

Abstract
The Erbb2 receptor is activated by UV irradiation, the primary cause of non-melanoma skin cancer. We hypothesized that Erbb2 activation contributes to UV-induced skin tumorigenesis by suppressing cell cycle arrest. Consistent with this hypothesis, inhibition of Erbb2 in v-rasHa transgenic mice before UV exposure resulted in both 56% fewer skin tumors and tumors that were 70% smaller. Inhibition of the UV-induced activation of Erbb2 also resulted in milder epidermal hyperplasia, S-phase accumulation, and decreased levels of the cell cycle regulator Cdc25a, suggesting altered cell cycle regulation on inhibition of Erbb2. Further investigation using inhibition or genetic deletion of Erbb2 in vitro revealed reduced Cdc25a levels and increased S-phase arrest in UV-irradiated cells lacking Erbb2 activity. Ectopic expression of Cdc25a prevented UV-induced S-phase arrest in keratinocytes lacking Erbb2 activity, demonstrating that maintenance of Cdc25a by Erbb2 suppresses cell cycle arrest. Examination of checkpoint pathway activation upstream of Cdc25a revealed Erbb2 activation did not alter Ataxia Telangiectasia and Rad3-related/Ataxia Telangiectasia Mutated activity but increased inhibitory phosphorylation of Chk1-Ser280. Since Akt phosphorylates Chk1-Ser280, the effect of Erbb2 on phosphatidyl inositol-3-kinase (PI3K)/Akt signaling during UV-induced cell cycle arrest was determined. Erbb2 ablation reduced the UV-induced activation of PI3K while inhibition of PI3K/Akt increased UV-induced S-phase arrest. Thus, UV-induced Erbb2 activation increases skin tumorigenesis through inhibitory phosphorylation of Chk1, Cdc25a maintenance, and suppression of S-phase arrest via a PI3K/Akt-dependent mechanism.
Activation of signaling pathways following UV radiation, known as the UV response, resembles the response of cells to growth factors. Interestingly, the receptor tyrosine kinase Erbb2 (HER2/Neu) is rapidly activated following UV exposure of skin and cultured keratinocytes.1 Erbb2 activity is increased following UV irradiation by two distinct mechanisms: reactive oxygen species-dependent inactivation of tyrosine phosphatases2,3,4 and an activator protein 2α-dependent increase in Erbb2 expression.5 Our recent gene profiling and cell biology experiments demonstrated that the UV-induced activation of Erbb2 increases inflammation and cell proliferation,1 suggesting Erbb2 may contribute to UV-induced skin tumorigenesis. Indeed, transgenic overexpression of Erbb2 in the skin results in epidermal and follicular hyperplasia and spontaneous tumor formation.6,7 In addition, Erbb2 overexpression is detected in some human non-melanoma skin cancers, where it is associated with more aggressive disease.8,9 Investigation of the influence of the UV-induced activation of Erbb2 on skin tumorigenesis, however, has not been previously reported.
Identification of the importance of the Erbb2 family member and signaling partner epidermal growth factor receptor (EGFR) on UV-induced skin tumors strongly supports a role for Erbb2 in skin tumorigenesis as well. The UV-induced activation of EGFR blocks cell cycle arrest, increases cell proliferation, suppresses apoptotic cell death, and increases skin tumorigenesis.10,11,12,13,14 The effects of EGFR on apoptosis and cell cycle arrest result, at least in part, from its activation of phosphatidyl inositol-3-kinase (PI3K)/Akt signaling.11,13 For example, EGFR-dependent PI3K/Akt activation blocks the activation of signaling downstream from ataxia telangiectasia and Rad3-related (ATR) to block cell cycle arrest.11,15,16,17
Activation of the ATR cell cycle checkpoint following UV-induced DNA damage allows time for DNA repair (reviewed in18). ATR phosphorylates and activates Chk1, and to a lesser extent Chk2, kinases that phosphorylate the cell cycle regulator Cdc25a.19,20 Phosphorylation by Chk1/2 inactivates Cdc25a and targets it for rapid, ubiquitin-directed degradation.19,21 The Cdc25a phosphatase activates cyclin-dependent kinase (CDK)2 by removal of inhibitory phosphate groups at CDK2-Tyr15 and CDK2-Thr14.19,21,22 Loss of Cdc25a activity following ATR activation thus blocks activation of cyclin/CDK complexes, resulting in cell cycle arrest.22 Cell cycle arrest allows time for the repair of DNA damage and reduces mutagenesis. If cell cycle arrest and DNA repair mechanisms are inadequate, cells acquire mutations that lead to cancer. EGFR promotes G2/M-phase progression by blocking the activation of this cell cycle checkpoint through PI3K/Akt-dependent inhibitory phosphorylation of Chk1,11 a potential mechanism for its role in promoting UV-induced skin tumorigenesis.14
In contrast to the extensive literature documenting the effects of EGFR activation on the response of the skin to UV, little investigation of the importance of other EGFR family members in UV-induced skin carcinogenesis has been undertaken. We hypothesized that repeated activation of the Erbb2 receptor resulting from chronic exposure to UV might also contribute to UV-induced skin tumorigenesis by deregulating cell cycle checkpoint control. This paradigm does not require oncogenic activation of Erbb2, but rather depends on repeated cycles of activation of normal physiological levels of proto-oncogenic Erbb2. We found that inhibition of the UV-induced activation of Erbb2 substantially reduced skin tumorigenesis in a transgenic mouse model. Using both mouse skin and cell culture models, we also determined that UV-induced skin tumorigenesis was associated with Erbb2-dependent inhibitory phosphorylation of Chk1, maintenance of Cdc25a, and decreased cell cycle arrest. These data demonstrate that activation of Erbb2 on UV irradiation increases UV-induced skin tumorigenesis by suppressing a DNA damage-induced cell cycle checkpoint.
Materials and Methods
Animals
The dorsal hair of homozygous v-rasHa transgenic Tg.AC mice on an FVB/N background was clipped one day before treatment using electric clippers (Wahl, Sterling, IL) and on the day of treatment with a Remington Microscreen shaver (Madison, NC). Four mg AG825 (AG Scientific, San Diego, CA and Calbiochem, San Diego, CA) dissolved in 200 μl dimethyl sulfoxide (DMSO), or the vehicle alone, was applied topically to the shaved back of the mice 2 hours before exposure to 10 kJ/m2 UV or sham irradiation. This concentration of AG825 applied over the shaved dorsal surface of the mouse did not significantly absorb UV. The UV-B TL 40W/12 RS bulbs (Philips, Somerset, NJ) used emitted approximately 30% UVA, 70% UVB, and <1% UVC, with a total output of 470 μW/cm2, as measured with radiometric photodetector probes (Newport, Irvine, CA). Tumor number, tumor volume using calipers, and skin-fold thickness using calipers (Mitutoyo, Aurora, IL) were assessed weekly. All animal procedures were performed in accordance with American Association of Laboratory Animal Care guidelines and approved by Creighton University’s Institutional Animal Care and Use Committee.
