

Abstract
Introduction
Accumulating evidence has demonstrated an association between periodontal infectious agents, such as Porphyromonas gingivalis, and vascular disease. Tissue factor (TF) and its specific tissue factor pathway inhibitor (TFPI) are produced by vascular smooth cells and are important regulators of the coagulation cascade.
Materials and Methods
To assess the role of P. gingivalis in atherothrombosis, we infected primary human aortic smooth cells (HASMC) with either P. gingivalis 381, its non-invasive mutant DPG3, or heat-killed P. gingivalis 381. Levels and activity of TF and TFPI were measured 8 and 24 hours after infection in cell extracts and cell culture supernatants.
Results
P. gingivalis 381 did not affect total TF antigen or TF activity in HASMC, but it significantly suppressed TFPI levels and activity compared to uninfected control cells, and those infected with the non-invasive mutant strain or the heat-killed bacteria. Further, P. gingivalis' LPS (up to a concentration of 5 μg/ml) failed to induce prothrombotic effects in HASMC, suggesting a significant role for the ability of whole viable bacteria to invade this cell type.
Conclusion
These data demonstrate for the first time that infection with a periodontal pathogen induces a prothrombotic response in HASMC.
Keywords: infection, periodontitis, P. gingivalis, thrombosis, atherosclerosis, smooth muscle cell
Vascular smooth muscle cells (VSMC) are the major cellular component of the vessel wall and play a pivotal role in vascular function. They contribute to the short-term regulation of the blood pressure by altering the luminal vessel diameter, but also contribute to long-term adaptation, by structural remodeling. They also constitute a significant portion of the atherosclerotic lesion and proliferation, migration and apoptosis of VSMC are essential to atherogenesis, plaque progression and rupture [1].
Accumulating evidence suggests that inflammation and coagulation are essential in the pathogenesis of vascular disease: inflammation leads to activation of coagulation, and coagulation considerably affects inflammatory activity [2]. In addition, multiple studies have shown that certain infections are implicated in atherogenesis and may serve as inflammatory stimuli that also contribute to acute events via plaque destabilization [3]. Periodontal diseases, chronic oral infections leading to destruction of tooth supporting structures and eventually tooth loss, are very common. Mild to moderate forms affect the majority of adults and severe forms have a prevalence of 10% to 15% [4]. Several studies have demonstrated a link between periodontal infections and atherosclerosis in human subjects [5-8]. It has also been postulated that since several infectious agents are likely involved in atherogenesis, the risk relates to the aggregate pathogen load [9]. As periodontal diseases are largely preventable and treatable, periodontal therapy with resultant reduction of the oral infection burden may contribute to the efforts to reduce the risk for vascular disease [10].
Porphyromonas gingivalis, a principal etiologic agent of chronic periodontitis in humans, can gain access into the bloodstream and has been identified in human atherosclerotic plaques [11, 12]. Frequent low-level bacteremias, such as those resulting from chewing, brushing and flossing in periodontitis patients [13], may provide a chronic insult to the vasculature that could initiate and/or exacerbate atherogenesis. P. gingivalis has been shown to enhance foam cell formation in human macrophages [14] and invade vascular cells, including endothelial and smooth muscle cells [15, 16]. Further, P. gingivalis has been shown to accelerate atherosclerosis in animal model studies [17-19]. We recently demonstrated that infection with P. gingivalis induces increased leukocyte adhesion to human aortic endothelial cells (HAEC), enhances procoagulant responses in HAEC and promotes up-regulation of adhesion molecule expression and pro-inflammatory cytokine production by this cell type [20, 21].
Although P. gingivalis has been shown to invade VSMC, its effects on VSMC phenotype and function have not been previously explored. Therefore, we sought to investigate P. gingivalis' ability to modulate human aortic smooth muscle cell (HASMC) expression of tissue factor (TF), a principal initiator of the coagulation cascade. In addition, we aimed to assess P. gingivalis' modulation of tissue factor pathway inhibitor (TFPI), the primary physiological inhibitor that regulates TF-dependent blood coagulation.
Materials and methods
Cells, bacterial strains, and culture conditions
Clonetics® HASMC were purchased and maintained in smooth muscle cell medium (SmGM-2, Lonza Walkersville Inc, Walkersville, MD) with supplements provided by the supplier. Confluent 4th-6th passage cells were used in all experiments. P. gingivalis FDC381 was grown on blood agar plates (Anaerobe Systems, Morgan Hill, CA) in anaerobic chambers at 37°C. DPG3, its fimbriae-deficient mutant constructed by insertional inactivation of the fimA gene [22], was grown on blood agar plates supplemented with erythromycin (Anaerobe Systems) in anaerobic chambers at 37°C. Bacterial suspensions were prepared in phosphate buffered saline without Mg2+/Ca2+ (PBS) using established growth curves and spectrophotometric analysis. In some experiments, heat-killed P. gingivalis 381 and ultrapure P. gingivalis lipopolysacharide (LPS) (Invivogen, San Diego, CA) were used as controls. The endotoxin activity of the P. gingivalis LPS preparation was 17.3 ± 3.96 EU/ng, as determined by the Limulus Amebocyte lysate method (Lonza Walkersville Inc, Walkersville, MD).
