

Abstract
The neuronal Ca2+-sensor protein VILIP-1, known to affect clathrin-dependent receptor trafficking, has been shown to interact with the cytoplasmic loop of the α4-subunit of the α4β2 nicotinic acetylcholine receptor (nAChR), which is the most abundant nAChR subtype with high-affinity for nicotine in the brain. The α4β2 nAChR is crucial for nicotine addiction and the beneficial effects of nicotine on cognition. Its dysfunction has been implicated in frontal lobe epilepsy, Alzheimer’s disease and schizophrenia. Here we report that overexpression of VILIP-1 enhances ACh responsiveness, whereas siRNA against VILIP-1 reduces α4β2 nAChR currents of hippocampal neurons. The underlying molecular mechanism likely involves enhanced constitutive exocytosis of α4β2 nAChRs mediated by VILIP-1. The two interaction partners co-localize in a Ca2+-dependent manner with syntaxin-6, a Golgi-SNARE protein involved in trans-Golgi membrane trafficking. Thus, we speculate that regulation of VILIP-1-expression might modulate surface expression of ligand-gated ion channels, such as the α4β2 nAChRs, possibly comprising a novel form of physiological up-regulation of ligand-gated ion channels.
Introduction
Neuronal Ca2+-sensor (NCS) proteins are involved in a variety of Ca2+-dependent signal transduction processes, including exocytosis, modulation of receptor-, ligand-gated ion channel-and signal effector function (Ikura and Ames, 2006; Burgoyne, 2007). VILIP-1 (visinin-like protein-1, VSNL1), belonging to the subfamily of VILIPs, has been shown to affect several signaling pathways, such as cAMP- as well as cGMP-signaling. It has also been implicated in clathrin-dependent receptor trafficking of the transferin receptor and the receptor guanylyl cyclase GC-B (Braunewell and Gundelfinger, 1999; Brackmann et al., 2005). More recently, VILIP-1 was identified in a functional protein complex with glutamate receptors of the kainate subtype GluR6 (Coussen et al., 2005), and form a signaling complex with P2X2 receptors in which it regulates surface expression of the receptor and its sensitivity and peak response to ATP (Chaumont et al., 2008). Moreover, VILIP-1 is a modulator of another ligand-gated ion channel, the α4β2 nicotinic acetylcholine receptors (nAChRs) that increases their surface expression levels and agonist sensitivity in oocytes (Lin et al., 2002). The nAChRs are activated by the endogenous neurotransmitter acetylcholine (ACh) and are cation-permeable channels. Pentameric nAChRs arise from eight subunits (α2-4, 6-10) which form the ACh binding site, and four subunits (α5, β2-4) which have a structural function but also contribute to the binding site. The α4β2 is the most abundant nAChR subtype in the brain, which in rodents forms the high-affinity binding site for the agonist nicotine (Le Novere et al., 2002; Hogg et al., 2003). Like most brain nAChRs, α4β2 receptors appear to be localized primarily presynaptically where they facilitate the release of various neurotransmitters (Dani et al., 2001). The α4β2 nAChR has been implicated in nociception, nicotine dependence and learning and memory (Mansvelder et al., 2005; Tapper et al., 2004; Maskos et al., 2005). An implication in attention deficit hyperactivity syndrome (ADHD, Levin et al., 2006) and in schizophreniform diseases (Bresse et al., 2000; De Luca et al., 2006) have been documented, and losses in numbers of α4β2 nAChRs in the cortex and the hippocampus in Alzheimer’s disease have been correlated with cognitive deficits (Gotti and Clementi, 2004).
Studies with nAChR knockouts for the α4 and the β2 subunits and theα4 knock-in mouse, which is hypersensitive to nicotine, identify α4β2 nAChR subunits as responsible for the essential features of nicotine-addiction (Tapper et al., 2004; Maskos et al., 2005; Piciotti et al., 1998). Chronic exposure of α4β2 receptors to nicotine results initially in receptor desensitization (Giniatullin et al., 2005), followed by subsequent functional up-regulation of high affinity nicotine binding sites (Schwartz and Kellar, 1983), thus most likely enhancing release of dopamine in the mesolimbic rewarding system (Dani et al., 2001). The molecular mechanisms of the nicotine-induced functional up-regulation relevant for addiction are under investigation (Buisson and Bertrand, 2002), and may depend on high affinity receptor stabilization, intracellular maturation, enhanced trafficking or reduced endocytosis (Peng et al., 1994, Harkness et al., 2002; Vallejo et al., 2005; Sallette et al., 2005; Pakkanen et al., 2005 Darsow et al., 2005). However, nicotine-induced up-regulation has not only been found in brain reward areas, but also in several learning and memory related brain areas such as the hippocampus (Hernandez and Terry, 2005). Interestingly, agonists for the α4β2 receptor produce long-lasting cognitive effects in animal models, which can outlast the actual presence of the agonists for several weeks. The basis for this discrepancy may reside in mechanisms similar to the long-lasting changes following nicotine-dependent induction of long-term potentiation and/or long-term depression (LTP, LTD) (Buccafusco et al. 2005). Since the neuronal Ca2+ sensor VILIP-1 is up-regulated during hippocampal LTP induction (Braunewell et al., 2002, Brackmann et al., 2004), is a functional modulator of α4β2 nAChRs (Lin et al., 2002), and is co-expressed with α4β2 nAChRs in subpopulations of hippocampal neurons (Zhao and Braunewell, 2008; Gierke et al., 2008), the up-regulation of α4β2 nAChRs by endogenous modulators such as VILIP-1 might be a physiological mechanism relevant for hippocampal plasticity. Here we describe the existence of VILIP-1-dependent up-regulation of functional α4β2 nAChRs in hippocampal neurons, and the initial analysis of the underlying molecular mechanisms.
Results
Expression of VILIP-1 enhances cell surface expression of endogenous and co- transfected α4β2 nAChR in hippocampal neurons
To investigate whether the neuronal Ca2+ sensor VILIP-1 act as modulator of α4β2 nAChR in hippocampal neurons leading to up-regulation of functional receptors, we transfected hippocampal cell cultures with VILIP-1-cDNA. In ELISA using monoclonal antibody specific for the α4 subunit of the nAChR (mAb299) the relative amount of receptor at the cell surface was determined. The enhanced protein expression of VILIP-1 led to an enhanced surface expression of endogenous α4β2 receptor in primary neurons (Fig. 1A, left panel). Similarly, when VILIP-1 was co-expressed with α4β2-flag-cDNA (α4β2 cDNA ratio 1:10 to yield high affinity receptor) a 1.8-fold enhancement of surface expression was measured (Fig. 1A, right panel), which is of comparable magnitude as the VILIP-1 effect previously observed in the oocyte expression system and tsA 201 cells (Lin et al., 2002). In controls, when hippocampal neurons co-transfected with flag-tagged α4β2 nAChR and GFP (Fig. 1B) were examined by immunofluorescence microscopy, no or very weak surface expression of the receptor was noticed (Fig. 1C). However, when the receptor was co-transfected with VILIP-1-GFP cDNA (Fig. 1D) instead of GFP a significant surface expression was noticeable (Fig. 1E). When the exact number of neurons expressing α4β2 nAChR at the surface was quantified in the co-transfected cultures a nearly threefold increase in surface-expressing neurons from 25% to 74% was observed (Fig. 1F). Similar numbers of neurons were used in control and VILIP-1-transfected cultures, no changes in cell number after transfection occurred, and in neurons transfected with α4β2 nAChR cDNA an effect of VILIP-1 on gene expression can be ruled out. Thus, these results suggest that increased protein expression of VILIP-1 leads to strong up-regulation of cell surface expression of endogenous and transfected α4β2 nAChR in primary hippocampal neurons.
Fig. 1. Expression of VILIP-1 enhances surface expression of endogenous and co-transfected α4β2 nACh receptor in hippocampal neurons.

A. VILIP-1 cDNA (pOPR-V1) or control vector (pOPR) was transfected alone (left) or co-transfected with flag-tagged α4β2 nAChR (right) into two week old primary hippocampal cultures. 16h following transfection, where no changes in cell numbers were observed, the surface expression of the endogenous α4β2 nAChR (left) or of the flag-tagged α4β2 nAChR (right) was quantified using mab299 against the α4-subunit (left) or an anti-flag antibody (right) in ELISA. B–E. Control GFP vector (B, C) or VILIP-1-GFP cDNA (D, E) were co-transfected with flag-tagged α4β2 nAChR (B–E) into 2 week old primary hippocampal cultures. After 40h in culture surface expression of the flag-tagged α4β2 nAChR (C, E) was monitored with anti-flag antibody by confocal immunofluorescence microscopy. Bar in E is 20 μm. F. Quantification of the number of cells expressing flag-tagged α4β2 nAChR at the cell surface in GFP (C) and VILIP-1-GFP (E) co-transfected hippocampal neurons. Mean values ± S.D. are from five experiments for A and three experiment for F carried out in triplicate.
