;79:291–302. doi: 10.1016/j.clpt.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Linder MW, et al. Warfarin dose adjustments based on CYP2C9 genetic polymorphisms. J. Thromb. Thrombolysis. 2002;14:227–232. doi: 10.1023/a:1025052827305. [DOI] [PubMed] [Google Scholar]
- 18.Shine D, et al. A randomized trial of initial warfarin dosing based on simple clinical criteria. Thromb. Haemost. 2003;89:297–304. [PubMed] [Google Scholar]
- 19.Sconce EA, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood. 2005;106:2329–2333. doi: 10.1182/blood-2005-03-1108. [DOI] [PubMed] [Google Scholar]
- 20.Herman D, Peternel P, Stegnar M, Breskvar K, Dolzan V. The influence of sequence variations in factor VII, gamma-glutamyl carboxylase and vitamin K epoxide reductase complex genes on warfarin dose requirement. Thromb. Haemost. 2006;95:782–787. [PubMed] [Google Scholar]
- 21.Wajih N, Hutson SM, Owen J, Wallin R. Increased production of functional recombinant human clotting factor IX by BHK cells engineered to overexpress VKORC1, the vitamin K 2,3–epoxide reducing enzyme of the vitamin K cycle. J. Biol. Chem. 2005;280:31603–31607. doi: 10.1074/jbc.M505373200. [DOI] [PubMed] [Google Scholar]
- 22.Wu A. Use of genetic and non-genetic factors in warfarin dosing algorithms. Pharmacogenomics. 2007;8:865–872. doi: 10.2217/14622416.8.7.851. [DOI] [PubMed] [Google Scholar]
- 23.King BP, Khan TI, Aithal GP, Kamali F, Daly AK. Upstream and coding region CYP2C9 polymorphisms: correlation with warfarin dose and metabolism. Pharmacogenetics. 2004;14:813–822. doi: 10.1097/00008571-200412000-00004. [DOI] [PubMed] [Google Scholar]
- 24.D'Ambrosio RL, et al. Polymorphisms in factor II and factor VII genes modulate oral anticoagulation with warfarin. Haematologica. 2004;89:1510–1516. [PubMed] [Google Scholar]
- 25.Shikata E, et al. Association of pharmacokinetic (CYP2C9) and pharmacodynamic (vitamin K-dependent protein-Factors II, VII, IX, and X, proteins S and C, and {gamma}-glutamyl carboxylase) gene variants with warfarin sensitivity. Blood. 2004;103:2630–2635. doi: 10.1182/blood-2003-09-3043. [DOI] [PubMed] [Google Scholar]
- 26.Kidd RS, Curry TB, Gallagher S, Edeki T, Blaisdell J, Goldstein JA. Identification of a null allele of CYP2C9 in an African-American exhibiting toxicity to phenytoin. Pharmacogenetics. 2001;11:803–808. doi: 10.1097/00008571-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 27.DeLozier TC, Lee SC, Coulter SJ, Goh BC, Goldstein JA. Functional characterization of novel allelic variants of CYP2C9 recently discovered in southeast Asians. J. Pharmacol. Exp. Ther. 2005;315:1085–1090. doi: 10.1124/jpet.105.091181. [DOI] [PubMed] [Google Scholar]
- 28.Caraco Y, Blotnick S, Muszkat M. CYP2C9 Genotype-guided warfarin prescribing enhances the efficacy and safety of anticoagulation: a prospective randomized controlled study. Clin. Pharmacol. Ther. 2008;83:457–467. doi: 10.1038/sj.clpt.6100316. [DOI] [PubMed] [Google Scholar]
- 29.Anderson JL, et al. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation. 2007;116:2563–2570. doi: 10.1161/CIRCULATIONAHA.107.737312. [DOI] [PubMed] [Google Scholar]
- 30.Millican E, et al. Genetic-based dosing in orthopaedic patients beginning warfarin therapy. Blood. 2007;110:1511–1515. doi: 10.1182/blood-2007-01-069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridker PM, et al. Long-term, low-intensity warfarin therapy for the prevention of recurrent venous thromboembolism. N. Engl. J. Med. 2003;348:1425–1434. doi: 10.1056/NEJMoa035029. [DOI] [PubMed] [Google Scholar]
- 32.Lenzini PA, et al. Optimal initial dose adjustment of warfarin in orthopedic patients. Ann. Pharmacother. 2007;41:1798–1804. doi: 10.1345/aph.1K197. [DOI] [PubMed] [Google Scholar]
- 33.Marsh S, King CR, Porche-Sorbet RM, Scott-Horton TJ, Eby CS. Population variation in VKORC1 haplotype structure. J. Thromb. Haemost. 2006;4:473–474. doi: 10.1111/j.1538-7836.2006.01759.x. [DOI] [PubMed] [Google Scholar]
- 34.Aquilante CL, Lobmeyer MT, Langaee TY, Johnson JA. Comparison of cytochrome P450 2C9 genotyping methods and implications for the clinical laboratory. Pharmacotherapy. 2004;24:720–726. doi: 10.1592/phco.24.8.720.36074. [DOI] [PubMed] [Google Scholar]
- 35.Langaee T, Ronaghi M. Genetic variation analyses by pyrosequencing. Mutat. Res. 2005;573:96–102. doi: 10.1016/j.mrfmmm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 36.King CR, et al. Performance of commercial platforms for rapid genotyping of polymorphisms affecting warfarin dose. Am J. Clin. Pathol. doi: 10.1309/1E34UAPR06PJ6HML. in press. [DOI] [PubMed] [Google Scholar]
- 37.DuBois D, DuBois E. Clinical calorimetry; a formula to estimate the approximate surface area if height and weight be known. Arch. Int. Med. 1916;17:863–871. [Google Scholar]
- 38.Efron B, Tibshirani R. An Introduction to the Bootstrap. Chapman & Hall; New York: 1993. [Google Scholar]