Cell Culture
Primary newborn mouse keratinocytes from CD-1 mice were isolated as described previously.23 In brief, the skins were floated overnight on 0.25% trypsin at 37°C, the epidermis separated, minced, centrifuged in S-MEM (Invitrogen, Carlsbad, CA) with 8% fetal bovine serum (Gemini Bio-Products, West Sacramento, CA), and plated in this medium. The next day, cells were refed S-MEM with 8% chelexed serum and a calcium concentration adjusted to 0.05 mmol/L. The cells were grown in this medium to 70% to 80% confluence, the medium replaced with a thin layer of PBS containing 0.05 mmol/L calcium, and exposed to 600 J/m2 UV or sham irradiated as described previously.1 Some keratinocytes were treated with 45 μmol/L AG825 (or with the concentrations indicated elsewhere)(AG Scientific, San Diego, CA), 15 μmol/L PI3K inhibitor LY294002 (Calbiochem, La Jolla, CA), 15 μmol/L Akt inhibitor IL-6-hydroxymethyl-chiro-inositol-2–20-methyl-3-O-ocadecylcarbomate (Calbiochem) or the vehicle alone. Plasmids with Cdc25a24 and enhanced green fluorescent protein (GFP) cDNA were cotransfected with Lipofectamine Transfection Reagent (Invitrogen, Carlsbad, CA). Transfection efficiency was quantified by determining the proportion of the cells that incorporated the GFP 1 day post-transfection. To obtain keratinocytes with genetic ablation of Erbb2, loxP sites were inserted flanking exon 1 of Erbb2, as described in.25 Keratinocytes homozygous for the Erbb2 loxP mutation were infected with Cre recombinase-expressing or empty adenoviral vectors in polybrene (Sigma, St. Louis, MO).
Cell Cycle
For in vivo analysis of cell cycle distribution, sections of formalin-fixed skin from paraffin embedded blocks were digested in PBS containing 0.5% pepsin (Sigma, St. Louis, MO). Cultured keratinocytes or pepsin-digested sections were suspended in Vindelov’s solution (3.5 mmol/L Tris base, pH 7.6, 10 mmol/L NaCl, 10 μg/ml ribonuclease A, 75 μg/ml propidium iodide, 1.0 μl/ml Ipegal),26 run on a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ), and analyzed using ModFit LT 3.1 software (Verity Software House, Topsham, ME). Some cells were treated with 10 μmol/L 5-bromo-2′-deoxyuridine (BrDU, Sigma) before harvest, treated with hydrochloric acid and trypsin, incubated with a fluorescein isothiocyanate-conjugated anti-BrDU antibody (BD Biosciences, San Diego, CA), and propidium iodide added before analysis. Data from at least 10,000 cells from each sample were collected using the flow cytometer.
Immunoblotting
Flash frozen skin was ground with a mortar and pestle on dry ice then homogenized with a polytron, or cells lysed in buffer containing 10 mmol/L Tris (pH 7.4), 150 mmol/L NaCl, 10% glycerol, 1% Triton X-100, 1 mmol/L EDTA, Complete Protease Inhibitor Cocktail (Roche, Germany), 1 mmol/L Na3VO4, 1.5 μmol/L EGTA, and 10 μmol/L NaF. Protein was measured using a BioRad assay (Sigma) and equal amounts of protein run on SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. Equal loading and transfer were confirmed using Ponceau S staining (Sigma). Membranes were immunoblotted with antibodies recognizing actin (Sigma), phosphorylated ATR/M substrates (Cell Signaling, Danvers, MA), Cdc25a (Santa Cruz Biotechnology, Santa Cruz, CA), Cdc25b (Cell Signaling), Cdc25c (Santa Cruz Biotechnology), phospho-Chk1-Ser296 (Cell Signaling), phospho-Chk1-Ser280 (gift of Ramon Parsons), phospho-Chk1345 (Santa Cruz), phospho-Chk2387 (Cell Signaling), and phospho-Akt (Cell Signaling, Beverly, MA), horseradish peroxidase-conjugated secondary antibodies (Cell Signaling), and visualized using chemiluminescent reagents (Pierce, Rockford, IL).
Microscopy
H&E staining was performed on paraffin-embedded sections following standard protocols. Following antigen retrieval, paraffin-embedded sections were incubated with antibodies to keratin 1, keratin 6, or filaggrin (all from Covance, Princeton, NJ), Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, Carlsbad, CA), and 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Apoptotic cells were identified using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (Promega, Madison, WI).
Statistical Methods
The tumor experiment was analyzed using two-way analysis of variance (ANOVA). Statistical significance in other experiments was assessed using two-way ANOVA or Student’s t-test with Bonferroni post-test. All experiments, excluding the tumor experiment, were replicated several times and consistent results obtained.