Expression of VILIP-1 enhances cell surface expression of transiently and stably transfected α4β2 nAChR in HEK cells
To set up an eukaryotic expression system which is useful for biochemical studies and for testing whether VILIP-1 can affect functional up-regulation of the α4β2 nAChR, we examined HEK cells transiently or stably transfected with α4β2 nAChR (Fig. 2). VILIP-1-GFP was co-transfected with flag-tagged α4β2 receptor and compared to co-transfection with GFP or without co-transfection (Fig. 2A–F). When α4β2 nAChR subunits were transfected alone only little surface expression of the receptor was observed by immunofluorescence staining (Fig. 2A). However, under permeabilizing conditions in the presence of the detergent TX-100 strong intracellular staining was detected, indicating the presence of receptor protein inside the cells (Fig. 2B). In GFP control transfected HEK cells (Fig. 2C) surface expression of α4β2 nAChR was weak (Fig. 2D), whereas co-expression with VILIP-1-GFP (Fig. 2E) led to a significant increase in surface expression as shown by immunofluorescence surface staining (Fig. 2F). It has been previously described that α4β2 nAChR although expressed at significant levels within transfected HEK cells, often need additional treatment in form of a temperature shift from 37° to 30 °C to reach the cell surface (Lin et al., 2002). Interestingly, VILIP-1 expression alone seems to be sufficient to lead to surface expression without a temperature shift, although a big fraction of the receptor most likely remains in the ER and Golgi (Fig. 2B). Counting the number of cells with surface expression (Fig. 2K) a significant increase of cells with surface expression was observed in VILIP-1-GFP co-transfected cells, 64% of the cells, in comparison to 25% in GFP alone co-transfected HEK cells. Finally we examined whether the effect also occurs in HEK cells stably transfected with human cDNA of α4β2 nAChR (Samochocki et al., 2003). In this case, compared to the effect of GFP control expression (Fig. 2G) on receptor expression (Fig. 2H), we observed a pronounced effect of VILIP-1-GFP expression (Fig. 2I) on the magnitude of surface expression of the receptor (Fig. 2J). Most cultured cells showed surface expression due to the nature of stable expression (Samochocki et al., 2003), therefore the magnitude of surface expression of nAChR in the stably transfected cells was quantified from the immunostainings with mAb299 against the α4 subunit using the ImageJ program. A significant increase in membrane-associated surface expression of the receptor was observed in VILIP-1-GFP-transfected compared to GFP transfected cells (Fig. 2L). Again in these experiments with transfected α4β2 nAChR cDNA an effect of VILIP-1 on gene expression can be ruled out. Thus, our data suggest that VILIP-1 not only increases the number of cells showing surface expression in HEK cells transiently transfected with rat α4β2 nAChR, but also increases the amount of receptor at the surface in HEK cells stably transfected with rat and human α4β2 nAChRs.
Fig. 2. Expression of VILIP-1 enhances surface expression of transiently transfected α4β2 nAChR in HEK cells and of human α4β2 nAChR in stably transfected HEK cells. A–F.
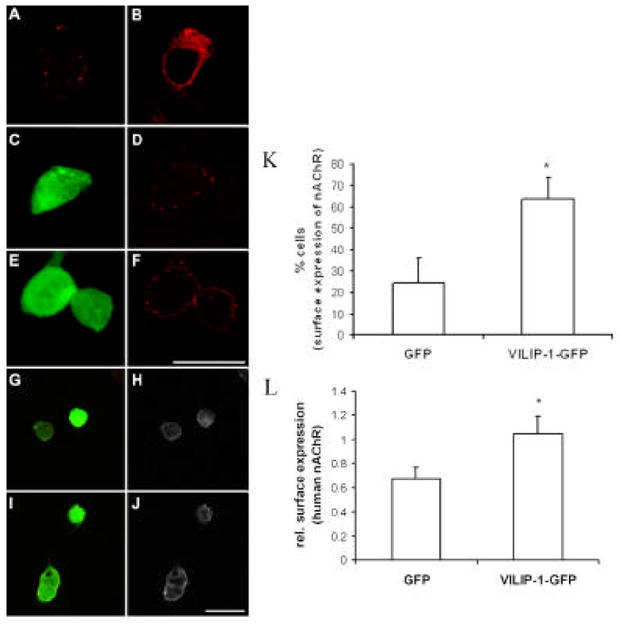
Flag-tagged α4β2 nAChR was transfected alone (A, B) or co-transfected with GFP (C, D), or with VILIP-1-GFP cDNA (E, F) into HEK cells. After 48h in culture only little surface expression of the flag-tagged α4β2 nAChR was monitored (A), but the receptor was strongly expressed intracellularly when stained in the presence of TX100 (B). Surface expression in GFP (C, D) and VILIP-1-GFP (E, F) co-transfected HEK cells was monitored with anti-flag antibody by confocal immunofluorescence microscopy (D, F). Bar in F is 20 μm. G–J. GFP (G, H) or VILIP-1-GFP cDNA (I, J) was transfected into HEK cells stably expressing the human α4β2 nAChR. After 48h in culture surface expression of human α4β2 nAChR (H, J) was monitored by confocal immunofluorescence microscopy in GFP (G, H) and VILIP-1-GFP (I, J) transfected cells with mAb299 antibody against the α-subunit of nAChR (H, J). Cross-bleeding of channels was avoided by sequential analysis of the red and green channel. Bar in J is 20 μm. K. Quantification of the number of cells expressing flag-tagged α4β2 nAChR in GFP (D) and VILIP-1-GFP (F) co-transfected HEK cells. No differences in cell numbers between GFP- and GFP-VILIP-1-transfected cells was observed. L. Quantification of the surface expressing of human α4β2 nAChR in GFP (H) and VILIP-1-GFP (J) transfected HEK cells in mAb299 stained cultures using the NIH ImageJ program. Mean values ± S.D. are from three experiments carried out in triplicate.
Molecular mechanisms of the VILIP-1 induced upregulation of functional α4β2 nicotinic acetylcholine receptor in hippocampal neurons
VILIP-1 performs a Ca2+-myristoyl switch which enhances membrane association of VILIP-1 in an ionomycin/Ca2+-dependent manner through the exposition of a hydrophobic myristoyl residue (Spilker et al., 2002). When we stimulated hippocampal neurons with carbachol or ionomycin (data not shown), which both can increase intracellular Ca2+ concentrations, we detected, in comparison to controls (Fig. 3A, G), translocation of VILIP-1 to the cell surface (Fig. 3D, J). When in the same experiment the Ca2+-dependent co-localization with the ER was examined (Fig. 3A–F), VILIP-1 (in red, Fig. 3A, D) and the ER marker calnexin (in green, Fig. 3B, E) did not co-localize (Fig. 3C, F, VILIP-1: red, calnexin: green), irrespective of Ca2+ stimulation. However, when co-staining with the Golgi marker syntaxin-6 was performed (Fig. 3G–L), VILIP-1 (in red; Fig. 3G, J) was found to show co-localization with syntaxin-6 (in green; Fig. 3H, K) even under unstimulated conditions (Fig. 3I, co-localization in yellow). Following carbachol treatment (Fig. 3L) a large increase in co-localization was observed, which is perfectly in agreement with the previously observed Ca2+-dependent co-localization of VILIP-1 with the trans-Golgi marker syntaxin6 (Spilker et al., 2002). Our results on the apparent lack of ER co-localization render the ER compartment as a less likely place for modulation of nAChR trafficking by VILIP-1. These findings, however, indicate that VILIP-1, which localizes to the Golgi as well as to the cell surface membrane, may regulate α4β2 nAChRs trans-Golgi trafficking.
Fig. 3. Ca2+-dependent localization of VILIP-1 to cell surface and the intracellular membranes of Golgi and ER in hippocampal neurons.

A–L. Hippocampal neurons were unstimulated (A–C, G–I) or stimulated with carbachol (D–F, J–L), which increases intracellular Ca2+ concentrations to enhance the membrane association of VILIP-1 (compare A to D and G to J), as described in detail previously for ionomycin treatment (28) A–F. Co-localization with the ER marker calnexin (B, E) could not be found in the merged images (C, F, VILIP-1: red, calnexin: green). G–L. Co-staining with the trans-Golgi marker syntaxin-6 (H, K) was observed in yellow in the merged images (I, L), and was enhanced after carbachol treatment to increase intracellular calcium (L), as previously described in detail for ionomycin treatment (Spilker et al., 2002). Representative stainings out of at least three experiments carried out in triplicate are shown. Bar in M is 20 μm.
Co-localization of α4β2 nAChR with ER and Golgi marker and Ca2+-dependent co-localization of VILIP-1-GFP, α4β2 nAChR and Golgi marker syntaxin-6 in hippocampal neurons
To simultaneously track the localization of α4β2 nAChR within the intracellular membranes of the ER and Golgi, we performed triple staining experiments (Fig. 4). When hippocampal neurons transfected with flag-tagged α4β2 nAChR were stained with an anti-flag antibody and antibodies against syntaxin-6 (Fig. 4A–C) and against calnexin (Fig. 4D–F), we found a significant co-localization of the α4β2 nAChR with the ER marker calnexin, seen in purple in the merged image (Fig. 4C), similar to the previously described data (Ren et al., 2005). In the case of the trans-Golgi marker syntaxin-6, we observed less, but still consistent co-localization with the receptor in yellow (Fig. 4F). As expected, no co-localization of the three markers together in white was noticeable in the merged image (data not shown).
