

Abstract
Wound healing is a crucial regenerative process in all organisms. We examined expression, integrity, and function of the proteins in the hepatocyte growth factor (HGF)/c-Met signaling pathway in normally healing and non-healing human skin wounds. Whereas in normally healing wounds phosphorylation of c-Met was most prominent in keratinocytes and dermal cells, in non-healing wounds phosphorylation of c-Met was barely detectable, suggesting reduced c-Met activation. In wound exudates obtained from non-healing, but not from healing wounds, HGF protein was a target of substantial proteolytic processing that was different from the classical activation by known serine proteases. Western blot analysis and protease inhibitor studies revealed that HGF is a target of neutrophil elastase and plasma kallikrein during skin repair. Proteolytic processing of HGF by each of these proteases significantly attenuated keratinocyte proliferation, wound closure capacity in vitro, and c-Met signal transduction. Our findings reveal a novel pathway of HGF processing during skin repair. Conditions in which proteases are imbalanced and tend toward increased proteolytic activity, as in chronic non-healing wounds, might therefore compromise HGF activity due to the inactivation of the HGF protein and/or the generation of HGF fragments that ultimately mediate a dominant negative effect and limit c-Met activation.
Tissue damage in skin induces a complex network of signaling systems such as growth factors, their receptors, extracellular matrix molecules, and different classes of proteases.1 The stringently regulated interaction of these mediators directs the restoration of the epidermis by epithelialization and of dermal structures by granulation tissue formation and matrix deposition. Many experimental studies have identified the essential functions of specific growth factors, their receptors and downstream signaling components in cutaneous repair2,3 and based on these studies much insight has been gained into human wound physiology. In contrast, mechanisms leading to impaired of healing are poorly understood and currently no efficient therapy is available for chronic wounds, which are often associated with diabetes or venous insufficiency.4 A better understanding of the pathogenic mechanisms leading to impaired healing is fundamental for the development of effective therapies for wound healing disorders.
Hepatocyte growth factor (HGF), also named scatter factor, is a pleiotropic growth factor that plays an essential role in cell growth, motility, and morphogenesis in different organ systems.5 Genetic deletion in mice demonstrated that HGF is critical for embryonic development.6,7 Comprehensive experimental evidence suggests that HGF is also a pivotal factor in postnatal processes such as cancer development, tissue repair and regeneration.5 HGF is a multidomain protein, synthesized and secreted as a biologically inert 90-kd monomeric precursor, that is converted into its bioactive form in the extracellular environment through a single cleavage at the Arg494-Val495 peptide bond by tightly regulated serine proteases (henceforth named classical HGF activation pathway). This proteolytic cleavage of HGF induces conformational changes to allow productive binding, dimerization, and activation of its receptor, c-Met. The mature HGF molecule is a heterodimer consisting of disulfide-linked α- and β-chains.8,9 The α-chain (60 kd) is composed of an N-terminal hairpin loop (N), homologous to the plasminogen activation peptide and four kringle domains (K1–K4), triple-looped cystein-rich motives involved in protein-protein interactions.8,9 The β-chain (36 kd) has strong homology to the protease domain of serine proteases, but it is devoid of any enzymatic activity. Several pro-HGF-converting enzymes and their inhibitors have been identified, including HGF activator,10 the blood coagulation factors XI11 and XII,12 the membrane-bound matriptase,13 as well as the urokinase-type plasminogen activator.14 A common feature of all known pro-HGF activators is the requirement for proteolytic conversion into an active enzyme, a process that is mediated by another set of proteases. Thus, the HGF/c-Met pathway is highly regulated by proteases and involves a complex network of activating enzyme and inhibitors. A more detailed characterization of protease activities and their impact on HGF function during tissue remodeling will contribute to a better understanding of the role of the HGF/c-Met axis in wound healing.
HGF mediates its biological functions on binding to its receptor c-Met, a tyrosine kinase encoded by the c-Met proto-oncogene.5 Due to the complex modular architecture of the HGF protein and different experimental designs, functional studies on ligand-receptor interactions have been complicated and results are contradictory. Progress in structure/function analysis of HGF has been achieved through protein engineering experiments and the determination of crystal structures for different domains.8 Earlier studies indicated that HGF mutants containing deletions of the N-, K1-kringle- or β-chain domains are biologically inactive, whereas mutants lacking the K2-, K3- or K4-kringle domains retain varying degrees of biological activity.15,16 These results have been partially supported by recent findings demonstrating that the K1-, K2- and K4-kringle domains of HGF are sufficient for c-Met activation, whereas the N- and β-chain domains are not essential, although the latter domains contribute additional binding sites necessary for receptor activation by full length HGF.8,9 A NK4 fragment composed of the first 447 residues of the α-chain has been shown to be a potent antagonist of c-Met-mediated HGF effects.17 Yet, the exact structural basis for c-Met activation remains incompletely understood.
HGF is widely expressed in different tissues, predominantly by mesenchymal cells, and acts as a paracrine effector on several cells of epithelial origin,18 on endothelial cells19 and on cells of the macrophage/monocyte lineage.20 Due to these properties, HGF has been implicated as a crucial molecule coordinating and facilitating cellular events in tissue repair, including re-epithelialization, angiogenesis, and granulation tissue formation. Circulating HGF levels rise markedly in several types of tissue injury, such as liver damage21 or arterial thrombosis.22 Neutralization of HGF in mice leads to retarded cutaneous healing associated with decreased neovascularization and granulation tissue formation.23 Consistently, vascularization and granulation tissue formation was increased in transgenic mice overexpressing HGF.24 A recent study demonstrated that keratinocytes deficient for c-Met were unable to contribute to the epithelialization of skin wounds and in conditional c-Met deficient mice wound closure was attenuated.25 Furthermore, diabetes impaired healing in mice was accelerated by topical application of recombinant human HGF (rhHGF), pointing to a unique potential of this molecule for the therapy of chronic non-healing wounds.23,26 To date, the impact of the HGF/c-Met pathway on human skin repair and its failure has not been comprehensively analyzed. Furthermore, detailed knowledge on the proteolytic processing of HGF at the wound site in skin repair is lacking.
Several classes of proteinases, as well as their inhibitors, have been implicated in the repair response in skin and genetic evidence obtained in mice indicates their crucial function.27 Cardinal functions of proteases in repair include regulation of cell motility, matrix remodeling, host defense, and modulation of cytokine activation. Dysregulation of wound proteases and their inhibitors is considered to be one of the major pathogenic mechanism underlying chronic non-healing wounds in man.4,28 The chronic non-healing wound environment of venous stasis ulcers, the most common cause of non-healing wounds at the lower leg in man, is the paradigm pathological condition were disintegrating inflammatory cell subsets release active peptidases and overwhelm local antipeptidase defenses. Substantial experimental evidence indicates that this process leads to uncontrolled destruction of factors promoting wound healing.29,30,31,32 However, detailed knowledge on the mechanisms by which uncontrolled proteases cause disturbed repair responses is lacking. Here we demonstrate for the first time that during skin repair HGF protein is a target of substantial proteolytic processing, which is different from the classical activation by known serine proteases and has major functional impact on the HGF/c-Met pathway in skin repair or its failure.
Materials and Methods
Wound Tissue
Biopsies were taken by consent from patients presenting chronic ulcera crura (n = 8) of more than 6 months duration, due to primary or secondary venous insufficiency (size of ulcer 10 cm2 to 30 cm2; mean age of patients 67 years; wounds showed no clinical sign of infection). The biopsies (spindle shaped 1 cm length × 0.3 cm width × 0.5 cm depth) were obtained from the wound edge of chronic wounds. Tissue of normally healing wounds (n = 9) (mean age of patients 56 years) was taken by consent from healthy volunteers. Wounds were created by performing a punch biopsy (0.6 cm diameter × 0.5 cm depth) at the lower back, and at indicated time points following wounding, the wound was excised. Wound tissues were embedded in OCT compound (Tissue Tek, Miles, IN), immediately frozen in liquid nitrogen, and stored at −80°C. The study adhered to the Declaration of Helsinki Principles and skin biopsies were collected according to a protocol approved by the ethics committee at the University of Cologne.
Wound Exudates
Wound exudate was obtained from patients presenting with ulcera crura due to venous insufficiency (n = 13; mean age of patients 67 years) or from patients with normally healing acute cutaneous wounds (n = 9, excision wounds of the lower leg awaiting wound closure by secondary intention; mean age of patients 65 years). For this purpose the wound was covered with a semipermeable polyurethane film (Hyalofilm, Hartmann, Heidelberg, Germany) for a maximum of 8 hours. Following collection (usually 1 ml was obtained), fluids were centrifuged (10 minutes, 13,000 × g, 4°C) to remove insoluble material, and supernatants were frozen at −80°C until use.