Results
Inhibition of Erbb2 Suppresses UV-Induced Skin Tumorigenesis
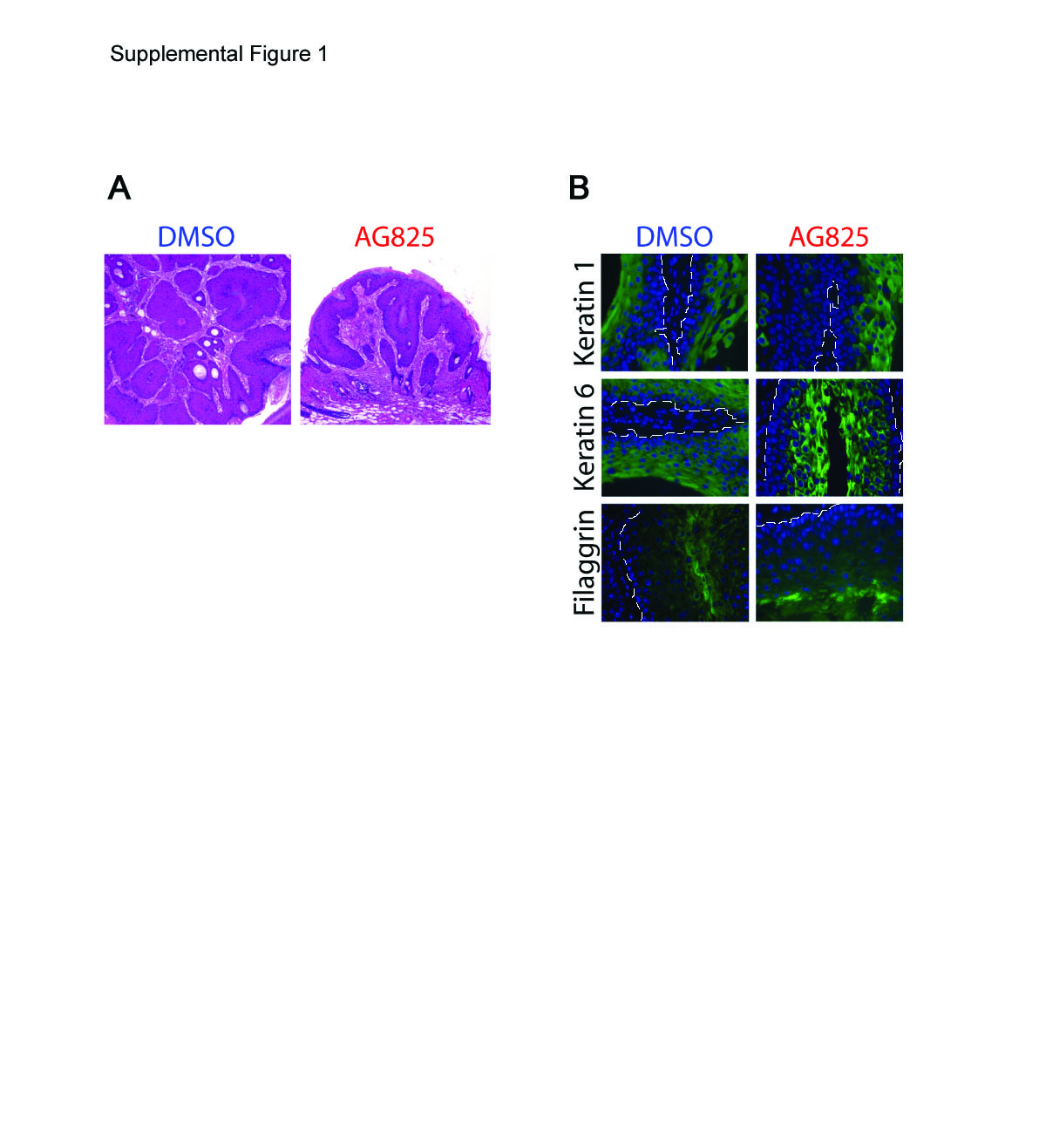
To test our hypothesis that the inhibition of Erbb2 prevents UV-induced skin tumorigenesis, groups of mice were topically treated with the Erbb2 inhibitor AG825 before UV irradiation using a published method.1 This skin tumorigenesis experiment was performed in v-rasHa transgenic Tg.AC mice, because of their enhanced sensitivity to UV-induced skin tumorigenesis,14,27 according to the timeline shown in Figure 1A. Inhibition of Erbb2 before UV irradiation blocked the development of more than half of the tumors, with 34 tumors per vehicle-treated mouse and only 15 tumors per inhibitor-treated mouse by the end of the experiment (Figure 1B). While the vehicle-treated mice continued to accrue tumors throughout the duration of the experiment, the inhibitor-treated group reached a plateau in tumor number by 7 weeks after the first UV exposure (Figure 1B). By the end of the experiment, mean tumor volume was also 70% less in the AG825-treated group when compared with the vehicle control (Figure 1B, axis on right). Representative tumors from each treatment group were examined histologically and all were benign squamous papillomas (see supplemental Figure S1A at http://ajp.amjpathol.org) with similar differentiation status (see supplemental Figure S1B at http://ajp.amjpathol.org). These data demonstrate that blocking the UV-induced activation of Erbb2 suppresses mouse skin tumorigenesis.
Figure 1.

Inhibition of Erbb2 suppresses UV-induced skin tumorigenesis. A: Experimental design showing treatment of mice in the tumor experiment. Groups of 10 mice were pretreated with AG825 or the vehicle DMSO alone 2 hours before 10 kJ/m2 UV or sham irradiation. This treatment was repeated one week later. At 12 weeks after the initial UV exposure, mice were euthanized and skin or tumors was removed for analysis. Hash marks on the timeline indicate the spacing is not correct to scale. B: Mean tumor number per mouse ± SEM (left axis) and tumor volume ± SEM (right axis) are shown. *Significantly different when compared with the vehicle-treated and UV-exposed group using a two-way ANOVA, where P ≤ 0.05.
Inhibition of Erbb2 Suppresses UV-Induced Epidermal Hyperplasia and Cell Proliferation
Because AG825 only transiently inhibits Erbb2 activity, the effects of the inhibitor were examined during the first few days after inhibitor treatment and UV irradiation. The influence of the Erbb2 inhibitor on epidermal hyperplasia was measured during the first 2 weeks of the tumor experiment regimen outlined in Figure 1A. Little hyperplasia was induced in the first week after UV irradiation, and the effect of the Erbb2 inhibitor on this response was minimal (Figure 2A, left panel). Following the second UV irradiation, epidermal hyperplasia was significantly suppressed by inhibition of Erbb2 (Figure 2A, left and right panels, day 9). Thus, the UV-induced activation of Erbb2 augments epidermal hyperplasia, a response that becomes more pronounced with multiple UV exposures.
Figure 2.

Inhibition of Erbb2 suppresses UV-induced hyperplasia and proliferation. Groups of AG825-and DMSO-pretreated mice were exposed to 10 kJ/m2 UV twice with an interval of 1 week, as described in Figure 1A. Mice were euthanized at the indicated time points following irradiation. A: Quantification of epidermal hyperplasia from at least three mice per group was averaged and the mean graphed. Vertical dashed lines indicate UV exposure. Representative H&E sections shown on right. Scale bar = 100 μm. B: The percent basal cells positive for terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling was quantified in skin sections at the indicated time points following the second UV irradiation. N ≥3. C: DNA content flow cytometry was performed using sections of skin from mice euthanized 2 days after the second UV exposure and the percentage of cycling cells graphed. N ≥3 mice. Treatment is significantly different using a Bonferroni posttest, where *P ≤ 0.05.