Fig. 4. Co-localization of α4β2 nAChR, ER and Golgi marker in a triple co-stained hippocampal neuron.

A–F. Hippocampal neurons cultured for two weeks transfected with flag-tagged α4β2 nAChR were simultaneously stained with anti-flag antibody and antibodies against syntaxin-6 and calnexin. A–C. Co-localization of α4β2 nAChR (A) with the ER marker calnexin (B) is seen in purple in the merged picture (C). D–F. Occasional co-localization of α4β2 nAChR (D) with the Golgi marker syntaxin-6 (E) is seen in yellow in the merged picture (F). Note, that no co-localization of Golgi marker syntaxin-6 can be found with α4β2 nAChR and the ER marker calnexin which would be observed as white in merged pictures (data not shown). Bar in F is 20 μm. Ca2+-dependent co-localization of VILIP-1-GFP, α4β2 nAChR and the Golgi marker syntaxin-6 in a triple co-stained hippocampal neuron. G, H. Hippocampal neurons cultured for two weeks were transfected with flag-tagged α4β2 nAChR and VILIP-1-GFP and simultaneously stained with anti-flag antibody and antibodies against syntaxin-6. G. Co-localization of VILIP-1-GFPα4β2 nAChR with the Golgi marker syntaxin-6 is seen occasionally (arrow) without stimulation as white co-staining in the merged picture (G, upper inset: VILIP-1, middle inset: α4β2 nAChR, lower inset: syntaxin-6, white box shows the magnified area above). H. In carbachol treated cultures, substantial co-localization of all three markers is observed as white co-staining in distinct spots within the hippocampal neuron (H, arrows point at white co-localization spots, white box shows the magnified area with highest density of co-localization). Bar in A, B is 5 μm. I. Quantification of the co-localization of VILIP- 1-GFP, α4β2 nAChR and syntaxin-6 as number of white spots in control (G) carbachol-treated (H) triple co-stainings. Mean values ± S.D. are from three experiments carried out in duplicate.
Thus, results in Fig. 3 and 4 clearly show that both interaction partners can be detected at Golgi membranes and point to the Golgi as a possible place of interaction of VILIP-1 with α4β2 nAChRs. To directly test the novel hypothesis that VILIP-1 following Ca2+ stimulation can interact with α4β2 nAChR at intracellular membranes of the Golgi, we performed triple co-localization experiments to detect α4β2 nAChR, VILIP-1 and the Golgi marker syntaxin-6 simultaneously (Fig. 4GH). Even without stimulation occasional co-localization of the three markers was observed as white co-staining in the merged picture (Fig. 4G, upper inset: VILIP-1, red, middle inset: α4β2 green, lower inset: syntaxin-6, blue). In carbachol treated cultures, an enhanced association of VILIP-1 with the membranes was noticeable (compare Fig. 4G and H, upper inset: VILIP-1, red) similar to the observation in Fig. 3. More importantly, a substantial increase in co-localization of all three markers was observed as white co-staining in distinct spots within the hippocampal neurons (Fig. 4H), as highlighted in the magnified box, representing the place of highest level of white co-localization of the three markers. To verify this assumption, we quantified the number of white spots showing co-localization (Fig. 4I), which revealed a significant ~fivefold increase in the number of spots per cells from an average of 9 in vehicle-treated versus 44 in carbachol-treated cultures.
Thus, data from Fig. 3 and 4 collectively show that the ER might not be the likely place where α4β2 nAChR and VILIP-1 co-localize and interact. However, from the triple staining data (Fig. 4) the novel notion arises that the two interaction partners α4β2 nAChR and VILIP-1 also partially, probably transiently, co-localize at the level of trans-Golgi membranes. This co-localization of VILIP-1 with syntaxin-6, a SNARE involved in membrane trafficking processes in the trans-Golgi and the endocytotic compartment is strongly enhanced in a Ca2+-dependent manner following carbachol treatment.
VILIP-1 affects membrane transport of the α4β2 nAChR at the level of exocytosis in transfected HEK cells
We have previously shown that VILIP-1 affects clathrin-dependent trafficking and receptor recycling of the natriuretic peptide receptor GC-B and the transferrin receptor at the level of endo- and/or exocytosis (Brackmann et al., 2005). We now wanted to directly address whether VILIP-1 can affect Golgi exit and therefore membrane trafficking at the level of constitutive exocytosis. We aimed for biochemical proof using biotinylation assays with HEK cells co-transfected with α4β2 nAChR and VILIP-1 (Fig. 5A). Input controls (Fig. 5A, left panel) showed that in cells co-transfected with vector or with VILIP-1 and α4β2 nAChR similar amounts of flag-tagged α4β2 nAChR can be found in whole cell precipitates. Next, we confirmed the effect of VILIP-1 on enhanced surface expression by surface biotinylation of proteins and detection of flag-tagged α4β2 nAChR in streptavidin-precipitates from the cell extracts. Under these conditions the expression of VILIP-1, compared to vector controls, leads to increased staining intensity of flag-immunoreactive band at ~65 on Western blots, representing enhanced surface expression of α receptor subunit (Fig. 6A, middle panel). We next used the α receptor subunit as biochemical marker for the surface expression of the α4β2 nAChR. When surface proteins were enzymatically digested before the biotinylation, the observed reappearance of the α4–subunit signal (~65kDa) of the nAChR, which showed the strongest signal, in the streptavidin-precipitates is a measure of the transport from the Golgi to the surface over time. When the re-appearance of the α4–subunit was compared in VILIP-1 versus empty vector transfected HEK cells, it became obvious that more receptor re-appeared and therefore a higher rate of exocytosis occurred within 10 minutes of measurement in VILIP-1-transfected cells, compared to vector control (Fig. 5A, right panel). Quantification of the Western blots (Fig. 5A, middle and right panel) showed a significant 50% increase in surface expression (Fig. 5B) and a 2,5 fold increase in constitutive exocytosis (Fig. 5) of α4β2 nAChR in VILIP-1-transfected HEK cells (pOPR-V1) compared to pOPR vector controls. The biotinylation experiments indicate that VILIP-1 did not influence the expression level of the receptor, but most likely at the level of the syntaxin-6-positive Golgi and/or transGolgi/endosomal compartment, can affect α4β2 nAChR surface expression by influencing trafficking between Golgi and surface, namely constitutive exoctosis, in transfected HEK cells.
Fig. 5. Expression of VILIP-1 affects surface expression via accelerating exocytosis of the α4β2 nAChR in double-transfected hippocampal neurons and in HEK cells.

A. In HEK cells co-transfected with flag-tagged α4β2 nAChR and pOPR-VILIP-1 or pOPR vector alone, biotinylation of cellular proteins and of surface proteins was performed and the flag-tagged α4β2 nAChR was detected in Western blot analysis of streptavidin-precipitates from the cell extracts. For input controls (left panel) following lysis and biotinylation, or for surface protein determination (middle panel) following biotinylation and then lysis, the flag-immunoreactive bands in streptavidin-precipitates at about 70 and 55kDa representing α4 and β2 subunits are monitored by Western blot analysis of control vector (pOPR) and VILIP-1-transfected (pOPR-V1) HEK cell extracts. Exocytosis was determined after enzymatic digest of surface proteins and subsequent biotinylation and lysis. The re-appearance of the α4–subunit signal in the streptavidin-precipitates was measured by biotinylation 10min after enzymatic treatment (right panel). B, C. Quantification of the Western blot in A, middle panel, indicates (B) a significant 50% increase in relative surface expression and (C) a 2,5 fold increase in relative level of exocytosis, in A, right panel, of the α4–subunit of the nAChR in VILIP-1-transfected HEK cells.
Fig. 6. Potentiation of ACh-induced whole-cell currents by expression of VILIP-1-GFP in HEK293 cells stably expressing human α4β2 nAChR (A) and in hippocampal neurons transfected with rat α4β2 nAChR (B).