Enzyme-Linked Immunosorbent Assay
The concentration of HGF in wound exudates was determined using a commercially available enzyme-linked immunosorbent assay that detects human HGF (R&D Systems, Minneapolis, MN). HGF levels were normalized to the total protein concentration, which was determined by the Bradford procedure (Bio-Rad Protein Assay, Bio-Rad, München, Germany).
Detection of Plasma Kallikrein Activity
Activity of plasma kallikrein in wound exudates and blood serum was detected by processing of a plasma kallikrein-specific substrate, Chromozym PK (Roche Diagnostics, Indianapolis, IN). Wound exudates and blood serum were diluted 10-fold with reaction buffer (Tris/imidazol buffer; 15 mmol/L, pH 7.9) and incubated with Chromozym PK in a 96-well plate at room temperature. Chromozyme PK is cleaved by plasma kallikrein into a residual peptide and free 4-nitraniline, which was measured at 405 nm, following a 5-minute incubation time.
Immunohistochemistry
To process tissue sections for the immunodetection of HGF or c-Met (non-healing wounds, n = 8; healing wounds, n = 3 per time point: day 1, day 8, day 14 after injury; non-wounded skin, n = 4) 5 μm of cryosection was fixed in 4% PFA, rinsed, and then blocked with 10% fetal calf serum/phosphate-buffered saline to reduce nonspecific antibody binding. Sections were incubated (1 hour at room temperature) with polyclonal rabbit antibody against HGF (raised against a recombinant protein corresponding to amino acids 32–176 mapping near the amino terminus of HGF α-chain) (1:100, Santa Cruz Biotechnology, Santa Cruz, CA) or with polyclonal rabbit antibody against c-Met (raised against a peptide mapping within a C-terminal cytoplasmic domain of Met) (1:50, Santa Cruz Biotechnology, Santa Cruz, CA) or with polyclonal rabbit antibody against phospho-c-Met (raised against a synthetic phosphopeptide corresponding to residues surrounding Y1234/Y1235) (1:50, R&D Systems, Minneapolis, MN). Macrophages were stained using a monoclonal mouse antibody against CD68 (Dianova, Augst, Switzerland). Bound primary antibodies were detected using an horseradish peroxidase-conjugated secondary antibody against rabbit IgG (1 hour at room temperature) (Dako Cytomation EnVision+System-HRP, Denmark), an Alexa 488-conjugated antibody against mouse IgG or an Alexa 594-conjugated antibody against rabbit IgG (Molecular Probes, Cambridge, UK). Hemalaun was used for counterstaining. Specificity of primary antibodies was demonstrated by omitting these or replacing them by an irrelevant isotype-matched rabbit or mouse antibody.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
SDS-PAGE was performed following the protocol of Laemmli. To analyze processing of HGF by plasma kallikrein or neutrophil elastase, recombinant human HGF (rhHGF) protein produced in a baculovirus/insect cell system (R&D Systems, Minneapolis, MN) was incubated at 37°C with plasma kallikrein (160 nmol/L, time as indicated) or neutrophil elastase (34 nmol/L, 2 hours) (Calbiochem, Germany) and reaction buffer (20 mmol/L Hepes, 150 mmol/L NaCl, 5 mmol/L CaCl2, pH 7.5). rhHGF was biotinylated (rhHGFbiotin) using sulfo-N-hydroxy succinimido-biotin-long chain (Pierce Chemical Co, Rockford, IL) for 30 minutes at room temperature and the reaction mixture was dialyzed against phosphate-buffered saline overnight at 4°C to eliminate free biotin; processing of biotinylated HGF was performed as outlined above. To analyze stability of rhHGF or rhHGFbiotin spiked into wound exudate, rhHGF (2.5 ng) or rhHGFbiotin (100 ng), wound exudate (3 μl) and reaction buffer were incubated at 37°C in a total volume of 30 μl. At indicated time points, reactions were terminated by the addition of Pefabloc (Roche Diagnostics, Indianapolis, IN). To identify HGF-degrading enzymes, protease inhibitor studies were performed using neutrophil elastase inhibitor IV (200 μmol/L) (Calbiochem, Germany),33 Pefabloc (5 mmol/L) (Roche Diagnostics, Indianapolis, IN), and EDTA (5 mmol/L) (Merck, Darmstadt, Germany). Wound exudates or protease solutions were incubated with protease inhibitors for 30 minutes at room temperature before adding rhHGF or rhHGFbiotin. Protease processed HGF and wound exudates were resolved on 10% reducing and non-reducing SDS-PAGE gels and transferred to nitrocellulose (Hybond C-super, Amersham Bioscience Europe GmbH, Freiburg, Germany). Integrity of HGF was determined by detecting immunoreactive products with polyclonal goat antibody against HGF (raised against rhHGF) (1:1000, R&D Systems, Minneapolis, MN) or with a streptavidin-horseradish peroxidase conjugate (1:1000, GE Health Care Lifesciences Europe). Detection was accomplished using the enhanced chemiluminescence Western blot detection system (ECL, Amersham Bioscience Europe GmbH).
Cell Culture
Primary human keratinocytes derived from neonatal foreskin were isolated and cultured following the method described earlier.34 Briefly, to initiate primary cultures, keratinocytes were cocultivated with 3T3-J2 mouse fibroblasts, pretreated with mitomycin C (Sigma, Germany). Keratinocyte growth medium was changed every 3–4 days with a 3:1 mixture of Dulbecco’s modified Eagle’s medium (Gibco, Karlsruhe, Germany) and Ham’s F12 (Gibco, Karlsruhe, Germany), supplements were added as previously described.34 Cells were subcultured by first removing the feeder layer cells with a brief EDTA wash and then treating keratinocytes with trypsin-EDTA. Madin-Darby canine kidney cells were cultured in Dulbecco’s modified Eagle’s medium (low glucose) (Gibco, Karlsruhe, Germany) containing 10% fetal calf serum (Perbio, Bonn, Germany) and penicillin/streptomycin (Biochrom AG, Berlin, Germany).
In Vitro Cell Culture Assays
Mitogenic Assay
Primary human keratinocytes (5 × 104 per well) were plated onto an eight-well chamber slide (Nalge Nunc International, NY) in keratinocyte growth medium, or keratinocyte starvation medium (3:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12, 1% fetal calf serum) supplemented with classically activated rhHGF (100 ng/ml), plasma kallikrein-digested rhHGF (100 ng/ml HGF in plasma kallikrein 160 nmol/L for 4 hours at 37°C), neutrophil elastase-digested rhHGF (100 ng/ml HGF in neutrophil elastase, 34 nmol/L at 37°C for 2 hours), plasma kallikrein (160 nmol/L) or neutrophil elastase (34 nmol/L). Simultaneously 5-bromo-2′-deoxyuridine (1 μmol/L) (Roche Diagnostics, Indianapolis, IN) was added to the cells and 5-bromo-2′-deoxyuridine incorporation was allowed to proceed for 30 minutes. At 37°C. Cells were fixed (30 minutes. with 70% ethanol) and 5-bromo-2′-deoxyuridine incorporation was visualized with a primary antibody against 5-bromo-2′-deoxyuridine (1:5, Becton Dickinson, Belgium) (45 minutes, room temperature). Detection of the primary antibody was performed using an Alexa 488-conjugated antibody against mouse IgG1 (1:1000, Molecular Probes, UK); propidium iodide (1:1000) was used as counterstain. 5-Bromo-2′-deoxyuridine positive cells were counted and expressed as percentage of the total cell number in the chamber (minimum 300 cells). Experiments were performed in duplicates and the analysis was repeated five times with keratinocytes isolated from five different donors. Pictures were taken with a Nikon Eclipse E800 microscope and images were analyzed with ImageJ (Rawak Software).
Wound Healing Assay
Primary human keratinocytes (106) were plated in a 6-well plate and grown to confluence in keratinocyte growth medium. Before scratching cells were starved for 24 hours in starvation medium (3:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F12, 1% fetal calf serum) and treated with mitomycin C (0.4 ng/ml, 2 hours, 37°C) (Sigma, Germany). Cell layers were scratched using a Pasteur pipette tip and cultured in keratinocyte starvation medium (negative control), 10% fetal calf serum (positive control), or starvation medium supplemented with classically activated rhHGF (100 ng/ml), plasma kallikrein-digested rhHGF (100 ng/ml HGF in plasma kallikrein 160 nmol/L for 4 hours at 37°C), neutrophil elastase-digested rhHGF (100 ng/ml HGF in neutrophil elastase 34 nmol/L at 37°C for 2 hours), plasma kallikrein (160 nmol/L) or neutrophil elastase (34 nmol/L). Closure of the scratch wound by migrating keratinocytes was recorded every 10 minutes with an Olympus microscope XI 81 (Olympus, Germany) and photographs edited with Adobe Photoshop CS2. To calculate keratinocyte migration at indicated time points after scratch wounding, the area of the scratch covered by cells was determined and expressed as percentage of the cell-covered scratch area observed with classically activated HGF. The analysis was repeated five times with keratinocytes isolated from five different donors. For cell migration analysis the cellR software (Olympus, Germany) was used.