The influence of Erbb2 on both apoptosis and cell proliferation was assessed in vivo to determine the mechanisms of Erbb2’s effect on hyperplasia in the skin after UV irradiation. The percentage of basal cells positive for terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling was increased at 24 hours after UV in vehicle-treated skin (Figure 2B), consistent with previous observations in this animal model (14). Inhibition of Erbb2 did not increase apoptotic cell death at this time point (Figure 2B). Erbb2 reportedly suppresses keratinocyte differentiation,28 a process leading to keratinocyte death that is mechanistically and morphologically distinct from apoptosis. Inhibition of Erbb2 before UV irradiation did not alter the localization of expression of early (keratin 5) or late (loricrin) markers of differentiation in the skin (see supplemental Figure S2A at http://ajp.amjpathol.org). These data demonstrate that Erbb2’s influence on hyperplasia and tumor development does not result from the suppression of cell death via apoptosis or terminal differentiation following UV exposure but rather must be a consequence of an effect on cell proliferation. Mean proliferating cell nuclear antigen expression, a marker of cell proliferation, was less in Erbb2 inhibitor treated sham-irradiated skin and in UV-exposed skin when compared with the vehicle-treated controls (see supplemental Figure S2B at http://ajp.amjpathol.org). Cell cycle analysis revealed that the percentage of cells in S-phase increased on inhibition of Erbb2 before UV irradiation (Figure 2C). These data suggest that Erbb2 affects cell cycle regulation in the skin, a hypothesis that was further investigated using both inhibition and genetic ablation of the receptor in cultured keratinocytes.
Abrogation of Erbb2 Activity Blocks DNA Synthesis, Resulting in S-Phase Arrest after UV Exposure
Inhibition and genetic ablation of Erbb2 in cultured keratinocytes were used to determine the mechanisms through which Erbb2 contributes to skin tumorigenesis. To test our hypothesis that the UV-induced activation of Erbb2 blocks cell cycle arrest after UV irradiation, DNA synthesis was examined after UV irradiation of cultured primary keratinocytes lacking Erbb2 activity. Forty-five μmol/L AG825 was required to block the UV-induced activation of Erbb2 in primary keratinocytes, as shown in Figure 3A. BrDU incorporation was significantly reduced in sham-irradiated keratinocytes lacking Erbb2 activity (Figure 3, B and C). By 12 hours after UV irradiation, BrDU incorporation was reduced to nearly zero in keratinocytes lacking Erbb2 activity while substantial BrDU incorporation occurred in the irradiated controls with intact Erbb2 signaling (Figure 3, B and C). Thus, Erbb2 activation was necessary for DNA synthesis following UV irradiation. While apoptosis was significantly increased in the inhibitor-treated and sham-irradiated cells compared with the vehicle-treated controls (see supplemental Figure S3 at http://ajp.amjpathol.org), the decrease in DNA synthesis after UV irradiation was not an indirect result of increased apoptosis. Inhibition of Erbb2 did not increase UV-induced apoptosis (see supplemental Figure S3 at http://ajp.amjpathol.org).
Figure 3.

Abrogation of Erbb2 signaling increases S-phase arrest after UV irradiation. A: Keratinocytes were treated with the indicated concentrations of AG825 (numbers indicate the inhibitor concentration in μM) or with the vehicle alone 2 hours before UV or sham irradiation. Protein lysate was collected 30 minutes later and immunoblotted for phospho-Erbb2 (P-Erbb2) or actin. B–D: Primary keratinocytes were pretreated with 45 μmol/L AG825 or the vehicle DMSO and sham-irradiated or exposed to 600 J/m2 UV irradiation 2 hours later. B–C: Ten hours after irradiation, cells were pulsed with BrDU and collected 2 hours later. Cells were incubated with anti-BrDU-fluorescein isothiocyanate and propidium iodide. Cells were run through a flow cytometer and single cells were analyzed. At least 10,000 events were measured for each sample. Representative profiles are shown in (B) and the mean number of BrDU-positive cells versus all single cells ± SEM for N ≥3 is shown in (C). Significantly different from the corresponding DMSO treated sample, where *P ≤ 0.05. D: DNA content was determined using propidium iodide fluorescence in cells run through a flow cytometer 24 hours after sham- or 600 J/m2 UV-irradiation. Mean cells ± SEM for N ≥20 shown. Bars with asterisk indicate significant differences between groups using a Bonferroni post–test on a two-way ANOVA (*P ≤ 0.05). E–F: Erbb2fl/fl keratinocytes were infected with Cre recombinase expressing or empty adenoviral vector. E: Protein extracts from the cells 1 day postinfection were immunoblotted as indicated. F: Cells were sham- or 600 J/m2 UV-irradiated 24 hours after infection and DNA content flow cytometry was performed 24 hours later. Bars with asterisk indicate significant differences between groups using a Bonferroni post-test on a two-way ANOVA (*P ≤ 0.05). N ≥3.
UV irradiation caused S-phase accumulation of keratinocytes (Figure 3, D and F), consistent with previous reports.29 The influence of Erbb2 activation on S-phase progression was determined using both pharmacological and genetic models to interfere with Erbb2 signaling. Pharmacological inhibition of Erbb2 before UV exposure increased the percentage of cells in S-phase by 10% (Figure 3D). Erbb2 was also ablated by Cre recombinase expression in Erbb2fl/fl cultured primary keratinocytes (Figure 3E). Genetic ablation of Erbb2 increased UV-induced S-phase arrest by 18% after UV irradiation (Figure 3F). These data demonstrate that abrogation of Erbb2 signaling contributes to S-phase arrest in both sham-irradiated keratinocytes and after UV irradiation.
Erbb2 promotes cell cycle progression by maintaining Cdc25a To understand how Erbb2 regulates S-phase progression, signaling pathways known to regulate progression into and through S-phase were examined. As a key mediator of S-phase arrest after UV irradiation, the ATR DNA damage response pathway was examined. Cdc25a is degraded in response to ATR activation and its degradation, in turn, triggers S-phase arrest.19,21 Cdc25a immunoreactivity disappeared 18 hours after exposure of vehicle-treated cells to UV (Figure 4A). Several different anti-Cdc25a antibodies produced a similar appearance of multiple Cdc25a bands on the immunnoblots (Figure 4 and data not shown). This timing of Cdc25a loss is later than has been documented elsewhere,21 but no explanation for this difference in timing is readily apparent. Inhibition or genetic ablation of Erbb2 decreased Cdc25a immunoreactivity in sham irradiated and UV-exposed cells when compared with the appropriate sham-or UV-exposed controls (Figure 4, A and B). The decrease in Cdc25a was observed in both sham-and UV-irradiated cells at all time points between 30 minutes and 12 hours post-UV (Figure 4, A and B). Consistent with this timing, we have previously documented the activation of Erbb2 by 30 minutes after UV irradiation of keratinocytes in culture.1 In contrast to the effect of Erbb2 abrogation on Cdc25a, Cdc25b and Cdc25c were not reduced on abrogation of Erbb2 (not shown).