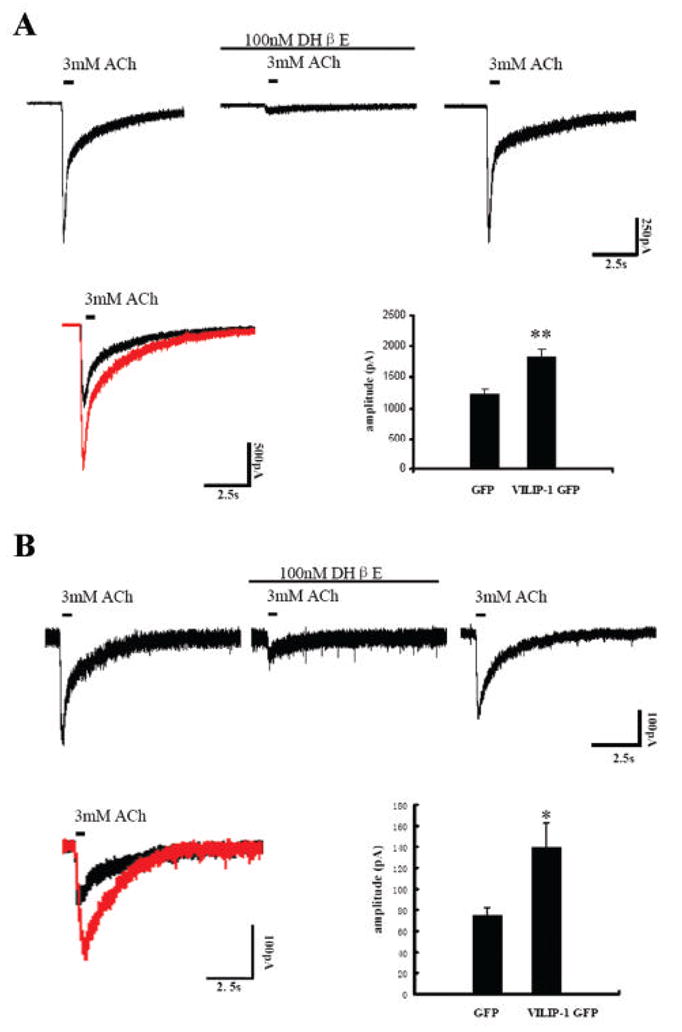
A. Whole-cell responses to ACh in the absence and presence of 100nM DHβE recorded from a single cell of the Ha4b2Lx/1 cell line, which was held at -60mV. The ACh concentration applied (3mM) was in the range of the saturation response. ACh evoked current was blocked completely by 100nM of the α4β2 nAChR antagonist DHβE (second trace). The current recovered after DHβE wash out. Representative traces of whole-cell responses following ACh stimulation in GFP (black trace) and GFP-VILIP-1 (red trace) transfected HEK cells are shown in the lower left panel. The response to ACh in VILIP-1-GFP transfected cells (lower right panel, n=33, from two experiments) is highly significant increased in amplitude (p<0.01) compared to the responses to ACh in GFP-transfected cells (n=18, from two experiments). B. Whole-cell responses to ACh, in the absence and presence of 100nM DHβE recorded from a single cultured hippocampal neuron which was held at -60mV. The evoked current partially recovered after DHβE wash out. Representative traces of whole-cell responses following ACh stimulation in GFP (black trace) and GFP-VILIP-1 (red trace) transfected neurons are shown in the left panel. The response to ACh in the VILIP-1-GFP transfected cells (lower right panel, n=13, from two experiments) shows highly significant increase in amplitude (p<0.05), as compared to GFP-transfected neurons (n=14, from three experiments).
VILIP-1 affects responsiveness of α4β2 nAChR to ACh in transiently and stably transfected HEK cells and in transfected hippocampal neurons
To test whether the effect of VILIP-1 on surface expression of α4β2 nAChR also leads to functional up-regulation of the expressed receptor we performed whole-cell patch-clamp studies (Fig. 6). First, recordings from α4β2 nAChR and GFP or VILIP-1-GFP-cotransfected HEK cells and from GFP or VILIP-1-GFP-transfected HEK cells stably expressing the human α4β2 nAChR (cell line Ha4b2Lx/1) were analyzed. ACh evoked whole-cell responses at saturating concentration of 3mM, which could be completely blocked by 100nM DHβE, an antagonist of α4β2 receptors, indicating the presence of functional α4β2 receptor in the Ha4b2Lx/1 cell lines ectopically expressing the human α4β2 nAChR. Following antagonist wash out the current could be recovered (Fig. 6A, upper panel). These data are comparable to those previously reported (Samochocki et al., 2003). Similar results were obtained using transiently transfected HEK293 cells (data not shown). Under the same saturation conditions (3 mM ACh) the response to ACh was significantly increased in amplitude in the VILIP-1-GFP co-transfected cells compared to the responses in GFP-transfected HEK cells expressing human α4β2 nAChR (Fig. 6A, compare representative traces in red, VILIP-1-GFP, and black, GFP), and in the transiently expressing HEK cells (data not shown).
Next, we looked at α4β2 nAChR and GFP or VILIP-1-GFP-cotransfected primary hippocampal neurons in which nicotinic currents had been isolated by blocking GABAergic, glutamatergic and muscarinergic transmission with bicuculline, CNQX, TTX, APV and atropine. Again at saturation conditions (3 mM ACh) the response to ACh could be blocked by the receptor antagonist DHβE (Fig. 6B, upper panel). To reduce possible side effects of desensitization, only the first current obtained from each cell was plotted for further statistic analysis. Only healthy looking neurons moderately expressing VILIP-1-GFP were used and showed a significant increase in the amplitude of ACh currents compared to healthy GFP-control neurons of comparable size and morphology (Fig. 6B, compare representative traces in red, VILIP-1-GFP, and black, GFP). When siRNA against VILIP-1 was co-transfected with GFP, we could observe a downregulation in VILIP-1 immunoreactivity in the GFP co-transfected cells, when compared to neurons co-transfected with GFP and scrambled siRNA (compare Fig. 7A, B to C, D). When hippocampal neurons were transfected with siRNA against VILIP-1 and later co-transfected with EGFP and α4β2 nAChR a reduced activation of α4β2 nAChR currents was observed (Fig. 7E), which reached highly significant levels. These results support the notion that enhanced VILIP-1 expression not only increases surface expression, but at the same time increase the responsiveness of cells expressing the rat and human α4β2 nAChR for its ligand ACh. In contrast, application of siRNA specific for VILIP-1 leading to the knockdown of endogenous VILIP-1 expression, was able to significantly attenuate the ACh-induced currents. The average α4 nicotinic current amplitude of 140 pA was strongly reduced to about 34 pA following co-transfection of α4β2 nAChRs with VILIP-1 siRNA. Thus, we conclude that VILIP-1 is able to up-regulate functional α4β2 nAChRs in hippocampal neurons.
Fig 7. SiRNA knockdown of VILIP-1 in GFP co-transfected hippocampal neurons (A–D) leads to reduction of ACh-induced whole-cell α4β2 nAChR currents.
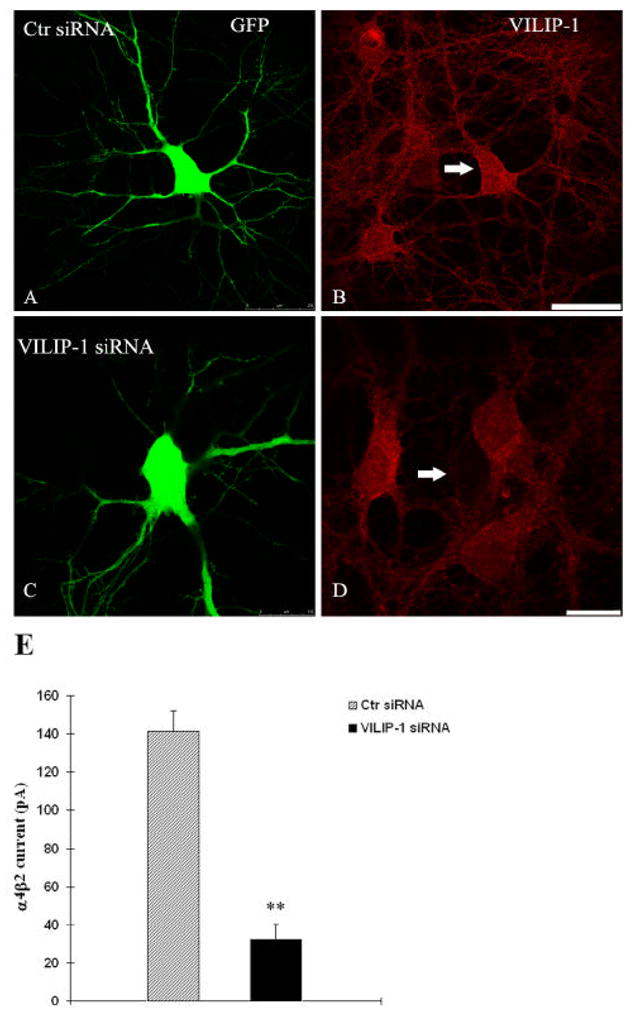
Representative confocal image showing control siRNA and GFP co-transfected hippocampal neuron (A) and expression of endogenous VILIP-1 (B). VILIP-1-siRNA/GFP-coexpressing neurons (C) show strongly reduced expression of endogenous VILIP-1 (D). In the representative experiment shown an average of 13 out of 15 co-transfected neurons per dish showed strongly reduced VILIP-1 expression levels, whereas 29 out of 33 neurons in the control siRNA group did not show changes in expression. Bar in A, B is 30μm, bar in C,D is 15 μm. E. In hippocampal neurons co-transfected with α4β2 nAChRs and control siRNA (Ctr siRNA) the average α4 nicotinic current amplitude was strongly reduced following co-transfection with VILIP-1 siRNA (VILIP-1 siRNA). Data are presented as mean ± SEM; ** p < 0.01 compared with control group.