Scatter Assay
Scatter assays were performed as previously described.35 Briefly, Madin-Darby canine kidney cells (100 cells/well) were plated in a 96-well plate and cultured in keratinocyte starvation medium for 24 hours. Cells were treated with keratinocyte starvation medium supplemented with classically activated rhHGF (100 ng/ml), plasma kallikrein-digested rhHGF (100 ng/ml HGF in plasma kallikrein 160 nmol/L for 4 hours at 37°C) or plasma kallikrein (160 nmol/L). Cell scatter activity was recorded every ten minutes with an Olympus microscope XI 81 (Olympus, Germany) and photographs edited with Adobe Photoshop CS2.
Signaling in Keratinocytes
Primary human keratinocytes were cultured and stimulated for indicated time periods with processed rhHGF as outlined above. Activation of c-Met was assessed by Western blot analysis (performed as outlined above) for phospho Erk1/2, total Erk1/2, phospho Akt and total Akt (Cell Signaling Technology, Inc.).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism5 (GraphPad Software, Inc., San Diego, CA). Significance of difference was analyzed using the unpaired student t-test. All data are presented as mean ± SD. P ≤ 0.05 was considered significant.
Results
Expression of HGF and c-Met during Wound Healing in Skin
To assess the distribution of HGF and c-Met protein during wound repair in skin, we performed immunohistochemistry studies on cryosections obtained from normally healing or non-healing human skin wounds caused by venous insufficiency. Whereas in non-wounded normal skin c-Met and HGF were not detectable, c-Met and HGF expression was strongly up-regulated in both healing and non-healing wound tissue (Figure 1). In normally healing wounds already at day 1 after wounding basal keratinocytes of the wound edge stained positive for c-Met (Figure 1, a and b). As the healing response advanced c-Met expression was also present in suprabasal layers of hyperplastic epidermal wound edges (Figure 1, c and d) and in small vessels (Figure 1e) and mononuclear infiltrating cells (Figure 1f) within the papillary dermis and the granulation tissue (Figure 1d). HGF expression was visible in mononuclear (Figure 1h) and spindle shaped cells within the granulation tissue, identified as fibroblasts by light microscopic criteria (Figure 1i), as well as less pronounced in the hyperproliferative epithelium of wound margins (Figure 1g). In non-healing chronic skin wounds c-Met expression was strongly up-regulated in basal and suprabasal layers of the hyperplastic epidermis, as well as in small vessels and in mononuclear and spindle shaped cells within the papillary dermis in close vicinity to the lesional epithelium at the wound edges (Figure 1, j and k). In contrast, dermal tissue in the center of the ulcer, not covered by epithelium, revealed only weak staining for c-Met. HGF staining in chronic non-healing wounds was predominantly localized in the papillary dermis at wound edges (Figure 1m) as well as in cell clusters within the reticular dermis in the center of the ulcer (indicated by arrowheads, Figure 1l). As HGF is a secreted molecule, HGF immunohistochemical staining may not exclusively reflect newly expressed HGF, but also HGF protein bound by c-Met to cell surfaces and/or extracellular matrix molecules.36
Figure 1.

Expression of HGF and c-Met is up-regulated in healing and non-healing human wounds. A: Scheme of a wound section several days following injury. Keratinocytes at the wound margin proliferate and migrate on the provisional dermal wound matrix and form the hyperproliferative epithelium. Analysis of c-Met (B-F, J,K) and HGF (G-I, L, M) expression by immunohistochemistry on cryosections of healing (B-I) and non-healing (J-M) human wounds several days after injury as indicated; brown staining indicates c-Met or HGF positive cells. Note that in early healing wounds (day 1 post injury, B) c-Met is strongly expressed in basal keratinocytes at the wound edge, and as the healing response advances to day 8 (C) and day 14 (D) after injury) also in suprabasal layers of the hyperplastic epithelium and in small vessels E and mononuclear cells (F, arrows) within the papillary dermis and granulation tissue. E and F: Magnifications of D; inset in C shows membrane staining of c-Met in keratinocytes within the HE. HGF is predominantly expressed in mononuclear cells (H, arrows) and spindle shaped cells (I, arrows) within the dermal compartment of healing (G-I) and non-healing (J-M) wounds. K and M are magnifications of J and L, respectively. Note that in J, dark brown color within the reticular dermis reveals fibrotic tissue and not specific c-Met staining (arrowheads). HE, hyperproliferative epithelium; e, epidermis; d, dermis; gr, granulation tissue; es, eschar. Arrows in A-C, G, J and L indicate wound margin.
Stability of HGF Protein in Healing and Non-Healing Wounds
Endogenous HGF protein in normally healing or chronic non-healing wounds was quantified in wound exudates by enzyme-linked immunosorbent assay. Wound exudate is the extracellular fluid of wounded tissue and contains numerous soluble mediators, extracellular matrix molecules, proteases and degradation products synthesized by the wounded tissue. Therefore, wound exudate reflects in part molecular and cellular processes within the microenvironment of the wound tissue. Cells at the wound surface are in direct contact with the wound exudate and the fluid is very likely to control their function. HGF protein concentration was significantly increased in wound exudates obtained from healing (P < 0.01) or non-healing (P < 0.003) wounds when compared with blood serum, in which HGF was not detected (detection limit <40 pg/ml) (Figure 2). These findings demonstrate the local synthesis and release of HGF into the wound environment. The mean HGF protein levels were significantly increased in non-healing wounds (n = 13) when compared with normally healing wounds (n = 8) (P < 0.004), reaching a mean concentration of 140 ng/ml and 77 ng/ml, respectively.
Figure 2.
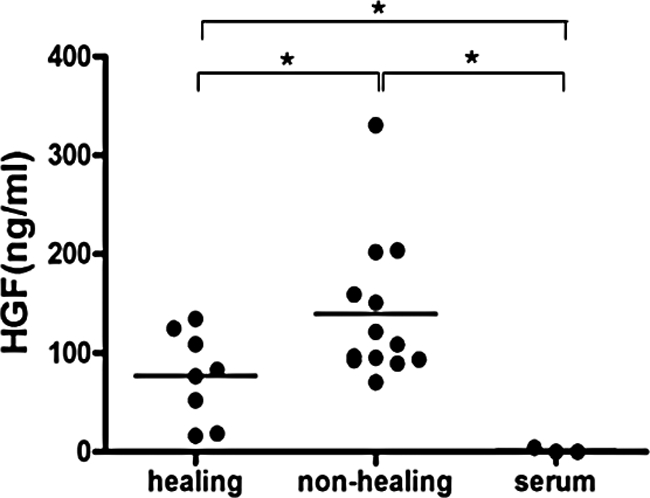
HGF protein levels in wound exudates of healing and non-healing human wounds. Enzyme-linked immunosorbent assay was used for quantification of HGF protein in wound exudates obtained from healing (n = 8) and non-healing (n = 13) human wounds and blood serum (n = 3); healing versus non-healing *P < 0.004; healing versus serum *P < 0.01; non-healing versus serum *P < 0.003.