Figure 4.

Decreased Cdc25a on abrogation of Erbb2 signaling increases S-phase arrest. A–B: Cdc25a and actin immunoblotting after abrogation of Erbb2 and UV irradiation. Keratinocytes were treated with 45 μmol/L AG825 or DMSO 2 hours before UV (A) or Erbb2fl/fl keratinocytes were infected with Cre recombinase-expressing or empty viruses 1 day before UV irradiation (B). Keratinocytes were exposed to 600 J/m2 UV or sham-irradiated, cell lysates collected at the indicated times after UV, and samples immunoblotted as indicated. C: Ectopic expression of Cdc25a repairs the cell cycle defect in keratinocytes lacking Erbb2 activity. Keratinocytes were transfected with plasmids expressing Cdc25a and GFP or with a GFP vector alone. One day later, cells were treated with AG825 or the vehicle DMSO alone and irradiated 2 hours later. Twenty-four hours after UV or sham-irradiation, DNA content was determined using propidium fluorescence in cells run through a flow cytometer. N ≥3. Experiment is representative of three performed. Significantly different compared with the corresponding GFP-transfected samples using a Bonferroni post–test on a two-way ANOVA where *P ≤ 0.01. D: AG825-and DMSO-pretreated mice were exposed to 10 kJ/m2 UV twice with an interval of 1 week. Epidermal protein homogenate was obtained 24 hours following the second UV or sham irradiation. Protein extracts were immunoblotted as indicated. Numbers below the panels indicate densitometric results for Cdc25a relative to actin immunoblotting.
To confirm that the loss of Cdc25a was the cause of S-phase arrest in cells lacking Erbb2 activity, keratinocytes were cotransfected with a Cdc25a expression vector24 and a GFP vector, treated with the Erbb2 inhibitor or vehicle alone, and the cell cycle distribution assessed after UV exposure. Consistent with the results shown in Figure 3, inhibition of Erbb2 before UV exposure resulted in the accumulation of keratinocytes in S-phase (Figure 4C, GFP-only bars on left). Ectopic Cdc25a expression completely blocked this S-phase arrest (Figure 4C, right-hand bars). Thus, Cdc25a loss on abrogation of Erbb2 signaling increases S-phase arrest. In addition, consistent with the in vitro data, inhibition of Erbb2 in vivo also reduced Cdc25a after UV. As shown in Figure 4D, AG825 treatment of dorsal mouse skin before UV exposure reduced Cdc25a by 50% compared with vehicle-treated and UV-irradiated control epidermis. Taken together, these data suggest that inhibition of Erbb2 causes increased loss of Cdc25a, leading to increased S-phase arrest and suppressing UV-induced skin tumorigenesis.
Erbb2 Activation Results in Inhibitory Phosphorylation of Chk1 through a PI3K/Akt-Dependent Mechanism
Phosphorylation of Cdc25a by ATR-activated Chk1 triggers the degradation of Cdc25a.19,21 Surprisingly given the demonstration of decreased Cdc25a on ablation of Erbb2, abrogation of Erbb2 signaling did not significantly affect ATR activity in sham-irradiated or UV-exposed cells, as shown by phosphorylation of ATR/M substrates (Figure 5A). Another measure of ATR activation, phosphorylation of Chk1 on Ser345, was increased maximally at 6 hours after UV irradiation in both inhibitor and vehicle-treated samples, but was not significantly altered by inhibition of Erbb2 (Figure 5B). The lack of increased ATR activity in inhibitor treated cells after UV (Figure 5, A and B) suggests an effect of Erbb2 downstream from ATR. In cells lacking the tumor suppressor phosphatase and tensin homolog, AKT can phosphorylate Chk1 at an inhibitory site and block its activation by ATR.29,30 Consequently, we hypothesized that Erbb2 activation of PI3K/Akt signaling inhibits Chk1 activation. We found that the UV-induced activation of PI3K/AKT in primary keratinocytes is largely dependent on Erbb2, as is constitutive Akt activity in unirradiated cells (Figure 5D). Both inhibition and genetic ablation of Erbb2 reduced Akt activity, as measured by its phosphorylation on immunoblot, in sham-irradiated keratinocytes and at 30 minutes post-UV (Figure 5D). In addition, inhibition of either PI3K or Akt increased S-phase arrest after UV irradiation, also consistent with our hypothesis (Figure 5E). From these results, and from the results of others using EGFR,11 we hypothesized that UV-induced Erbb2 activation dampens the activation of the S-phase ATR checkpoint through a PI3K/Akt-dependent inhibitory phosphorylation of Chk1 on Ser280. Consistent with this hypothesis, phosphorylation of Chk1 on Ser280 was reduced by more than 50% in sham- and UV-irradiated cells on ablation of Erbb2 signaling by inhibitor treatment (Figure 5F). Also consistent with this hypothesis, Chk1 phosphorylation on Ser296, a putative autophosphorylation site necessary for fully active Chk1,30 was increased in keratinocytes lacking Erbb2 activity (Figure 5C, Sham and 3 hours). These results demonstrate a novel mechanism by which Erbb2 suppresses activation of the ATR-Chk1 S-phase checkpoint. Interestingly, ablation of Erbb2 signaling also increased the activation of Chk2 (Figure 5C, sham and 3-hour time points). The mechanism for Erbb2’s effect on Chk2 is unclear but may involve Akt as well.29
Figure 5.

Erbb2 blocks Chk1 activation through a PI3K/Akt dependent mechanism after UV irradiation. Keratinocytes in culture were treated with 45 μmol/L AG825 or the vehicle DMSO (A–D, F) or Erbb2fl/fl keratinocytes were infected with Cre recombinase expressing or empty virus (D) and sham-irradiated or exposed to 600 J/m2 UV 2 hours (for inhibitor) or 24 hours later (for Cre recombinase infection). Protein lysate was obtained at the indicated time points after UV irradiation (A–D, F). A: ATR/M activity was determined by immunoblotting for ATR/M substrate phosphorylation using an antibody specific for the phosphorylated consensus ATR/M substrate sequence and immunoblotting for actin. The sum of the signal from all bands detected with the ATR/M substrate phosphorylation antibody was normalized to actin levels using densitometry. The mean and SE for at least six samples at each time point and treatment is shown. No significant differences were detected between DMSO and AG825 treated samples at any time point. B: Chk1 phosphorylation on Ser345 was determined after immunoblotting with a phospho-Chk1-Ser345 specific antibody (inset). The signal for Chk1 phosphorylation relative to actin was determined using densitometry and the results from two replicate experiments averaged and graphed. Mean ± SEM is shown. C: Immunoblotting for Chk1 and Chk2 phosphorylation on abrogation of Erbb2 and UV exposure in cell lysates (“P” = phospho). D: Akt activation, measured by Akt phosphorylation (P-AKT), is dependent on Erbb2 activation after UV irradiation. “m” indicates minutes post-UV. E: Inhibition of PI3K or Akt, as described in Materials and Methods, causes S-phase arrest 24 hours after UV irradiation. N ≥4 experiments. Significantly different when compared with the vehicle-treated and UV-irradiated control, where *P ≤ 0.05. F: Immunoblotting for inhibitory phosphorylation of Chk1 on Ser280 on abrogation of Erbb2 activity and UV irradiation.