Discussion
Neuronal Ca2+-sensor protein VILIP-1 enhances surface expression leading to upregulation of functional α4β2 nAChR in hippocampal neurons
In addition to nicotine-induced functional up-regulation (Buisson and Bertrand, 2002) a physiological mechanisms for up-regulation of functional α4β2 nAChRs seem to exist. VILIP-1 is a myristoylated neuronal Ca2+-sensor protein which was identified as an interaction partner of α4β2 nAChRs in a yeast-two-hybrid interaction screen using the large intracellular loop of the α4 subunit as bait. Initial characterization in oocytes showed that co-expression of VILIP-1 with recombinant α4β2 nAChR up-regulated surface expression two-fold and increased the agonist-sensitivity to ACh three-fold. The modulation of the recombinant α4β2 nAChR by VILIP-1 was attenuated with myristoylation mutants or EF-hand mutants not able to bind Ca2+. We have put forward the hypothesis that VILIP-1, possibly via the mechanism of the Ca2+-myristoyl switch (Ames et al., 1997; Spilker et al., 2002), may serve as a modulator of α4β2 nAChR (Lin et al., 2002). Interestingly, the application of nicotine to hippocampal cultures leads to the Ca2+-dependent and reversible membrane-translocation of VILIP-1 via activation of α7- but not α4-containing nAChRs. This may provide a novel functional crosstalk between α4- and α7-containing nAChRs (Zhao et al., 2008). Here we examine the potential physiological importance of the effect. VILIP-1 enhances surface expression of flag-tagged α4β2 nAChRs in hippocampal neurons leading to increased ACh evoked currents, thus, functionally up-regulating these α4β2 nAChRs in hippocampal neurons. Thus, we postulate that VILIP-1 is part of a novel physiological mechanism for up-regulation of functional α4β2 nAChRs in the brain.
Neuronal Ca2+-sensor protein VILIP-1 is excluded from ER, but localizes to Golgi, indicating a role in α4β2 nAChR trafficking via the Golgi compartment in hippocampal neurons
An ER export motif, important for cargo selection into COPII-coated vesicles that transport proteins from the ER to the Golgi, was identified within the cytoplasmic loop of both subunits and are critical structural determinants for ER export of fully assembled nAChRs (Ren et al., 2005). One possible hypothesis to explain the effect of VILIP-1 would be that it interacts with the ER export motif and accelerates exit of the α4β2 nAChR from the ER. To test this hypothesis, we have performed co-localization studies in hippocampal neurons in culture. VILIP-1 did not localize to the ER in the presence or absence of Ca2+, which makes it less likely that VILIP-1 influences ER export. One remaining possibility could be that Ca2+ release causes rapid association between VILIP-1 and α4β2 nAChR in the ER, but that once this association occursα4β2 nAChR are rapidly released from the ER. However, we were able to show localization of VILIP-1 with markers of the trans-Golgi network, which was strongly enhanced by increasing the intracellular Ca2+ level. Thus, we postulate that VILIP-1 plays a role in modulating exit from the Golgi rather than the ER, thereby leading to enhanced surface expression of the receptor in primary hippocampal neurons.
The Ca2+-myristoyl switch of VILIP-1 and co-localization with α4β2 nAChR in syntaxin-6 positive Golgi structures define a novel signalling mechanism
We have previously postulated that the Ca2+-myristoyl switch of VILIP-1 is part of a signalling mechanism which enables a receptor system to respond to a Ca2+ stimulus in living hippocampal neurons (Spilker et al., 2002; Brackmann et al., 2005), and thereby possibly acts as a coincidence detection complex. The molecular mechanism underlying coincidence detection in such a protein complex seems not to involve direct and fast Ca2+-dependent activation of the receptor at the cell surface, but depending on activity of the neuron and intracellular Ca2+-levels appears to rely on the slow modulation of receptor trafficking, occuring in the minute range (Lin et al., 2002; Brackmann et al., 2005). This model is supported by previous reports showing that VILIP-1 interacts with membrane transport intermediates isolated from rat brain, such as synaptic vesicles and clathrin-coated vesicles (CCV) (Blondeau et al., 2004; Morciano et al., 2005), and the fact that VILIP-1 affects clathrin-dependent membrane trafficking of the transferrin receptor and the receptor guanylyl cyclase B (Brackmann et al., 2005). Receptor trafficking is mediated by a complex machinery of trafficking and sorting proteins. CCVs and the trans-Golgi network (TGN) represent key sorting sites for many transmembrane proteins (Bonifacio and Traub, 2003). In this study we have found co-localization of VILIP-1 and α4β2 nAChR with one component of the transport machinery, the trans-Golgi and endosomal SNARE syntaxin-6. The primary role of SNAREs (soluble NSF attachment receptor) is to mediate fusion of cellular transport vesicles with a target compartment. In hippocampal neurons the SNARE syntaxin-6 is present in complexes with SNAPs (soluble NSF attachment protein) and is primarily found on the trans-Golgi network, where it partially co-localizes with the adapter protein AP-1 on clathrin-coated membranes (Bock et al., 1997). Moreover, syntaxin-6 is part of a SNARE complex which is enriched on clathrin-coated and synaptic vesicles in rat brain (Kreykenbohm et al., 2002). Syntaxin-6 is also found in endosomes and regulates the delivery of microdomain-associated lipids and proteins to the cell surface, which are required for caveolar endocytosis (Choudhury et al, 2006). VILIP-1 is able to affect the clathrin-dependent receptor recycling of a set of receptors including the transferin receptor, the receptor guanylyl cyclase GC-B (Brackmann et al., 2005). These findings support an involvement of VILIP-1 in clathrin-dependent transport of syntaxin-6-positive vesicles from the trans-Golgi network to the cell surface and/or in endosomal trafficking. Moreover, VILIP-1 was identified to interact with several ligand-gated ion channels, including the α4β2 nAChR (Lin et al., 2002), glutamate receptors of the kainate subtype GluR6 (Coussen et al., 2005) and P2X2 receptors (Chaumont et al., 2008). VILIP-1 forms a signaling complex with P2X2 receptors and proteins such as N-ethylmaleimide-sensitive factor (NSF), tubulin1α, vesicle amine transport protein 1 homolog (VAT1), glutamic acid decarboxylase (GAD) and synapsin IIb (Chaumont et al., 2008), indicating that VILIP-1 might play a general role in trafficking of these ligand-gated ion channels via cellular transport vesicles.
Biochemical experiments reveal that the neuronal Ca2+-sensor protein VILIP-1 affect α4β2 receptor trafficking at the level of constitutive exocytosis
Based on these literature data the co-localization of VILIP-1 and α4β2 nAChR with the trans-Golgi SNARE syntaxin-6 is functionally linked to membrane transport via post-Golgi pathways. To prove this assumption we have performed biotinylation assays, which together with enzymatic cleavage of newly synthesized receptors at the cell surface allowed us to precisely address the constitutive exocytotic pathway in cells co-transfected with flag-tagged α4β2 nAChR and VILIP-1. These results clearly demonstrated biochemically that not only VILIP-1 did significantly enhance surface protein expression of α4β2 nAChR, but that within 10 minutes following complete removal of all surface receptors, the re-appearance of surface expression of the tagged receptor was strongly promoted in VILIP-1 transfected cells. These findings support an involvement of VILIP-1 in clathrin-dependent transport of syntaxin-6-positive vesicles from the trans-Golgi network to the cell surface and possibly in endosomal trafficking. Other members of the NCS protein family seem to perform related functions in receptor trafficking, but at different trafficking compartments. KChIP stimulates traffic of Kv4 K+ channels from a post-ER but pre-Golgi vesicular compartment (Hasdemir et al., 2005). In contrast, NCS-1, like VILIP-1, stimulates constitutive traffic from the TGN (Haynes et al., 2005).
Possible implications for hippocampal network activity and synaptic plasticity: a working hypothesis
The nAChRs are known to enhance GABAergic neurotransmission in different brains structures, underlying the cholinergic modulation of synaptic plasticity and network activity (Couey et al., 2007; Nashmi et al., 2007). A variety of evidence has been provided for a link between nicotinic AChR enhancement, cognitive function (Mansvelder et al., 2005) and disease (Hogg et al., 2003; Gotti and Clement, 2004). Amino acid mutations in the human α4β2 nAChR associated with autosomal dominant nocturnal frontal lobe epilepsy, which when are engineered in the corresponding α4β2 nAChR in mice, lead to disturbances of inhibitory synchronization of cortical networks by GABAergic interneurons (Klaassen et al., 2006). Since VILIP-1 is misregulated in hippocampal pyramidal neurons and in CA1 and denate gyrus interneurons in schizophrenic brains (Bernstein et al., 2002, 2003), and is up-regulated following induction of hippocampal LTP in vivo (Braunewell et al., 2004; Brackmann et al., 2004), we postulate that VILIP-1 in conjunction with the α4β2 receptor plays an important role in modulating network activity in healthy and diseased hippocampus (Braunewell, 2005). When looking for the possible functional impact of pathologically increased VILIP-1 expression, we have observed nAChR-dependent enhancement in IPSCs in rat hippocampal cell culture transfected with VILIP-1-GFP, likely mediated by the α4β2 nAChR (Gierke et al., 2008). This is supported by the fact that functional expression of the high-affinity nicotine-binding α4β2 nAChR appears to be restricted to certain interneuron populations (Alkondon and Albuquerque, 2001Alkondon and Albuquerque, 2002). The α4β2 nAChRs predominantly contribute to IPSCs generated by interneurons of stratum oriens and stratum radiatum in the CA1 region of rat hippocampal slices in vitro (Alkondon and Albuquerque, 2001Alkondon and Albuquerque, 2002) and in rat hippocampal cultures, where 65% of IPSCs were identified to be dependent on α4β2 nAChR (Braga et al., 2004). From these different sets of data it appears that VILIP-1 in cultured hippocampal neurons induces an enhancement in surface expression of α4β2 nAChRs and, in turn, enhancement of ACh-induced currents via α4β2 nAChR. Thus, we can put forward a hypothesis (Fig. 8) for the molecular mechanism of VILIP-1-mediated up-regulation of α4β2 nAChR in hippocampal neurons, which might serve as a novel physiological mechanism underlying plasticity of nicotinergic neurotransmission.