To assess the integrity of the endogenous HGF protein during repair, wound exudates derived from healing (n = 5) or non-healing wounds (n = 7) were subjected to reducing and non-reducing SDS-PAGE and HGF Western blot analysis. To assess the sensitivity of this analysis we first performed Western blot analysis of recombinant human HGF (rhHGF) only. Western blot analysis revealed recombinant human HGF (rhHGF) protein in reducing SDS-PAGE analysis as three immunoreactive bands which migrated with a molecular weight of 90 kd, 60 kd, and 36 kd, representing the single chain proform, the α-chain and the β-chain, respectively (Figure 3A, left lane). This finding indicates that rhHGF protein used in this study was already partially activated following the classical activation pathway. In non-reducing SDS-PAGE analysis rhHGF heterodimer and monomer (pro-HGF) migrated with a molecular weight of approximately 75 kd (Figure 3A, right lane). Furthermore, these studies revealed a limited sensitivity of the Western blot under non-reducing conditions. In fact, due to a low concentration of endogenous HGF in wound exudates and limits of the sensitivity of western blotting under non-reducing conditions, integrity of endogenous HGF could not be assessed under these conditions. Therefore, rhHGF was spiked into wound exudates obtained from healing or non-healing wounds for defined time periods and its integrity assessed by non-reducing Western blot analysis. Incubation of rhHGF with wound exudate obtained from non-healing wounds, but not from healing wounds, resulted in rapid proteolytic degradation of HGF (Figure 3B). Two hours incubation resulted in a prominent immunoreactive product, which migrated with an approximate molecular weight of 45 kd (Figure 3B). This band had exactly the same electrophoretic mobility as a proteolytic product obtained following incubation of rhHGF protein with neutrophil elastase. In accordance with previous findings,37 incubation of rhHGF with neutrophil elastase and non-reducing SDS-PAGE analysis, resulted in the generation of a 45-kd band consistent with the α-chain and an approximately 30-kd band consistent with the α/β remnant (Figure 3B). In none of the wound exudates analyzed an immunoreactive band was detected that was consistent with the α/β remnant. Direct evidence for a role of neutrophil elastase in HGF cleavage was provided by partial rescue of HGF degradation by preincubation of the wound exudate with a specific neutrophil elastase inhibitor (Figure 3C). In addition to neutrophil elastase, other serine proteases might be involved in rhHGF degradation, because Pefabloc (inhibitor of a broad spectrum of serine proteases) resulted in a more effective protection of rhHGF from proteolytic degradation (Figure 3D). In contrast EDTA, a classical inhibitor of matrix metalloproteinases, did not protect rhHGF from degradation in wound exudate obtained from non-healing wounds (Figure 3D), suggesting that matrix metalloproteinases are not significantly involved in this process. To substantiate our findings on neutrophil elastase-mediated processing of HGF in wound exudate obtained from non-healing wounds, we assessed HGF proteolysis by a technique independent of HGF immunoreactivity, ie, detection of biotinylated HGF (HGFbiotin) by streptavidin. These studies revealed HGFbiotin fragments in wound exudates obtained from non-healing wounds consistent with cleavage by neutrophil elastase (Figure 3E). Furthermore, proteolysis of HGFbiotin in wound exudates obtained from healing wounds was minimal and thereby confirmed the HGF-specific Western blot analysis.
Figure 3.

The immunoreactive 45-kd HGF fragment present in wound exudate analyzed under non-reducing conditions corresponds to the 45-kd fragment obtained by neutrophil elastase processing of rhHGF. A: rhHGF protein (5 ng, left lane, reducing conditions; 20 ng, right lane, nonreducing conditions). B: rhHGF (10 ng) was incubated with neutrophil elastase (NE) or wound exudate obtained from non-healing (wound 4) or healing (wound 5) wounds for increasing time periods as indicated. C: rhHGF (10 ng), neutrophil elastase, neutrophil elastase inhibitor (NEI), and wound exudate obtained from non-healing (wound 4) wounds were incubated for increasing time periods as indicated. D: rhHGF (10 ng) was incubated with neutrophil elastase (NE) and neutrophil elastase inhibitor (NEI) or wound exudate obtained from non-healing wounds (wound 4) preincubated with NEI, Pefabloc (Pefa), or EDTA (5 mmol/L) for 2 hours. Samples were subjected to non-reducing SDS-PAGE analysis and integrity of HGF protein was determined by detecting immunoreactive products with a HGF-specific antibody. E: Biotinylated HGF (HGFbiotin) was incubated with neutrophil elastase or wound exudate obtained from non-healing (wound 4) or healing (wound 3) wounds for increasing time periods as indicated. Samples were subjected to non-reducing SDS-PAGE analysis and HGF fragments were detected by a streptavidin-horseradish peroxidase conjugate.
Increased sensitivity of western-blot analysis under reducing conditions enabled us to assess the integrity of endogenous HGF protein in wound exudates. Reducing SDS-PAGE analysis of wound exudates revealed a prominent immunoreactive product, which migrated above 50 kd and below 60 kd, in wound exudates obtained both from healing and non-healing wounds (Figure 4A). In none of the wound exudates analyzed an immunoreactive band was detected that was consistent with the β-chain. Interestingly, the 50- to 60-kd band detected in wound exudates under reducing conditions had exactly the same electrophoretic mobility as a proteolytic product obtained following incubation of rhHGF protein with plasma kallikrein (Figure 4B). As recently demonstrated by Peek and colleagues,11 limited digestion of rhHGF for 4 hours with plasma kallikrein produced the canonical α-chain and in addition a second α-chain fragment (α2), whose apparent molecular mass of 54 kd was about 6 kd lower than the canonical α-chain (Figure 4C). Extended plasma kallikrein processing of rhHGF for 8 hours did not lead to a more complete cleavage of the canonical α- or α2-chain, but rather resulted in the complete digestion of the β-chain (Figure 4C). Incubation of rhHGF protein for 8 hours at 37°C without any proteases did not affect rhHGF stability (Figure 4C). To analyze whether in healing wounds also the canonical HGF activation process occurs, we spiked rhHGF (Figure 4D) or HGFbiotin (Figure 4E) in wound exudates and analyzed its proteolytic processing. The kinetics of HGF proteolysis in both approaches revealed initially a shift of the pro-HGF precursor (90 kd) to a pronounced α- (60 kd) and weaker β-chain (36 kd), demonstrating canonical activation. In addition, with increasing time of incubation also an α2-chain (54 kd) became apparent, indicating alternative plasma kallikrein-mediated cleavage (Figure 4, D and E).
Figure 4.

The immunoreactive 54-kd HGF fragment present in wound exudate analyzed under reducing conditions corresponds to the 54-kd fragment (α2-chain) obtained by plasma kallikrein processing of rhHGF. A: Wound exudates (dilution 1:10 or 1:5 in sample buffer) obtained from healing (wound 3) or non-healing (wounds 1 and 2) wounds were subjected to reducing SDS-PAGE analysis; integrity of HGF protein was determined by detecting immunoreactive products with a HGF-specific antibody. B: rhHGF protein (5 ng) (lanes 1, 4, 7), rhHGF protein (5 ng) incubated with plasma kallikrein (160 nmol/L) for 4 hours (lanes 2 and 5) or wound exudate obtained from non-healing (lane 3, wound 1, or lane 6, wound 2) or healing (lane 8, wound 3) wounds were subjected to reducing SDS-PAGE analysis and HGF immunodetection. C: rhHGF protein (5 ng) was incubated with plasma kallikrein for increasing time periods as indicated; to demonstrate that HGF is not subjected to degradation by heat, HGF protein was incubated for 8 hours at 37°C without plasma kallikrein (right lane). D: rhHGF (5 ng) was incubated with wound exudate obtained from healing wound (wound 5) for increasing time periods as indicated. HGF degradation was monitored by SDS-PAGE under reducing conditions (pKallikrein, plasma kallikrein). E: Biotinylated HGF (HGFbiotin) was incubated with plasma kallikrein (pK) or wound exudate obtained from non-healing (wound 4) or healing (wound 3) wounds for increasing time periods as indicated. Samples were subjected to non-reducing SDS-PAGE analysis and HGF fragments were detected by a streptavidin-horseradish peroxidase conjugate.
Plasma Kallikrein Activity in Wound Exudates Obtained from Healing or Non-Healing Wounds
We and many others have determined activity levels of various classes of proteases during skin repair in humans. In those studies the non-healing condition has been associated with significantly increased protease activity levels when compared with the healing condition, including the activity of neutrophil elastase and mast cell chymase.29,30,31,38,39,40 In contrast, the activity of plasma kallikrein during skin repair in the murine or human system has not been reported. To assess the activity in wound exudates obtained from healing or non-healing wounds, we quantified plasma kallikrein activity by a specific chromogenic assay. Plasma kallikrein activity was present in wound exudates obtained from healing and non-healing wounds. In normally healing wounds, the activity was significantly increased during the early phase of repair (up to 4 days after injury) when compared with activity levels in blood serum (P < 0.001, early healing versus blood serum). However, activity levels decreased as the healing response advanced until day 25 after wounding, down to levels detected in blood serum (Figure 5). These results revealed a transient increase in plasma kallikrein activity during the early phase of normal repair indicative of a local down-regulation of plasma kallikrein activity as the healing response advances. In wound exudates obtained from non-healing wounds plasma kallikrein activity levels were significantly increased when compared with activity levels in blood serum (P < 0.05) and were similar to those of healing wounds during the early phase of repair (Figure 5).
Figure 5.