Discussion
Chronic exposure to UV is the main etiological factor contributing to non-melanoma skin cancer. The research presented here demonstrates that the UV-induced activation of Erbb2 promotes UV-induced skin tumorigenesis in a v-rasHa transgenic model. Erbb2 activation by UV occurs within the first 30 minutes after exposure to UV and then subsides by 40 minutes post-UV.1 With such a short pulse of receptor activation after UV irradiation and a short duration of the inhibitor’s effects, it is remarkable that many weeks later, both tumor multiplicity and tumor volume are decreased by more than half in Erbb2-inhibitor treated skin.
The mechanisms through which Erbb2 activation contributes to UV-induced skin tumorigenesis were investigated both in vivo and in vitro. Inhibition of Erbb2 in mouse skin before UV irradiation caused S-phase arrest and suppressed epidermal hyperplasia. In the absence of Erbb2 signaling, DNA synthesis largely stops by 12 hours after UV and keratinocytes accumulate in S-phase several hours later. In addition, inhibition of Erbb2 in sham-irradiated cells substantially decreased DNA synthesis and resulted in S-phase arrest without UV exposure. This response also certainly impacted the responses we observed following UV. Based on these data and previously published results with EGFR,11 we investigated whether Erbb2 dampens activation of the ATR DNA damage cell cycle checkpoint, known to cause S-phase arrest following UV exposure. On investigation, we demonstrated that Erbb2 does not affect the activation of ATR itself but rather the Chk1/2 kinases downstream from ATR.
Chk1 is activated by phosphorylation on Ser345 and Ser317 by ATM/R kinases and on Chk1 association with other proteins such as Claspin.18,30,31,32 We found no increase in Chk1 Ser345 phosphorylation in Erbb2 inhibitor treated cells. However, phosphorylation of Chk1 Ser296, an autophosphorylation site correlating with full activation of Chk1,32 was increased on inhibition of Erbb2 at the earliest time points examined. In addition, phosphorylation of Chk1 on Ser280 was decreased in sham-irradiated and UV-exposed keratinocytes at all time points examined when Erbb2 signaling was disrupted. Phosphorylation of Ser280 is inhibitory for Chk1 activation and both blocks its phosphorylation by ATR and the binding of Chk1 to other proteins, necessary for Chk1 activation.31 Akt phosphorylates Ser280 and thus blocks Chk1 activation, resulting in defective checkpoint activation and potentially in the accumulation of DNA damage and mutations.15,16,17,33 Our data are consistent with Erbb2 activation of PI3K/Akt signaling leading to inhibitory phosphorylation of Chk1 on Ser280, substantially reducing Cdc25a loss in both sham-irradiated and UV-exposed cells (Figure 6A). The mechanism by which Erbb2 regulates Chk2 activation may involve Akt activation as well, although this requires further investigation. A bioinformatics analysis revealed two regions of Chk2 with sequences very similar to the Akt consensus sequence that were centered on Ser124 and Ser144, consistent with the hypothesis that Akt can phosphorylate Chk2.
Figure 6.

Erbb2 regulates cell cycle progression following UV exposure by suppressing ATR checkpoint activation. A: In normal cells, UV-induced Erbb2 activation in turn activates PI3K/Akt signaling, which suppresses Chk1 activation and subsequent degradation of Cdc25a. B: On abrogation of Erbb2 activity, increased Chk1/2 activity leads to decreased Cdc25a and subsequently, to increased S-phase arrest. The net effect of decreased Erbb2 activity is decreased skin tumor development, presumably because of improved DNA repair and decreased mutagenesis.
Activated Chk1/2 phosphorylates the Cdc25 family of phosphatases, inactivating them and targeting them for rapid degradation. Among the Cdc25 family members, only Cdc25a was significantly reduced on loss of Erbb2 signaling. Decreased Cdc25a on abrogation of Erbb2 was observed in both UV-exposed mouse skin and in keratinocytes in culture. PI3K/Akt-dependent suppression of Chk1 activity and S-phase arrest have been documented in other models.15,16,17 In addition, in phosphatase and tensin homolog−/− cells that exhibit a defective cell cycle checkpoint response to ionizing radiation, elevated Akt activity led to inhibitory Chk1 phosphorylation on Ser280 and increased hypophosphorylated Cdc25a.34 The results presented here document that Erbb2 activation on UV irradiation modulates the ATR pathway by activating PI3K/Akt signaling.
PI3K signaling has previously been linked to Erbb2 and cell cycle progression in other organs. Overexpression of Erbb2 activates Akt in breast cancer cells to allow for S-phase progression.35 However, a connection between Erbb2 and a DNA damage checkpoint has not been previously established, except indirectly through its known interactions with EGFR. The results presented here and in our previous publications1 demonstrate that Erbb2 inhibition decreases proliferation after UV due to cessation of DNA synthesis rather than through an increase in cell death. These results contrast with previous publications using tumor cells overexpressing Erbb2 in which a block in Erbb2 primarily leads to apoptosis.36 The biological functions of Erbb2 when expressed at normal physiological levels may differ from its effects when overexpressed. Most published research has focused on signaling in tumor cell lines with oncogenic Erbb2. Our data suggest that further investigation of the normal biological functions of Erbb2 is also warranted.