Fig. 8. Hypothesis for a novel physiological mechanism of up-regulation of functional α4β2.
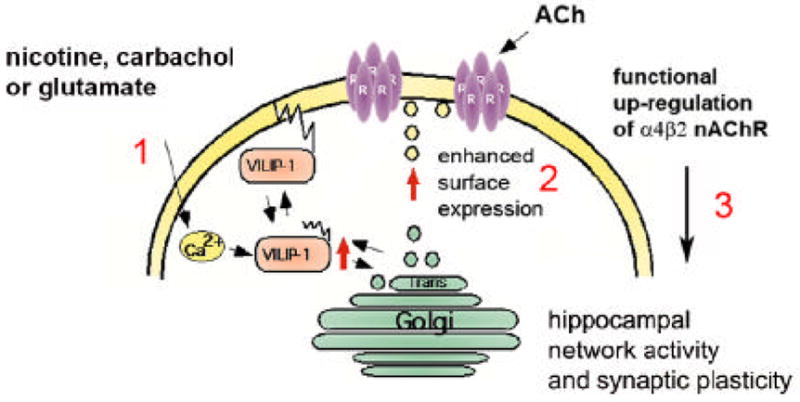
The cartoon shows the neuronal Ca2+-sensor VILIP-1, is activated following stimulation of a neuron by a signal, such as glutamate, carbachol or nicotine, which is acting on receptors increasing the intracellular Ca2+ level (1). VILIP-1 shuttles to cell surface and Golgi membranes as well as clathrin-coated vesicles, where it co-localizes with α4β2 nAChR and with syntaxin-6, a SNARE implicated in clathrin-dependent transport mechanisms in the trans-Golgi network. VILIP-1 causes enhanced exocytosis and surface transport of α4β2 nAChR (2), consequently increasing the surface expression and finally the sensitivity of the neuron towards ACh (3). The nicotine or glutamate induced translocation of VILIP-1 to cellular membranes (1, Zhao et al., 2008), and in turn the up-regulation of functional α4β2 nAChRs (2, Lin et al., 2002, this study) may contribute to plasticity of nicotinergic neurotransmission in principal neurons and/or in interneurons in the hippocampus, since the expression of VILIP-1 in conjunction with the α4β2 nAChR enhances the frequency of inhibitory postsynaptic currents (IPSCs) in hippocampal cultures (3, Gierke et al., 2008). Thereby, the physiological up-regulation of α4β2 nAChR by VILIP-1 might modulate hippocampal network activity and synaptic plasticity.
Experimental Methods
Materials
Nicotine, brefeldin A, carbachol, monensin for cell stimulation experiments were purchased from Sigma (St Louis, MO). Cell culture reagents were obtained from Gibco-Invitrogen (San Diego, CA). Unless otherwise specified, all other reagents were purchased from Sigma and Roth.
Antibodies
Rat polyclonal antibody, raised against recombinant VILIP-1 fusion protein, was affinity-purified on corresponding glutathion-S-transferase (GST)-tagged fusion proteins immobilized on N-hydroxysuccinimide ester coupled agarose columns (Bio-Rad, Hercules, CA) as previously described (Spilker et al., 2002). Antibodies against the α-subunit of the nAChR were mAb299 (Covance, Berkeley, CA, USA) and Guinea pig anti-α4 nAChR polyclonal antibody (Chemicon, Germany). Further polyclonal antibodies used in this study comprised rabbit anti-calnexin (Sigma and Stressgene, Victoria, BC, Canada), rabbit and goat anti-flag (Sigma, Abcam, Cambridge, UK) and mouse anti-syntaxin6 (Sigma). Cy3, Alexa Fluor™ 488 and Cy5 labeled secondary antibodies were purchased from Dianova (Hamburg, Germany), Molecular Probes (Eugene, OR) and Abcam.
Cell culture and transfection of HEK 293 cells and hippocampal neurons
Human embryonic kidney cells 293 were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10 % fetal bovine serum, 2mM L-glutamine and antibiotics at 37°C. Cells were maintained in a humidified 95% air containing 5% CO2. Hα4β2L-/1 (Samochocki et al., 2003) cells stably expressing the human α4β2 nAChR (H, human; α4β2; L-, HEK-293 cells that do not express the L 1C-bCa2+channel 1, clone 1) were grown in DMEM containing 13 % fetal calf serum in an incubator at 37°C, 10% CO2. To keep the cells in the exponential phase of growth, they were harvested every 3 days and plated at a density of 1.5 x 104 cells/cm2. For transient transfection the HEK cells were plated in 24-well dishes and 12 h later transfected using Lipofectamine 2000 (Invitrogen). To increase the transfection efficiency cells were incubated for 4–6 h in unsupplemented DMEM. α4β2 surface expression was detected after incubation at 37°C for 48 h. Hippocampal cultures were prepared from fetal wistar rat brains (E18) as previously described (Spilker et al., 2002). Cells were plated onto poly-D-lysin coated 12 well culture plates (12 mm in diameter) at a density of 120.000 cells for transient transfection experiments and in 24 well culture plates with glass coverslips at a density of 60.000 cells for immunocytochemistry in DMEM containing 10 % fetal calf serum, 2 mM L-glutamine and antibiotics. 24 hours after plating culture medium was exchanged for Neurobasal medium (Invitrogen) supplemented with 1x B27, 0.5 mM L-glutamine and antibiotics. Hippocampal neurons were transfected with cDNA or siRNA after 10–14 days in culture using the lipofectamin transfection method (Invitrogen). The cDNAs coding for GFP, VILIP-1-GFP were essentially as previously described Spilker et al., 2002. Rat α4-flag and β2-flag nAChR subunit cDNA was as described (Ren et al., 2005). Carbachol (0.1mM) and ionomycin (1μM) stimulation was applied 20 h after transfection for 2 minutes.
Enzyme-linked immunoassay for quantitating cell surface α4β2 nAChRs
Cell surface α4β2 nAChRs were measured in hippocampal neurons plated in 12-well dishes. Cells were washed once in PBS and fixed with 3% paraformaldehyde for 5 minutes at room temperature. After 4 washes with PBS, the cells were blocked with 3% BSA in PBS for 30 minutes, and then incubated for 1h at room temperature with a mAb against the β2 nAChR subunit (mAb 295) whose epitope is located in the extracellular domain of the subunit. After 6 washes with PBS, the cells were incubated with horseradish peroxidase-conjugated goat anti-rat secondary Ab for 1 h, washed 8 times with PBS, and finally incubated with the HRP substrate 3, 3′, 5, 5′-tetramethylbenzidine (Sigma) for 30minutes. The absorbance of the supernatant was then measured at 405nm in a Beckmann spectrophotometer. The nonspecific background binding of Abs to cells was determined using nontransfected cells and was typically <0.5% of the total binding observed for wild type α4β2 nAChRs.
Biotinylation assay for quantitating exocytosis of α4β2 nAChRs
To test the effect of VILIP-1 on surface expression, HEK cells were co-transfected with α4β2 nAChR and pOPR-V1 or as control with pOPR for 48h and were afterwards biotinylated using Sulfo-NHS-SS-Biotin (Pierce Rockford, IL.; 0.15 mg/ml) in PBS for 10 minutes on ice. Cells were washed three times with TBS (Tris 25 mM, pH 7.4, NaCl 137 mM, KCl 5 mM, CaCl2 2.3 mM, MgCl2 0.5 mM, Na2HPO4 0.143 g/l), lysed 10–30 minutes in extraction buffer (Tris-HCl 10 mM pH 7.5, EDTA 10 mM, Triton X-100 1%, SDS 0.1% and protease inhibitor cocktail 1x). For input controls, cell were first lysed and then biotinylated. After centrifugation supernatants containing equal amount of protein were incubated with streptavidin beads (Pierce Rockford, IL) for 1–2h, washed four times in extraction buffer (without detergents) and subjected to SDS-PAGE. Western Blot analysis using anti-flag antibodies was performed to detect α4β2 nAChR. To specifically test the effect on exocytosis cell surface was stripped before biotinylation using trypsin digestion (0.1 mg/ml) at 4°C for 15minutes. After a chase period of 10minutes at 37°C, biotinylation was done at 4°C. Following washing, cell lysis, precipitation with streptavidin beads as above Western Blot analysis was performed to detect α4β2 nAChR which had been exocytosed using anti-flag antibodies.