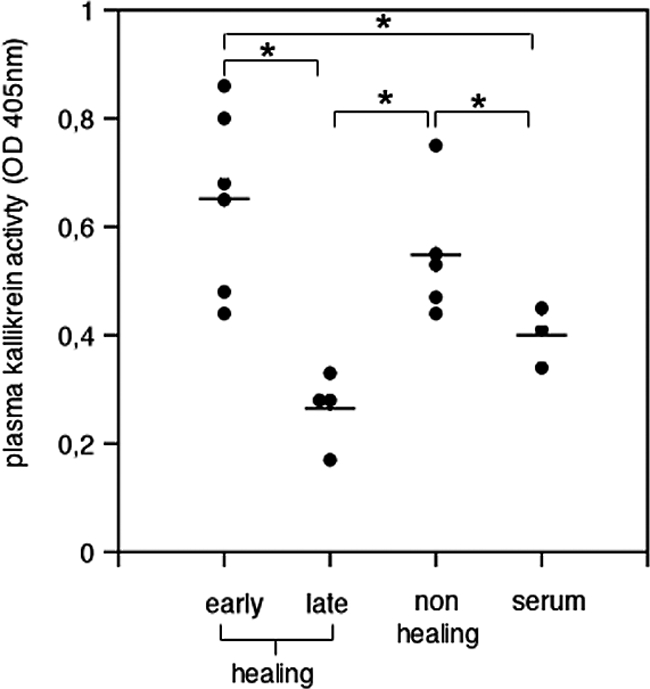
Plasma kallikrein activity in wound exudates of healing and non-healing human wounds. Plasma kallikrein activity was quantified in wound exudates obtained from normally healing wounds collected during the early (days 1–4 after injury) or late (days 8–25 after injury) phase of healing and from non-healing wounds as well as in blood serum of wounded patients; each dot represents plasma kallikrein activity in wound exudate or serum obtained from a different patient. Early healing versus blood serum, *P < 0.04; non-healing versus blood serum, *P < 0.05; early phase healing versus late phase healing, *P < 0.001; non-healing versus late phase healing, *P < 0.002.
Neutrophil Elastase or Plasma Kallikrein-Mediated Cleavage of rhHGF Attenuates its Mitogenic and Wound Closure Capacity
To assess functional consequences of alternative proteolytic processing of rhHGF, we exposed primary human keratinocytes either to neutrophil elastase- or plasma kallikrein-processed rhHGF or classically activated rhHGF (as provided by the manufacturer) and compared cell proliferation and closure of scratch wounds. To generate neutrophil elastase- or plasma kallikrein-processed rhHGF, enzymes and HGF were incubated for 2 and 4 hours at 37°C, respectively. Under these conditions the entire pro-HGF monomer and active heterodimer were subject to cleavage by neutrophil elastase (Figure 3). On plasma kallikrein-mediated processing, the entire pro-HGF monomer was subject to activation, resulting in the classically activated α/β-heterodimer, which to a major extent was additionally cleaved within the K4 domain (Figure 4C). Thus, cells exposed to plasma kallikrein-digested rhHGF were exposed to a mixture, consisting of classically activated α/β-heterodimer and classically activated α/β-heterodimer with additional cleavage within the K4 domain (referred to as alternatively or plasma kallikrein-activated rhHGF).
To quantify the mitogenic response of primary human keratinocytes to neutrophil elastase-, plasma kallikrein- or classically activated rhHGF, 5-bromo-2′-deoxyuridine incorporation was analyzed. The mitogenic activity of cells stimulated with neutrophil elastase- or plasma kallikrein-activated rhHGF was significantly attenuated when compared with exclusively classically activated rhHGF (Figure 6, A and B; P < 0.03 and P < 0.04, respectively). Furthermore, to examine the capacity of primary keratinocytes to close an epithelial wound, cell culture monolayers were scratch-wounded. In the presence of neutrophil elastase- or plasma kallikrein-activated HGF, scratch-wound closure was significantly delayed when compared with exclusively classically activated rhHGF (Figure 7, A and B). Scatter activity on Madin-Darby canine kidney cells was not affected in response to alternatively-activated rhHGF when compared with classically activated rhHGF (data not shown).
Figure 6.
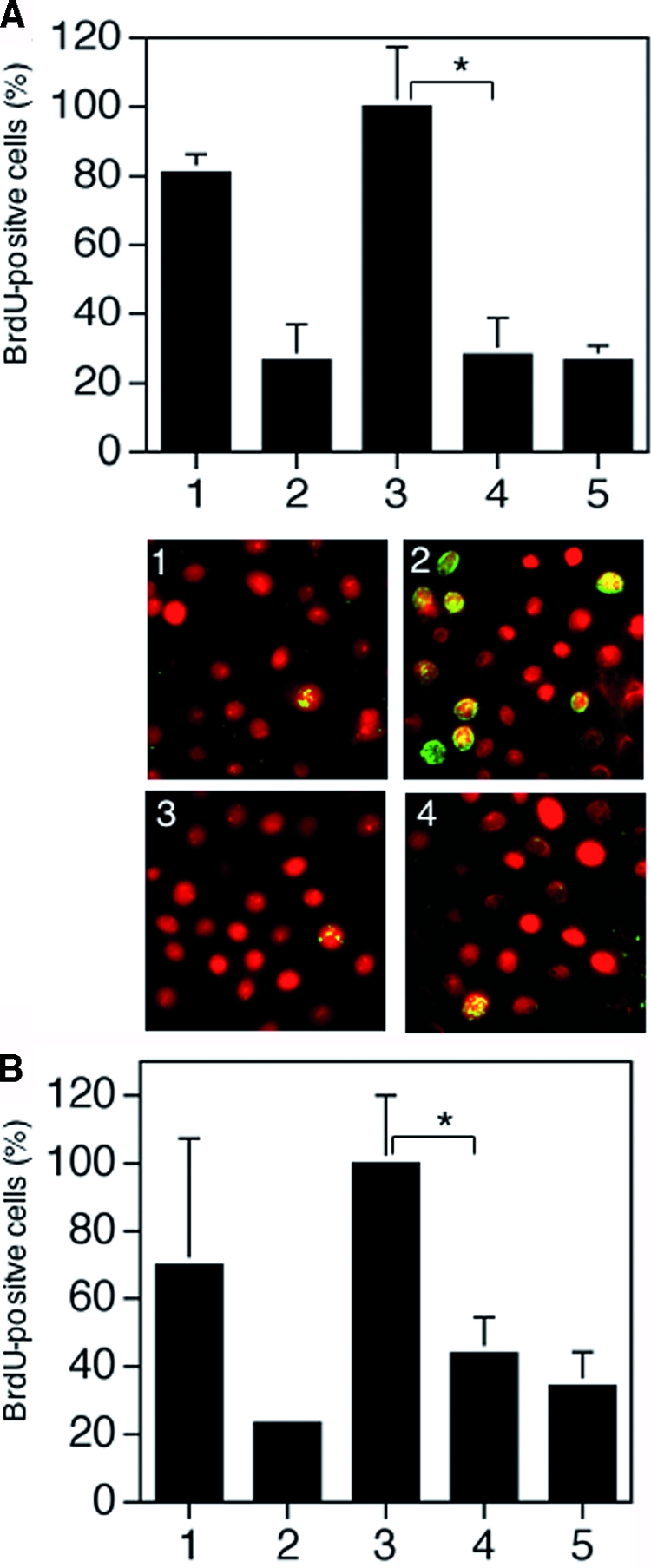
Alternatively mediated cleavage of rhHGF attenuates its mitogenic activity in human keratinocytes. Following starvation primary human keratinocytes were exposed as shown in A to growth medium (positive control) (1), starvation medium (negative control) (2), classically activated rhHGF (3), neutrophil elastase-digested rhHGF (4), or neutrophil elastase in keratinocyte starvation medium (5); or as shown in B to growth medium (positive control) (1), starvation medium (negative control) (2), classically activated rhHGF (3), plasma kallikrein-digested rhHGF (4), or plasma kallikrein in keratinocyte starvation medium (5). Following 30 minutes of stimulation, DNA synthesis was detected by 5-bromo-2′-deoxyuridine incorporation visualized by immunofluorescent staining (green nuclei) (shown only for NE digestion); cells were counterstained by propidium iodide. The experiments were performed in duplicate, and P is calculated from five independent experiments including data derived from keratinocytes isolated from five different donors; *P < 0.04.
Figure 7.

Alternatively mediated cleavage of rhHGF attenuates its wound closure capacity. Primary human keratinocytes were scratch-wounded and exposed as shown in A to starvation medium (1), classically activated rhHGF (2), neutrophil elastase-digested rhHGF (3), or neutrophil elastase in starvation medium (4); or as shown in B to growth medium (positive control) (1), starvation medium (2), classically activated rhHGF (100 ng/ml) (3), plasma kallikrein-digested rhHGF (4), or plasma kallikrein in starvation medium (5). 550 minutes (A) or 300 minutes (B) after scratch-wounding the area of the scratch covered by cells was determined and expressed as percentage of the cell covered scratch area observed in classically activated HGF conditions. P is calculated from five independent experiments including data derived from keratinocytes isolated from five different donors; *P < 0.01.
In addition, we also used primary human keratinocytes stimulated by neutrophil elastase-processed HGF to study signal transduction by proteins that are crucial for cell proliferation and cell migration. Erk 1/2 and Akt were phosphorylated, and thus activated, in control cells stimulated with classically activated rhHGF (Figure 8). In contrast, phosphorylation of these proteins in keratinocytes stimulated with neutrophil elastase-processed HGF was significantly attenuated. Taken together, these findings demonstrate that neutrophil elastase- or plasma kallikrein-processing of HGF significantly impairs mitogenic and wound closure capacities of primary human keratinocytes in vitro.