While extensive documentation of the role of EGFR in UV responses has been undertaken,10,13,14,37,38,39 much less is known about the importance of Erbb2 in UV-induced skin tumorigenesis. Investigation of signaling specificity downstream from EGFR family members demonstrates that activation and dimerization of these receptors in various combinations can produce quite distinct effects. For example, Erbb2-containing heterodimers have a broader specificity and higher affinity for ligands, bind to a larger set of phosphotyrosine-binding proteins, undergo slower endocytosis and more frequent recycling than do other family member homo- or heterodimers (reviewed in40). Consequently, signals initiated from Erbb2 dimers are more intense and sustained. In particular, Erbb2/Erbb3 heterodimers are especially effective at activating PI3K/Akt signaling (reviewed in40). These data predict that the functions of EGFR family members can be quite different. Our recent investigation of sensory innervation in Egfr null mice illustrates this point. Loss of EGFR expression results in disorganized and excessive nerve fiber branching in the skin,41 while ablation of Erbb2 eliminates cutaneous innervation entirely (reviewed in41). The results presented here reveal substantial overlap, but also distinct effects of EGFR and Erbb2, on the response of skin cells to UV. UV-induced EGFR activation similarly leads to suppression of cell cycle arrest through inhibitory phosphorylation of Chk1,11 decreased keratinocyte proliferation, and suppression of epidermal hyperplasia.10 The effect of EGFR on Cdc25 family members has not been reported, however. Abrogation of EGFR activity also suppressed UV-induced skin tumorigenesis through both a suppression of proliferation and by increasing apoptotic cell death.14 Thus, EGFR and Erbb2 have distinct effects on apoptosis following UV irradiation. Because of the importance of apoptosis in eliminating cells with DNA damage, these data suggest interfering with EGFR signaling may be more effective in preventing skin tumorigenesis than blockade of Erbb2. EGFR and Erbb2 also have distinct cell cycle effects following UV irradiation since inhibition of EGFR triggers G2/M-phase arrest rather than S-phase arrest.7 We have observed a similar response following UV exposure of Egfr mutant keratinocytes as well, precluding simply differences in experimental technique between labs. The mechanisms for signaling specificity downstream from EGFR and Erbb2 resulting in distinct effects on the cell cycle and apoptosis remain unexplored. The results presented here differ from previous publications by focusing for the first time on the influence of Erbb2 on UV-induced skin tumorigenesis. In contrast to much of the published literature on Erbb2 overexpression, the current paper reveals roles for normal physiological levels of Erbb2 during tumorigenesis. The interactions of Erbb2 with other family members, such as EGFR, during UV-induced skin tumorigenesis remain to be investigated.
This research places Erbb2 upstream of a DNA damage sensitive cell cycle checkpoint, revealing cross talk between Erbb2 and a DNA damage response pathway during tumorigenesis. Based on this research and previous publications,1,11 we propose the model shown in Figure 6 to explain our results. Erbb2 activation following UV irradiation activates PI3K/Akt signaling, resulting in inhibitory phosphorylation of Chk1 on Ser280 and blocking activation of Chk1 and Chk2 by ATR (Figure 6A). Without strong Chk1/2 activation, keratinocytes do not completely cease synthesizing DNA. Loss of Erbb2 in contrast, allows for full activation of Chk1/2, Cdc25a loss, increased S-phase arrest, and reduced tumor formation (Figure 6B). Because these conclusions are based on research using a tumor-prone transgenic mouse model, additional research is necessary to determine the role of Erbb2 in UV-induced human skin cancer.
Supplementary Material
Acknowledgments
We thank Greg Perry for his assistance with flow cytometry, Ramon Parsons for the gift of the phospho-Chk1-Ser280 antibody, Ingrid Hoffman for the Cdc25a plasmid, U. Mueller for the floxed Erbb2 mice, and Stuart Yuspa for adenoviruses and helpful critique of the manuscript.
Footnotes
Address reprint requests to Laura A. Hansen, Department of Biomedical Sciences, School of Medicine, Creighton University, 2500 California Plaza, Omaha, NE 68178. E-mail: lhansen@creighton.edu.
Supported in part by the Intramural Research Program of the National Institutes of Health (NIH), National Institute for Environmental and Health Sciences, NIH (grants P20 RR018759 and 1RO1ES015585) and by the State of Nebraska LB595. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number 1 CO6 RR17417-01 from the National Center for Research Resources, NIH.
Supplemental material for this article can be found on http://ajp.amjpathol.org.
References
- Madson JG, Lynch DT, Tinkum KL, Putta SK, Hansen LA. Erbb2 regulates inflammation and proliferation in the skin after ultraviolet irradiation. Am J Pathol. 2006;169:1402–1414. doi: 10.2353/ajpath.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffer PJ, Burgering BM, Peppelenbosch MP, Bos JL, Kruijer W. UV activation of receptor tyrosine kinase activity. Oncogene. 1995;11:561–569. [PubMed] [Google Scholar]
- Huang RP, Wu JX, Fan Y, Adamson ED. UV activates growth factor receptors via reactive oxygen intermediates. J Cell Biol. 1996;133:211–220. doi: 10.1083/jcb.133.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- Han CY, Lim SC, Choi HS, Kang KW. Induction of ErbB2 by ultraviolet A irradiation: potential role in malignant transformation of keratinocytes. Cancer Sci. 2008;99:502–509. doi: 10.1111/j.1349-7006.2007.00718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bol D, Kiguchi K, Beltran L, Rupp T, Moats S, Gimenez-Conti I, Jorcano J, DiGiovanni J. Severe follicular hyperplasia and spontaneous papilloma formation in transgenic mice expressing the neu oncogene under the control of the bovine keratin 5 promoter. Mol Carcinog. 1998;21:2–12. [PubMed] [Google Scholar]
- Xie W, Wu X, Chow LT, Chin E, Paterson AJ, Kudlow JE. Targeted expression of activated erbB-2 to the epidermis of transgenic mice elicits striking developmental abnormalities in the epidermis and hair follicles. Cell Growth Differ. 1998;9:313–325. [PubMed] [Google Scholar]
- Krahn G, Leiter U, Kaskel P, Udart M, Utikal J, Bezold G, Peter RU. Coexpression patterns of EGFR. HER2, HER3 and HER4 in non-melanoma skin cancer. Eur J Cancer. 2001;37:251–259. doi: 10.1016/s0959-8049(00)00364-6. [DOI] [PubMed] [Google Scholar]