Immunocytochemistry
Following fixation of hippocampal neurons with 3% paraformaldehyde in PBS, pH 7.4 for 5 minutes at room temperature (adapted from Ren et al., 2005), cells were washed twice with 25 mM glycine in PBS to quench background staining. The cells were blocked for surface staining in 3% bovine serum albumine, 10% horse serum in PBS (blocking solution) or blocked and permeabilized with 0.1% Triton X-100 added to blocking solution for 30 minutes, respectively. Cells were incubated with primary antibodies diluted in blocking solution at 4 °C overnight. In background controls the first antibodies were omitted. After washing three times with PBS secondary antibodies diluted in blocking solution without Triton X-100 were applied to the cells for 1 h at room temperature. After removal of unbound antibodies coverslips were mounted on slides with Mowiol (Calbiochem) including 1,4-diazobicyclo-[2.2.2]-octane (Merck, Darmstadt, Germany) to reduce fading. GFP, Alexa Green 488, Cy3 and Cy5 fluorescence were visualized using a Leica SP laser scanning microscope (Leica DM LFSA or DM 2500; Wetzlar, Germany) with Argon-ion (488 nm), Helium-Neon (543 nm) and Helium-Neon (633 nm) or solid-state (488, 532, 635 nm) laser excitation, respectively. The excitation light was coupled in via the main dichroic beam splitters (RSP500 for GFP and DD488/543 for Cy3 or DD488/635 for GFP, Alexa Green 488, Cy5 and DD405/532 for Cy3 fluorescence, respectively). The emitted light was collected in sequential scans in the range of 505 nm to 550 nm (GFP, Alexa Green 488), 550 nm to 600 nm (Cy3) and 650 nm to 750 nm (Cy5), respectively. The pinhole was set airy 1. For the experiments, Leica HCX PL APO 63x/1.4 and 100x/1.4 oil immersion objectives were used at the upright microscope. The spatial resolution was 0.37 μm/pixel (in the x- and y-axes) for the aquired 512 × 512 and 1024 × 1024 pixel images. To avoid photo damage, the lowest laser intensity necessary for an adequate signal-to-noise ratio was used. Conventional fluorescence microscopy was performed on a Leica DMR fluorescence microscope. Images were recorded digitally and processed using the Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) and the ImageJ 1.37 software (available at http://rsb.info.nih.gov/ij/).
Co-transfection of hippocampal neurons with siRNA and EGFP
To demonstrate the knock down of VILIP-1 using siRNA, 1 μl Lipofectamin-2000 (Invitrogen) was preincubated with 25 μl Opti-MEM I reduced serum medium (Gibco, Carlsbad, CA) at room temperature (20°C) for 5 min. Meanwhile, either 1μl (2 0μM) control siRNA or a mixture of three VILIP-1 antisense siRNAs in combination with 0.1μl EGFP vector was diluted to 25 μl Opti-MEM I reduced serum medium (Gibco) in a ratio of 10: 1. Allstars negative control siRNA and three different VILIP-1 siRNA sequences were obtained from (Qiagen, Hilden, Germany). The VILIP-1 sequences are: rat Vsnl1-5 sense r(AGC CGU UAG UCU GAA UUA A)dTdT, antisense r(UUA AUU CAG ACU AAC GGC U) dTdT, Vsnl1-6 sense r(CAA AGA UGA CCA GAU UAC A)dTdT, antisense r(UGU AAU CUG GUC AUC UUU G)dTdT, Vsnl1-8 sense r(GUG CGA CAU UCA GAA AUG A)dTdT, antisense r(UCA UUU CUG AAU GUC GCA C)dTdT. Lipofectamin 2000 and plasmid dilution were mixed and incubated at room temperature for 20 min. 50μl DNA and Lipofectamin 2000 complexes were added to each well of the 7-day-old hippocampal cells that had been cultured in 0.5 ml growth medium. 72 hours after transfection, the cells were used for subsequent experiments. To measure the α4 nAChR current after siRNA transfection, a second transfection was performed as follows: 1 μl Lipofectamin-2000 (Invitrogen) was preincubated with 25 μl Opti-MEM I reduced serum medium (Gibco, Carlsbad, CA) at room temperature (20°C) for 5 min. 0.1μg EGFP, 1 μg α4, and 5 μg β2 nAChR vector were diluted to 25μl Opti-MEM I reduced serum medium (Gibco). Then the Lipofectamin 2000 and plasmid dilution were mixed and incubated at room temperature for 20 min. 50 μl DNA and Lipofectamin 2000 complexes were added to each well. 72 hours after transfection, the cells were used for patching experiments.
Patch clamp analysis
Whole-cell current recordings from stably transfected HEK cells were performed 12 to 24 h after transfection using an EPC-7 patch-clamp system (HEKA Elektronik, GmbH, Germany). The bathing solution was composed of (mM) 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM D-glucose, and 10 mM HEPES (pH 7.3; 300 mOsM)), and the internal pipette solution contained 140 mM CsCl (equilibrated with CsOH), 11mM EGTA, 10 mM HEPES, and 1 mM MgCl2 (pH 7.3; 300 mOsM. In patch recordings of hippocampal neurons, whole-cell currents from transiently transfected cells were recorded 48 h after transfection. Cells of comparable size and morphology were used for the different groups (GFP vs VILIP-1-GFP). Sodium channels, NMDA, AMPA, and GABAA receptors were blocked by 0.5 μM TTX, 30μM AP-5, 30μM CNQX, 5μM bicuculline, 1nM MLA. ACSF (129 mM NaCl, 21 mM NaHCO3, 3 mM KCl, 1.6 mM CaCl2, 1.8 mM MgSO4, 1.25 mM NaH2PO4 and 10 mM glucose, pH 7.4) was used as extracellular buffer. The internal solution contained the following (in mM): potassium gluconate 136.5, KCl 17.5, NaCl 9, MgCl2 1, HEPES 10, EGTA 0.2, Mg-ATP 2, GTP, 0.3, pH 7.2. Microelectrodes (borosilicate glass) had an external diameter 2.0 mm, and the pipette resistance was 2–5 MΩ. Following formation of a high-resistance seal, capacitance transients in cells were minimized using the C-Fast facility. No compensation was made for series resistance. Whole-cell currents were induced by fast application of the test substances by means of a single patch pipette or home made multiple micropipettes with a joined tip, positioned near the cell. The cells were superfused with the bathing solution at a rate of 1.5–2.0 ml/minutes. In all experiments, atropine (1 μM) was included in the bathing solutions in order to inhibit intrinsic muscarinic responses. Signals were low-pass (Bessel) filtered at 3 kHz (whole-cell measurements) and analyzed on a PC using the TIDA software (Version3, HEKA Elektronik, GmbH, Germany). All data are presented as mean +/− SEM.
Acknowledgments
This work was conducted within the framework of the national priority program (Schwerpunktprogram) SPP1226 Nicotine: Molecular and physiological mechanisms in the central nervous system (www.nicotine-research.com) funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, grant Br1579/9-1) and DFG grant (Br1579/8-1) to K.H.B., and from the National Institutes of Health (DA 019675) and NARSAD (to R.A).