Figure 8.

Signaling is attenuated in keratinocytes stimulated with rhHGF processed by neutrophil elastase. Western blot analysis of phospho Erk1/2, total Erk1/2, phospho-Akt, and total Akt in keratinocytes stimulated with classically activated rhHGF or neutrophil elastase-processed HGF.
C-Met Phosphorylation in Wound Repair
To assess the activation of c-Met in wounded tissue we performed immunohistochemical analysis of wound cryosections using anti-phospho-c-Met antibodies. In healing wounds phosphorylation of c-Met was evident in the hyperproliferative epidermal wound edge and papillary wound dermis (Figure 9a). In contrast, in non-healing wounds, the hyperproliferative epithelium did not stain for c-Met phosphorylation, occasionally some dermal cells revealed weak staining (Figure 9b). In healing and non-healing wounds the dermal cells that stained positive for c-Met-phosphorylation were predominantly macrophages as identified by CD68 staining (Figure 9, c and d; see Supplemental Figure S1 at http://ajp.amjpathol.org). These results indicate attenuated phosphorylation and activation of c-Met in non-healing wounds.
Figure 9.

c-Met phosphorylation in wound repair. Immunohistological analysis of cryosections from healing (A, C, D, day 8 after wounding) and non-healing (B) wounds using an anti-phospho-c-Met antibody; brown staining indicates phospho-c-Met positive cells. Note that healing wounds show phosphorylation of c-Met in the hyperproliferative epidermal (HE) wound edge and papillary dermis, whereas non-healing wounds show staining exclusively in dermal cells. Arrows in (C) indicate cells positive for c-Met phosphorylation. D: Predominantly macrophages (CD68) (indicated by arrowheads) display phosphorylation of c-Met. e, epidermis; d, dermis.
Discussion
We have demonstrated that c-Met and HGF expression is elevated during tissue repair in normally healing skin wounds and chronic venous stasis ulcers. Both healing and non-healing wounds reveal a similar cellular expression pattern for c-Met and HGF within the epidermal and dermal cell compartment. Our data suggest that in cutaneous tissue repair the HGF/c-Met system acts primarily in a paracrine fashion and/or additionally through an autocrine loop, as has been suggested for other epithelial-mesenchymal tissues41,42 and recently in cutaneous wound healing in mice.25
In addition to a positive regulation of HGF expression at the transcriptional level, regulation of HGF activity following skin injury is probably controlled through extracellular proteolytic activation by proteases present at the wound site. Indeed, the latter event is considered the rate-limiting step for HGF/c-Met signaling in vivo and is thought to be a mechanism to localize HGF activities to injured tissues. Therefore, we assessed the integrity of the HGF protein in wound exudate, which reflects proteolytic processing of HGF protein within the wound microenvironment. Several pro-HGF activating enzymes have been identified by in vitro analysis, including HGF activator, urokinase-type plasminogen activator, matriptase and several blood clotting factors.11,13,14 Several of these proteases have been implicated in the cutaneous healing response and kinetics of rhHGF processing in wound exudate obtained from healing wounds supports their participation in classical pro-HGF conversion at the normally healing wound site43 (Figure 10). More important, our data strongly suggest that during skin repair the pro-HGF monomer and/or the classical HGF heterodimer is subjected to proteolytic pathways that are different to the classical pathway mediated by known serine proteases. SDS-PAGE and Western blot analysis for HGF of wound exudates obtained from healing or non-healing human wounds revealed three major facts: First, non-reducing SDS-PAGE and Western blot analysis of rhHGF incubated in wound exudate of non-healing wounds, but not of healing wounds, revealed a prominent immunoreactive 45-kd protein band that had the same electrophoretic mobility as a proteolytic product obtained following incubation of HGF with neutrophil elastase. This fragment was previously shown to antagonize c-Met activation.37 Second, under reducing conditions the endogenous HGF form present in wound exudates obtained from healing or non-healing wounds appeared as a prominent immunoreactive band of approximately 54 kd. This band had exactly the same electrophoretic mobility as a proteolytic product obtained following incubation of HGF with plasma kallikrein, which was composed of the N-terminal residues of the α-chain. Third, a HGF fragment representing the β-chain and/or a α/β remnant was barely detectable in all exudate samples, under either reducing or non-reducing conditions. Together, these findings demonstrate that during skin repair the pro-HGF single chain and/or the classical heterodimer are subjected to enzymatic cleavage that is different from the classical proteolytic activation pathway, mediated by known serine proteases.10,11,12,13,14
Figure 10.

Model for proteolytic processing of HGF during human wound repair. This diagram presents the domain organization of HGF and proposed pathways of proteolytic processing during skin repair. The major structural motifs include N-terminal hairpin loop (N), kringle (K1–K4), and serine peptidase (SP) domains. Classical activation (1): Pro-HGF is activated by hydrolysis of position Arg494-Val495 by several serine proteases, resulting in the bioactive disulfide-linked α/β-heterodimer. Alternative processing (2): hydrolysis by plasma kallikrein (pKallikrein) occurs in addition within the K4-domain at position Arg424-His425, resulting in a cleaved K4 domain, which is held together by the disulfide bond network in the K4 domain11 (3); hydrolysis by neutrophil elastase or chymase at vulnerable sites within an “inactivation segment”37 occurs at positions Val478-Asn479 or Leu480-Asp481 as indicated, resulting in a NK4-like fragment antagonizing actions of HGF and an α/β remnant. In the highly proteolytic microenvironment of non-healing wounds, HGF might be a target of several proteases, including classical HGF activators, plasma kallikrein, neutrophil elastase, and/or mast cell chymase. + and −, pathways leading to activation or inactivation ligands of c-Met signaling, respectively.
Interestingly, non-reducing SDS-PAGE and Western blot analysis of rhHGF incubated in wound exudate of non-healing wounds, but not of healing wounds, revealed a prominent immunoreactive 45-kd protein band. Recently, neutrophil elastase and mast cell chymase have been shown to effectively process classically activated HGF-heterodimer, resulting in NK4-like fragments consisting of the first 478 or 480 residues of the α-chain, respectively.37 Thus, the site of cleavage for both proteases lies within the α-chain on the N-terminal side of the cysteine linking the α- and β-chain and results in cleavage of the β-chain (Figure 10). We and others provided evidence that the activities of both proteases are highly up-regulated during skin repair in humans29,30,31,38,39,40 and our data strongly suggest that both proteases are involved in HGF processing during repair. This hypothesis is supported by the finding of a prominent immunoreactive band of approximately 45 kd consisting of the N-terminal residues of the α-chain that is consistent with HGF fragments resulting from neutrophil elastase and/or chymase cleavage. Furthermore, proteolytic cleavage of rhHGF was in part abolished by a specific inhibitor for neutrophil elastase, demonstrating an important role of neutrophil elastase in HGF processing in non-healing wounds.
The 54-kd protein band present in wound exudates analyzed under reducing conditions had exactly the same electrophoretic mobility as a proteolytic product obtained following incubation of rhHGF with plasma kallikrein. Recently, in in vitro studies plasma kallikrein and coagulation factor XIa were identified as novel serine proteases efficiently cleaving the pro-HGF monomer, not only at the canonical cleavage site Arg494-Val495, but in addition within the K4 domain at the site Arg424-His42511 (Figure 10). Quantitative analysis of plasma kallikrein activity in exudates from healing and non-healing wounds revealed significantly increased levels during the early phase of healing and in the chronic phase of non-healing wounds, when compared with activity levels in blood plasma and the late phase of healing wounds. Thereby, plasma kallikrein might contribute in the regulation of cellular events during repair, as indicated by our data in particular in the processing of pro-HGF monomer and/or the HGF heterodimer to modulate their biological activity. The lack of an immunoreactive signal that corresponds to the β-chain in wound exudates analyzed under non-reducing and reducing conditions is suggestive for additional extensive proteolysis of this HGF domain during cutaneous repair and consequent loss of function.
Taken together, we demonstrate that during skin repair the pro-HGF monomer and/or the classical HGF heterodimer is subjected to proteolytic pathways that are different from the classical pathway mediated by known serine proteases. Our findings indicate that plasma kallikrein and/or additional inflammatory peptidases, such as neutrophil elastase, participate in HGF processing during repair. Our data provide the first evidence for alternative HGF processing in the human system, in particular during skin repair in humans, and raises the question of what the functional consequences of these unconventional proteolytic processing events might be for HGF/c-Met-mediated pathways in repair.