- Maubec E, Duvillard P, Velasco V, Crickx B, Avril MF. Immunohistochemical analysis of EGFR and HER-2 in patients with metastatic squamous cell carcinoma of the skin. Anticancer Res. 2005;25:1205–1210. [PubMed] [Google Scholar]
- El Abaseri TB, Putta S, Hansen LA. Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis. 2006;27:225–231. doi: 10.1093/carcin/bgi220. [DOI] [PubMed] [Google Scholar]
- Jean C, Hernandez-Pigeon H, Blanc A, Charveron M, Laurent G. Epidermal growth factor receptor pathway mitigates UVA-induced G2/M arrest in keratinocyte cells. J Invest Dermatol. 2007;127:2418–2424. doi: 10.1038/sj.jid.5700863. [DOI] [PubMed] [Google Scholar]
- Peus D, Vasa RA, Meves A, Beyerle A, Pittelkow MR. UVB-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem Photobiol. 2000;72:135–140. doi: 10.1562/0031-8655(2000)072<0135:uiegfr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Wan YS, Wang ZQ, Shao Y, Voorhees JJ, Fisher GJ. Ultraviolet irradiation activates PI 3-kinase/AKT survival pathway via EGF receptors in human skin in vivo. Int J Oncol. 2001;18:461–466. doi: 10.3892/ijo.18.3.461. [DOI] [PubMed] [Google Scholar]
- El Abaseri TB, Fuhrman J, Trempus C, Shendrik I, Tennant RW, Hansen LA. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005;65:3958–3965. doi: 10.1158/0008-5472.CAN-04-2204. [DOI] [PubMed] [Google Scholar]
- Shtivelman E, Sussman J, Stokoe D. A role for PI 3-kinase and PKB activity in the G2/M phase of the cell cycle. Curr Biol. 2002;12:919–924. doi: 10.1016/s0960-9822(02)00843-6. [DOI] [PubMed] [Google Scholar]
- Bortul R, Tazzari PL, Billi AM, Tabellini G, Mantovani I, Cappellini A, Grafone T, Martinelli G, Conte R, Martelli AM. Deguelin. A PI3K/AKT inhibitor, enhances chemosensitivity of leukaemia cells with an active PI3K/AKT pathway. Br J Haematol. 2005;129:677–686. doi: 10.1111/j.1365-2141.2005.05504.x. [DOI] [PubMed] [Google Scholar]
- Box AH, Demetrick DJ. Cell cycle kinase inhibitor expression and hypoxia-induced cell cycle arrest in human cancer cell lines. Carcinogenesis. 2004;25:2325–2335. doi: 10.1093/carcin/bgh274. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- Lee CH, Chung JH. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem. 2001;276:30537–30541. doi: 10.1074/jbc.M104414200. [DOI] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, Zhang H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278:21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nature Protocols. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassepass I, Voit R, Hoffmann I. Phosphorylation at serine 75 is required for UV-mediated degradation of human Cdc25A phosphatase at the S-phase checkpoint. J Biol Chem. 2003;278:29824–29829. doi: 10.1074/jbc.M302704200. [DOI] [PubMed] [Google Scholar]
- Leu M, Bellmunt E, Schwander M, Farinas I, Brenner HR, Muller U. Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Development. 2003;130:2291–2301. doi: 10.1242/dev.00447. [DOI] [PubMed] [Google Scholar]
- Vindelov LL. Flow microfluorometric analysis of nuclear DNA in cells from solid tumors and cell suspensions. A new method for rapid isolation and straining of nuclei Virchows. Arch B Cell Pathol. 1977;24:227–242. [PubMed] [Google Scholar]
- Trempus CS, Mahler JF, Ananthaswamy HN, Loughlin SM, French JE, Tennant RW. Photocarcinogenesis and susceptibility to UV radiation in the v-Ha-ras transgenic Tg.AC mouse. J Invest Dermatol. 1998;111:445–451. doi: 10.1046/j.1523-1747.1998.00237.x. [DOI] [PubMed] [Google Scholar]
- De Potter IY, Poumay Y, Squillace KA, Pittelkow MR. Human EGF receptor (HER) family and heregulin members are differentially expressed in epidermal keratinocytes and modulate differentiation. Exp Cell Res. 2001;271:315–328. doi: 10.1006/excr.2001.5390. [DOI] [PubMed] [Google Scholar]
- Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO. Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res. 2005;65:4861–4869. doi: 10.1158/0008-5472.CAN-04-2633. [DOI] [PubMed] [Google Scholar]
- King FW, Skeen J, Hay N, Shtivelman E. Inhibition of Chk1 by activated PKB/Akt. Cell Cycle. 2004;3:634–637. [PubMed] [Google Scholar]
- Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and claspin in a human cell-free system. Biochem J. 2005;388:705–712. doi: 10.1042/BJ20041966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puc J, Parsons R. PTEN loss inhibits CHK1 to cause double stranded-DNA breaks in cells. Cell Cycle. 2005;4:927–929. doi: 10.4161/cc.4.7.1795. [DOI] [PubMed] [Google Scholar]
- Puc J, Keniry M, Li HS, Pandita TK, Choudhury AD, Memeo L, Mansukhani M, Murty VV, Gaciong Z, Meek SE, Piwnica-Worms H, Hibshoosh H, Parsons R. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Tuna M, Chavez-Reyes A, Tari AM. HER2/neu increases the expression of Wilms’ Tumor 1 (WT1) protein to stimulate S-phase proliferation and inhibit apoptosis in breast cancer cells. Oncogene. 2005;24:1648–1652. doi: 10.1038/sj.onc.1208345. [DOI] [PubMed] [Google Scholar]
- Xia W, Bisi J, Strum J, Liu L, Carrick K, Graham KM, Treece AL, Hardwicke MA, Dush M, Liao Q, Westlund RE, Zhao S, Bacus S, Spector NL. Regulation of survivin by ErbB2 signaling: therapeutic implications for ErbB2-overexpressing breast cancers. Cancer Res. 2006;66:1640–1647. doi: 10.1158/0008-5472.CAN-05-2000. [DOI] [PubMed] [Google Scholar]
- He YY, Huang JL, Chignell CF. Delayed and sustained activation of extracellular signal-regulated kinase in human keratinocytes by UVA: implications in carcinogenesis. J Biol Chem. 2004;279:53867–53874. doi: 10.1074/jbc.M405781200. [DOI] [PubMed] [Google Scholar]
- Wang HQ, Quan T, He T, Franke TF, Voorhees JJ, Fisher GJ. Epidermal growth factor receptor-dependent. NF-kappaB-independent activation of the phosphatidylinositol 3-kinase/Akt pathway inhibits ultraviolet irradiation-induced caspases-3, -8, and -9 in human keratinocytes. J Biol Chem. 2003;278:45737–45745. doi: 10.1074/jbc.M300574200. [DOI] [PubMed] [Google Scholar]
- Xu Y, Shao Y, Voorhees JJ, Fisher GJ. Oxidative inhibition of receptor-type protein-tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes. J Biol Chem. 2006;281:27389–27397. doi: 10.1074/jbc.M602355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Maklad A, Nicolai JR, Bichsel KJ, Evenson JE, Lee T-C, Threadgill DW, Hansen LA. The EGFR is required for proper innervation to the skin. J Invest Dermatol. 2009;129:690–698. doi: 10.1038/jid.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}