Abbreviations
- ACh
acetylcholine
- NCS
neuronal Ca2+ sensor
- nAChR
nicotinic acetylcholine receptor
- VSNL1
visinin-like protein-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ames JB, Ishima R, Tanaka T, Gordon JI, Stryer L, Ikura M. Molecular mechanics of calcium-myristoyl switches. Nature. 1997;389:198–202. doi: 10.1038/38310. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Becker A, Keilhoff G, Gorcyca WA, Braunewell KH, Grecksch G. Ketamine induced changes of the expression of neuronal calcium sensor proteins VILIP-1, -3 and hippocalcin in a partial schizophrenia model in rats. Neurosci Lett. 2003;339:95–98. doi: 10.1016/s0304-3940(02)01482-9. [DOI] [PubMed] [Google Scholar]
- Bernstein HG, Braunewell KH, Spilker C, Danos P, Baumann B, Diekmann S, Gundelfinger ED, Bogerts B. Hippocampal expression of the Ca2+ sensor protein VILIP-1 in schizophrenia. Neuroreport. 2002;23:393–396. doi: 10.1097/00001756-200203250-00006. [DOI] [PubMed] [Google Scholar]
- Blondeau F, Ritter B, Allaire PD, Wasiak S, Girard M, Hussain NK, Angers A, Legendre-Guillemin V, Roy L, Boismenu D, Kearney RE, Bell AW, Bergeron JJ, McPherson PS. Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proc Natl Acad Sci U S A. 2004;101:3833–3838. doi: 10.1073/pnas.0308186101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans- Golgi network vesicle trafficking. Mol Biol Cell. 1997;8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem. 2003;72:395–447. doi: 10.1146/annurev.biochem.72.121801.161800. [DOI] [PubMed] [Google Scholar]
- Brackmann M, Zhao C, Kuhl D, Manahan-Vaughan D, Braunewell KH. MGluRs regulate the expression of neuronal calcium sensor proteins NCS-1 and VILIP-1 and the immediate early gene arg3.1/arc in the hippocampus in vivo. Biochem Biophys Res Commun. 2004;322:1073–1079. doi: 10.1016/j.bbrc.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Brackmann M, Schuchmann S, Anand R, Braunewell KH. Neuronal Ca2+ sensor NCS protein VILIP-1 affects cGMP signalling of guanylyl cyclase B by regulating clathrin-dependent receptor recycling in hippocampal neurons. J Cell Sci. 2005;118:2495–2505. doi: 10.1242/jcs.02376. [DOI] [PubMed] [Google Scholar]
- Braunewell K-H, Brackmann M, Manahan-Vaughan D. Group I mGluRs regulate the expression of the intracellular neuronal calcium sensor protein VILIP-1 in vitro and in vivo, possible implications for mGluR-dependent hippocampal plasticity? Neuropharmacology. 2003;44:707–715. doi: 10.1016/s0028-3908(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Braunewell KH. The darker side of Ca2+ signaling by neuronal Ca2+-sensor proteins, from Alzheimer’s disease to cancer. Trends Pharmacol Sci. 2005;26:345–351. doi: 10.1016/j.tips.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Gundelfinger ED. Intracellular neuronal calcium sensor proteins, a family of EF-hand calcium binding proteins in search of a function. Cell Tissue Res. 1999;295:1–12. doi: 10.1007/s004410051207. [DOI] [PubMed] [Google Scholar]
- Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, Marks MJ, Collins AC, Leonard S. Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology. 2000;23:351–364. doi: 10.1016/S0893-133X(00)00121-4. [DOI] [PubMed] [Google Scholar]
- Buccafusco JJ, Letchworth SR, Bencherif M, Lippiello PM. Long-lasting cognitive improvement with nicotinic receptor agonists, mechanisms of pharmacokinetic-pharmacodynamic discordance. Trends Pharmacol Sci. 2005;26:352–360. doi: 10.1016/j.tips.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci. 2002;23:130–136. doi: 10.1016/S0165-6147(00)01979-9. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couey JJ, Meredith RM, Spijker S, Poorthuis RB, Smit AB, Brussaard AB, Mansvelder HD. Distributed Network Actions by Nicotine Increase the Threshold for Spike-Timing-Dependent Plasticity in Prefrontal Cortex. Neuron. 2007;54:73–87. doi: 10.1016/j.neuron.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Chaumont S, Compan V, Toulme E, Richler E, Housley GD, Rassendren F, Khakh BS. Regulation of P2X2 receptors by the neuronal calcium sensor VILIP1. Sci Signal. 2008;14(141):ra8. doi: 10.1126/scisignal.1162329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A, Marks DL, Proctor KM, Gould GW, Pagano RE. Regulation of caveolar endocytosis by syntaxin 6-dependent delivery of membrane components to the cell surface. Nat Cell Biol. 2006;8:317–328. doi: 10.1038/ncb1380. [DOI] [PubMed] [Google Scholar]
- Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- Darsow T, Booker TK, Pina-Crespo JC, Heinemann SF. Exocytic trafficking is required for nicotine-induced up-regulation of alpha 4 beta 2 nicotinic acetylcholine receptors. J Biol Chem. 2005;280:18311–18320. doi: 10.1074/jbc.M501157200. [DOI] [PubMed] [Google Scholar]
- De Luca V, Voineskos S, Wong G, Kennedy JL. Genetic interaction between alpha4 and beta2 subunits of high affinity nicotinic receptor, analysis in schizophrenia. Exp Brain Res. 2006;174:292–296. doi: 10.1007/s00221-006-0458-y. [DOI] [PubMed] [Google Scholar]
- Gierke P, Zhao C, Bernstein HG, Noack C, Anand R, Heinemann U, Braunewell KH. Implication of neuronal Ca2+-sensor protein VILIP-1 in the glutamate hypothesis of schizophrenia. Neurobiol Dis. 2008;32:162–175. doi: 10.1016/j.nbd.2008.07.008. [DOI] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci. 2005;28:371–378. doi: 10.1016/j.tins.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Harkness PC, Millar NS. Changes in conformation and subcellular distribution of {alpha}4{beta}2 nicotinic acetylcholine receptors revealed by chronic nicotine treatment and expression of subunit chimeras. J Neurosci. 2002;22:10172–10181. doi: 10.1523/JNEUROSCI.22-23-10172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasdemir B, Fitzgerald DJ, Prior IA, Tepikin AV, Burgoyne RD. Traffic of Kv4 K+ channels mediated by KChIP1 is via a novel post-ER vesicular pathway. J Cell Biol. 2005;171:459–469. doi: 10.1083/jcb.200506005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes LP, Thomas GM, Burgoyne RD. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J Biol Chem. 2005;280:6047–6054. doi: 10.1074/jbc.M413090200. [DOI] [PubMed] [Google Scholar]
- Hernandez CM, Terry AV., Jr Repeated nicotine exposure in rats: effects on memory function, cholinergic markers and nerve growth factor. Neuroscience. 2005;130:997–1012. doi: 10.1016/j.neuroscience.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1–46. doi: 10.1007/s10254-003-0005-1. [DOI] [PubMed] [Google Scholar]
- Ikura M, Ames JB. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: two ways to promote multifunctionality. Proc Natl Acad Sci U S A. 2006;103:1159–1164. doi: 10.1073/pnas.0508640103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen A, Glykys J, Maguire J, Labarca C, Mody I, Boulter J. Seizures and enhanced cortical GABAergic inhibition in two mouse models of human autosomal dominant nocturnal frontal lobe epilepsy. Proc Natl Acad Sci U S A. 2006;103:19152–19157. doi: 10.1073/pnas.0608215103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreykenbohm V, Wenzel D, Antonin W, Atlachkine V, von Mollard GF. The SNAREs vti1a and vti1b have distinct localization and SNARE complex partners. Eur J Cell Biol. 2002;81:273–280. doi: 10.1078/0171-9335-00247. [DOI] [PubMed] [Google Scholar]
- Le Novere N, Corringer PJ, Changeux JP. The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol. 2002;53:447–456. doi: 10.1002/neu.10153. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology (Berl) 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Lin L, Jeanclos EM, Treuil M, Braunewell KH, Gundelfinger ED, Anand R. The calcium sensor protein visinin-like protein-1 modulates the surface expression and agonist sensitivity of the alpha 4beta 2 nicotinic acetylcholine receptor. J Biol Chem. 2002;277:41872–41878. doi: 10.1074/jbc.M206857200. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, van Aerde KI, Couey JJ, Brussaard AB. Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology Berl. 2005;184:1–14. doi: 10.1007/s00213-005-0070-z. [DOI] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloez-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP. Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature. 2005;436:103–107. doi: 10.1038/nature03694. [DOI] [PubMed] [Google Scholar]
- Morciano M, Burre J, Corvey C, Karas M, Zimmermann H, Volknandt W. Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem. 2005;95:1732–1745. doi: 10.1111/j.1471-4159.2005.03506.x. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional alpha4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakkanen JS, Jokitalo E, Tuominen RK. Up-regulation of beta2 and alpha7 subunit containing nicotinic acetylcholine receptors in mouse striatum at cellular level. Eur J Neurosci. 2005;21:2681–2691. doi: 10.1111/j.1460-9568.2005.04105.x. [DOI] [PubMed] [Google Scholar]
- Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Mol Pharmacol. 1994;46:523–530. [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Ren X-Q, Cheng S-B, Treuil MW, Mukherjee J, Rao J, Braunewell KH, Lindstrom JM, Anand R. Structural Determinants of α4β2 Nicotinic Acetylcholine Receptor Trafficking. J Neurosci. 2005;25:6676–6686. doi: 10.1523/JNEUROSCI.1079-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallette J, Pons S, Devillers-Thiery A, Soudant M, Prado de Carvalho L, Changeux JP, Corringer PJ. Nicotine upregulates its own receptors through enhanced intracellular maturation. Neuron. 2005;46:595–607. doi: 10.1016/j.neuron.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Samochocki M, Hoffle A, Fehrenbacher A, Jostock R, Ludwig J, Christner C, Radina M, Zerlin M, Ullmer C, Pereira EF, Lubbert H, Albuquerque EX, Maelicke A. Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J Pharmacol Exp Ther. 2003;305:1024–1036. doi: 10.1124/jpet.102.045773. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science. 1983;220:214–216. doi: 10.1126/science.6828889. [DOI] [PubMed] [Google Scholar]
- Spilker C, Dresbach T, Braunewell KH. Reversible translocation and activity-dependent localization of the Ca2+-myristoyl switch protein VILIP-1 to different membrane compartments in living hippocampal neurons. J Neurosci. 2002;22:7331–7339. doi: 10.1523/JNEUROSCI.22-17-07331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Vallejo YF, Buisson B, Bertrand D, Green WN. Chronic nicotine exposure upregulates nicotinic receptors by a novel mechanism. J Neurosci. 2005;25:5563–5572. doi: 10.1523/JNEUROSCI.5240-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Braunewell KH. Expression of the neuronal calcium sensor visinin-like protein-1 in the rat hippocampus. Neuroscience. 2008;153:1202–1212. doi: 10.1016/j.neuroscience.2007.10.067. [DOI] [PubMed] [Google Scholar]
- Zhao C, Anand R, Braunewell KH. Nicotine-induced Ca(2+)-myristoyl Switch of Neuronal Ca(2+) Sensor VILIP-1 in Hippocampal Neurons: A Possible Crosstalk Mechanism for Nicotinic Receptors. Cell Mol Neurobiol. 2008 Oct 17; doi: 10.1007/s10571-008-9320-z. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]