Our in vitro data demonstrate that neutrophil elastase or plasma kallikrein-mediated processing of HGF significantly attenuates its mitogenic and migratory activities on primary human keratinocytes. As revealed by non-reducing Western blot analysis of wound exudates, the immunoreactive 45-kd HGF band consists predominantly of an NK4-like domain, which results from proteolytic cleavage by neutrophil elastase or chymase. NK4 is composed of the first 447 residues of the α-chain and was originally generated by fragmentation with pancreatic elastase.37 Consistently, the HGF fragments resulting from neutrophil elastase (first 478 residues of α-chain) or mast cell chymase (first 480 residues of α-chain) processing of HGF-heterodimer were named NK4-like fragments.37 Both NK4 and NK4-like fragments have been shown to antagonize c-Met-mediated HGF effects and recombinant NK4 or NK4-expressing vectors were successfully used to inhibit invasion, metastasis, and angiogenesis in tumor models.17,44 The underlying molecular mechanisms by which NK4 or NK4-like variants antagonize HGF effects have not yet been identified. Our data show that neutrophil elastase-mediated digestion of HGF attenuates signaling pathways crucial for cell proliferation and migration.
As demonstrated by our analysis and those of others, limited plasma kallikrein processing of HGF results in cleavage within the K4 domain and extensive processing in additional loss of the β-chain.11 These events provide the most likely molecular explanation for the observed loss of effects on primary keratinocytes in culture. Cleavage within the K4 domain might cause conformational alterations within the HGF structure, and hence interfere with HGF-c-Met interactions resulting in altered receptor binding, dimerization, and/or activation. Based on recent mutagenesis studies and crystallographic data, a mutant molecule consisting exclusively of the K4 domain binds c-Met with low affinity, which is reflected in its partial biological activity when compared with the activity of mutants containing the N-, K1-kringle and K2-kringle domain (N-K1-K2) or the mature HGF molecule.9 More importantly, the biological activity of the N-K1-K2 mutant is significantly suppressed by adding the K4 domain, providing ample evidence for an important role of the K4 domain to regulate interactions of HGF-domains with c-Met and finally the biological activity of the HGF heterodimer. Indeed, a mutant containing the N-K1-K2-K3-K4 domains (NK4) has been proposed as an effective HGF antagonist for cancer therapy, although the underlying molecular mechanisms are not yet fully understood.44 Besides interfering with direct HGF/c-Met interactions and activation, cleavage in K4 might influence the interactions of HGF with co-receptors that modulate the HGF/c-Met pathway. Along these lines, recent data demonstrate that integrins (α6β4)45 or co-stimulatory molecules such as neuropilins46 significantly enhance HGF-mediated activation of c-Met and modulate the cellular response to HGF. The functional impact of both receptors has been demonstrated in skin repair and impaired signaling through altered HGF-integrin or/and HGF-neuropilin interactions is conceivable. Overall, our data demonstrate that plasma kallikrein mediates HGF processing in repair, resulting in attenuated biological activities. The underlying molecular mechanisms for the observed loss of function remain to be clarified in more detail.
What is the impact of neutrophil elastase or plasma kallikrein processing of HGF in physiological or pathophysiological repair? During normal wound healing where the activities of proteases and their inhibitors are well balanced these events might present a physiological mechanism to protect epithelial or other cells from overwhelming HGF stimulatory effects. In contrast, in chronic inflammatory processes, as the non-healing wound microenvironment where proteases are significantly increased over protease inhibitors, a shift to the production of HGF fragments with attenuated or antagonistic HGF activities occurs, which impairs the healing response. Interestingly, in non-healing wounds HGF levels (or those of HGF cleavage products) were significantly increased over those in healing wounds and increased levels of antagonistic HGF fragments might result in a dominant negative effect, thus contributing to impaired HGF-mediated healing responses. Indeed, the attenuated signal of phosphorylated c-Met observed in non-healing versus healing wounds might at least in part be caused by lack of active HGF and/or antagonistic HGF fragments.
Overall, our findings provide the first evidence that during skin repair the pro-HGF monomer and/or the classical HGF heterodimer is subjected to proteolytic pathways, which are different from the classical pathway mediated by known serine proteases. Our results indicate that inflammatory peptidases, such as neutrophil elastase as well as plasma kallikrein, participate in HGF processing during repair. In vitro data demonstrate that this event attenuates activities on keratinocyte functions crucial for epithelialization during repair. Furthermore, cleavage by inflammatory peptidases leads to the generation of a HGF fragment, which might expose antagonistic effects on c-Met, as demonstrated earlier for the NK4 fragment. Therefore, our observations are of clinical importance. Our findings provide support to the idea that maintaining the balance of proteases and their inhibitors is essential to control HGF/c-Met signaling during skin repair. Furthermore, removing the NK4-like fragment from non-healing wounds might promote healing and interfere with the pathology of chronic ulcers. Finally, our findings might have direct implications for a protein engineering approach, generating protease resistant c-Met agonists to facilitate skin repair, or for a protease inhibition approach to rebalance uncontrolled proteases in chronic wounds.
Supplementary Material
Acknowledgments
We gratefully acknowledge Walter Birchmeier for providing rhHGF protein, Madin-Darby canine kidney cells, and helpful discussion of the data. We thank Michael Piekarek for technical assistance and Semra Özcelik and Ruth Pofahl for support with the keratinocyte culture. We thank Michael Gerharz and Gunnar Schütz for helpful discussion of the results.
Footnotes
Address reprint requests to Sabine A. Eming, M.D., Professor of Dermatology, Department of Dermatology, University of Cologne, Joseph-Stelzmann Str. 9, 50931 Köln, Germany. E-mail: sabine.eming@uni-koeln.de.
Supported by European Community grant LSHB-CT-2005-512102 (to S.A.E.), the Köln Fortune Program of the Faculty of Medicine at the University of Cologne (M.G., S.A.E.), and Deutsche Forschungsgemeinschaft grant SFB 829 (to S.A.E., T.K., C.N., M.P.).
N.B. and D.H. contributed equally to this work.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Gurtner GC, Werner S, Barrandon Y, Lomgaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Schäfer M, Werner S. Transcriptional control of wound repair. Annu Rev Cell Dev Biol. 2007;23:69–92. doi: 10.1146/annurev.cellbio.23.090506.123609. [DOI] [PubMed] [Google Scholar]
- Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- Fonder MA, Lazarus GS, Cowan DA, Aronson-Cook B, Kohli AR, Mamelak AJ. Treating the chronic wound: a practical approach to the care of nonhealing wounds and wound care dressings. J Am Acad Dermatol. 2008;58:185–206. doi: 10.1016/j.jaad.2007.08.048. [DOI] [PubMed] [Google Scholar]
- Trusolino L, Comoglio PM. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer. 2002;2:289–300. doi: 10.1038/nrc779. [DOI] [PubMed] [Google Scholar]
- Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376:768–771. doi: 10.1038/376768a0. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373:699–702. doi: 10.1038/373699a0. [DOI] [PubMed] [Google Scholar]
- Gherardi E, Sandin S, Petoukhov MV, Finch J, Youles ME, Ofverstedt LG, Miguel RN, Blundell TL, Vande Woude GF, Skoglund U, Svergun DI. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc Natl Acad Sci USA. 2006;103:4046–4051. doi: 10.1073/pnas.0509040103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes O, Pillozzi S, Deakin JA, Carafoli F, Kemp L, Butler PJ, Lyon M, Gherardi E. Insights into the structure/function of hepatocyte growth factor/scatter factor from studies with individual domains. J Mol Biol. 2007;367:395–408. doi: 10.1016/j.jmb.2006.12.061. [DOI] [PubMed] [Google Scholar]
- Miyazawa K, Shimomura T, Kitamura N. Activation of hepatocyte growth factor in the injured tissues is mediated by hepatocyte growth factor activator. J Biol Chem. 1996;271:3615–3618. doi: 10.1074/jbc.271.7.3615. [DOI] [PubMed] [Google Scholar]
- Peek M, Moran P, Mendoza N, Wickramasinghe D, Kirchhofer D. Unusual proteolytic activation of pro-hepatocyte growth factor by plasma kallikrein and coagulation factor XIa. J Biol Chem. 2002;277:47804–47809. doi: 10.1074/jbc.M209778200. [DOI] [PubMed] [Google Scholar]
- Shimomura T, Miyazawa K, Komiyama Y, Hiraoka H, Naka D, Morimoto Y, Kitamura N. Activation of hepatocyte growth factor by two homologous proteases, blood-coagulation factor XIIa and hepatocyte growth factor activator. Eur J Biochem. 1995;229:257–261. doi: 10.1111/j.1432-1033.1995.tb20463.x. [DOI] [PubMed] [Google Scholar]
- Lee SL, Dickson RB, Lin CY. Activation of hepatocyte growth factor and urokinase/plasminogen activator by matriptase, an epithelial membrane serine protease. J Biol Chem. 2000;275:36720–36725. doi: 10.1074/jbc.M007802200. [DOI] [PubMed] [Google Scholar]
- Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992;11:4825–4833. doi: 10.1002/j.1460-2075.1992.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Takehara T, Inoue H, Hagiya M, Shimizu S, Nakamura T. Deletion of kringle domains or the N-terminal hairpin structure in hepatocyte growth factor results in marked decreases in related biological activities. Biochem Biophys Res Commun. 1991;181:691–699. doi: 10.1016/0006-291x(91)91246-9. [DOI] [PubMed] [Google Scholar]
- Okigaki M, Komada M, Uehara Y, Miyazawa K, Kitamura N. Functional characterization of human hepatocyte growth factor mutants obtained by deletion of structural domains. Biochemistry. 1992;31:9555–9561. doi: 10.1021/bi00155a007. [DOI] [PubMed] [Google Scholar]
- Date K, Matsumoto K, Shimura H, Tanaka M, Nakamura T. HGF/NK4 is a specific antagonist for pleiotrophic actions of hepatocyte growth factor. FEBS Lett. 1997;420:1–6. doi: 10.1016/s0014-5793(97)01475-0. [DOI] [PubMed] [Google Scholar]
- Nusrat A, Parkos CA, Bacarra AE, Godowski PJ, Delp-Archer C, Rosen EM, Madara JL. Hepatocyte growth factor/scatter factor effects on epithelia. Regulation of intercellular junctions in transformed and nontransformed cell lines, basolateral polarization of c-met receptor in transformed and natural intestinal epithelia, and induction of rapid wound repair in a transformed model epithelium. J Clin Invest. 1994;93:2056–2065. doi: 10.1172/JCI117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, Gaudino G, Tamagnone L, Coffer A, Comoglio PM. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119:629–64. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilmann M, Odenthal M, Jung W, Vande Woude GF, Dienes HP, Schirmacher P. Neoexpression of the c-met/hepatocyte growth factor-scatter factor receptor gene in activated monocytes. Blood. 1997;90:4450–4458. [PubMed] [Google Scholar]
- Gohda E, Tsubouchi H, Nakayama H, Hirono S, Sakiyama O, Takahashi K, Miyazaki H, Hashimoto S, Daikuhara Y. Purification and partial characterization of hepatocyte growth factor from plasma of a patient with fulminant hepatic failure. J Clin Invest. 1988;81:414–419. doi: 10.1172/JCI113334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumori A. Roles of hepatocyte growth factor and mast cells in thrombosis and angiogenesis. Cardiovasc Drugs Ther. 2004;18:321–326. doi: 10.1023/B:CARD.0000041252.33870.74. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Yoshida S, Yamaguchi Y, Itami S, Yoshikawa K, Tabata Y, Matsumoto K, Nakamura T. Neutralization of hepatocyte growth factor leads to retarded cutaneous wound healing associated with decreased neovascularization and granulation tissue formation. J Invest Dermatol. 2004;120:355–343. doi: 10.1046/j.1523-1747.2003.12039.x. [DOI] [PubMed] [Google Scholar]
- Toyoda M, Takayama H, Horiguchi N, Otsuka T, Fukusato T, Merlino G, Takagi H, Mori M. Overexpression of hepatocyte growth factor/scatter factor promotes vascularization and granulation tissue formation in vivo. FEBS Lett. 2001;509:95–100. doi: 10.1016/s0014-5793(01)03126-x. [DOI] [PubMed] [Google Scholar]
- Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177:151–162. doi: 10.1083/jcb.200701086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan D, Gherardi E, Fan TP, Edwards D, Warn R. Diverse and potent activities of HGF/SF in skin wound repair. J Pathol. 2004;203:831–838. doi: 10.1002/path.1578. [DOI] [PubMed] [Google Scholar]
- Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med. 1996;2:287–292. doi: 10.1038/nm0396-287. [DOI] [PubMed] [Google Scholar]
- Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–525. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- Eming S, Smola H, Hartmann B, Malchau G, Wegner R, Krieg T, Smola-Hess S. The inhibition of matrix metalloproteinase activity in chronic wounds by a polyacrylate superabsorber. Biomaterials. 2008;29:2932–2940. doi: 10.1016/j.biomaterials.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Zhu M. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, alpha1-proteinase inhibitor, and alpha2-macroglobulin. J Invest Dermatol. 1996;106:335–341. doi: 10.1111/1523-1747.ep12342990. [DOI] [PubMed] [Google Scholar]
- Lauer G, Sollberg S, Cole M, Flamme I, Sturzebecher J, Mann K, Krieg T, Eming SA. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol. 2000;115:12–18. doi: 10.1046/j.1523-1747.2000.00036.x. [DOI] [PubMed] [Google Scholar]
- Roth D, Piekarek M, Paulsson M, Christ H, Bloch W, Krieg T, Davidson JM, Eming SA. Plasmin modulates vascular endothelial growth factor-A-mediated angiogenesis during wound repair. Am J Pathol. 2006;168:670–684. doi: 10.2353/ajpath.2006.050372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda M, Miyamoto T. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem Biophys Res Commun. 1991;177:814–820. doi: 10.1016/0006-291x(91)91862-7. [DOI] [PubMed] [Google Scholar]
- Eming SA, Lee J, Snow RG, Tompkins RG, Yarmush ML, Morgan JR. Genetically modified human epidermis overexpressing PDGF-A directs the development of a cellular and vascular connective tissue stroma when transplanted to athymic mice–implications for the use of genetically modified keratinocytes to modulate dermal regeneration. J Invest Dermatol. 1995;105:756–763. doi: 10.1111/1523-1747.ep12325550. [DOI] [PubMed] [Google Scholar]
- Weidner KM, Behrens J, Vandekerckhove J, Birchmeier W. Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J Cell Biol. 1990;111:2097–2108. doi: 10.1083/jcb.111.5.2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parr C, Jiang WG. Expression of hepatocyte growth factor/scatter factor, its activator, inhibitors and the c-Met receptor in human cancer cells. Int J Oncol. 2001;19:857–863. [PubMed] [Google Scholar]
- Raymond WW, Cruz AC, Caughey GH. Mast cell and neutrophil peptidases attack an inactivation segment in hepatocyte growth factor to generate NK4-like antagonists. J Biol Chem. 2006;281:1489–1494. doi: 10.1074/jbc.M511154200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Smola-Hess S, Kurschat P, Hirche D, Krieg T, Smola H. A novel property of povidon-iodine: inhibition of excessive protease levels in chronic non-healing wounds. J Invest Dermatol. 2006;126:2731–2733. doi: 10.1038/sj.jid.5700474. [DOI] [PubMed] [Google Scholar]
- Huttunen M, Harvima IT. Mast cell tryptase and chymase in chronic leg ulcers: chymase is potentially destructive to epithelium and is controlled by proteinase inhibitors. Br J Dermatol. 2005;152:1149–1160. doi: 10.1111/j.1365-2133.2005.06428.x. [DOI] [PubMed] [Google Scholar]
- Herrick S, Ashcroft G, Ireland G, Horan M, McCollum C, Ferguson M. Up-regulation of elastase in acute wounds of healthy aged humans and chronic venous leg ulcers are associated with matrix degradation. Lab Invest. 1997;77:281–288. [PubMed] [Google Scholar]
- Adams JC, Furlong RA, Watt FM. Production of scatter factor by ndk, a strain of epithelial cells, and inhibition of scatter factor activity by suramin. J Cell Sci. 1991;98 (Pt 3):385–394. doi: 10.1242/jcs.98.3.385. [DOI] [PubMed] [Google Scholar]
- Ferracini R, Di Renzo MF, Scotlandi K, Baldini N, Olivero M, Lollini P, Cremona O, Campanacci M, Comoglio PM. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene. 1995;10:739–749. [PubMed] [Google Scholar]
- Conway K, Price P, Harding KG, Jiang WG. The molecular and clinical impact of hepatocyte growth factor, its receptor, activators, and inhibitors in wound healing. Wound Repair Regen. 2006;14:2–10. doi: 10.1111/j.1743-6109.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Nakamura T. NK4 (HGF-antagonist/angiogenesis inhibitor) in cancer biology and therapeutics. Cancer Sci. 2003;94:321–327. doi: 10.1111/j.1349-7006.2003.tb01440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell. 2001;107:643–654. doi: 10.1016/s0092-8674(01)00567-0. [DOI] [PubMed] [Google Scholar]
- Hu B, Guo P, Bar-Joseph I, Imanishi Y, Jarzynka MJ, Bogler O, Mikkelsen T, Hirose T, Nishikawa R, Cheng SY. Neuropilin-1 promotes human glioma progression through potentiating the activity of the HGF/SF autocrine pathway. Oncogene. 2007;26:5577–5586. doi: 10.1038/sj.onc.1210348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.