Co-incubation of HASMC with P. gingivalis and its LPS
HASMC were seeded (105/well) in 24-well plates (Corning, Acton, MA) for 24 hours and were then infected with viable, or heat-killed, P. gingivalis 381 or DPG3 at a multiplicity of infection (MOI) of 1:100 (i.e. 100 bacteria per cell) for 90 minutes in a 37°C, 5% CO2 environment. In order to approximate in vivo conditions, the bacteria were not centrifuged onto the HASMC to promote intimate contact. For all experiments MOI was calculated based on the number of cells per well when seeded. After the incubation period, cells were washed with PBS, and maintained in SmGM-2 medium without antibiotics for 8 or 24 hours. The cell culture supernatant was collected and stored at -70°C until analyzed. In additional experiments, HASMC were co-incubated with various concentrations of P. gingivalis LPS for either 8 or 24 hours. After the incubation period, cell culture supernatants were harvested and kept frozen until further analyses.
Antibiotic protection assay and assessment of HASMC necrosis
In order to confirm the potential of P. gingivalis 381 to invade HASMC in our experiments, recovery of viable microorganisms from antibiotic-treated cells was assessed. Confluent HASMC on a 96-well plate (Corning) were infected with P. gingivalis 381 or DPG3 (MOI 1:100) for 90 min at 37°C, 5% CO2. After washing, metronidazole (200 mg/ml, Sigma) was added to each well for 1 hour. Control experiments confirmed that extracellular bacteria were killed at this antibiotic concentration. Following metronidazole treatment, cells were washed, lysed with sterile deionized water, and lysates were plated on blood agar plates. Colonies were counted after a three-day incubation under standard anaerobic conditions. To assess non-specific necrosis, smooth muscle cell membrane integrity was measured using a lactate dehydrogenase (LDH) assay (Roche, Mannheim, Germany). Data are given as percent specific LDH release.
Determination of TF levels, TF activity, TFPI levels, and TFPI activity
After the cell culture supernatants were removed, cells were lysed by 3 freeze-thaw cycles. TF was extracted for 30 minutes in TRIS buffered saline containing 0.1% Triton X-100 at 37°, samples were collected and stored at -70°C until analyzed. Expression of total TF in cell extracts was assessed by ELISA (American Diagnostica, Stamford, CT). TF procoagulant activity in cell extracts was determined using a two-stage chromogenic activity assay (American Diagnostica). In the first stage of this assay, the TF in the sample is allowed to complex with factor (F) VIIa to generate TF/FVIIa complexes and convert FX into FXa. In the second stage, FXa cleaves Spectrozyme® FXa, a highly specific FXa chromogenic substrate and a chromophore is released. Absorbance is read and compared to values from a standard curve generated using known amounts of active TF. Levels of total TFPI and TFPI activity were quantified in culture supernatants, using a commercially available ELISA kit and a three-stage chromogenic activity assay, respectively (American Diagnostica). The latter measures the ability of TFPI to inhibit the catalytic activity of the TF/FVIIa complex to activate Fx to FXa. After incubation of samples with TF/FVIIa and FX, the residual activity of the TF/FVIIa complex is measured using Spectrozyme® FXa. The TFPI activity present in the sample is interpolated from a standard curve constructed using known TFPI activity levels.
Data and statistical analysis
All experiments were performed in duplicate wells for each condition and repeated at least three times. Data are presented as mean ± SD and n represents the number of experiments. Statistical comparisons were performed using Student's t-tests and the resultant p values are reported. Under the Bonferroni adjustment for multiple comparisons, an individual p<0.0125 is necessary to achieve statistical significance at the 5% level.
Results
P. gingivalis interaction with HASMC
We first addressed the potential of P. gingivalis to adhere to and/or invade HASMC in our experimental setting using antibiotic protection assays. P. gingivalis 381 had the ability to adhere to/invade HASMC, which was significantly higher compared to its fimbriae-deficient mutant, DPG3. Mean counts of invading bacteria were 3.22 ± 0.35 × 103 for strain 381 vs 0.50 ± 0.18 × 103 for DPG3 (p=0.0013, n=3).
Since infection of eukaryotic cells with an intracellular pathogen may lead to cell death, we carefully observed our infected cells under phase-contrast microscopy. At no time did the infected HASMC show any signs of cell death, such as morphologic shrinking, rounding or detachment at the MOI used in our experiments. To further assess cell death, we employed an LDH release assay. As depicted in Fig. 1A, infection of HASMC with P. gingivalis 381 at MOI 1:100 did not increase LDH release after 8h (19.22 ± 0.90 %) compared to non-infected control (16.77 ± 2.09 %), DPG3 (18.10 ± 3.87 %), or heat-killed P. gingivalis 381 (17.73 ± 1.64 %). This was also true for the 24h time point (Fig. 1B; non-infected 21.01 ± 3.10 %, P. gingivalis 381 20.62 ± 2.91 %, DPG3 22.23 ± 6.37 %, and heat-killed P. gingivalis 381 18.98 ± 1.47 %).
Figure 1. HASMC necrosis assessed by lactate dehydrogenase (LDH) release.
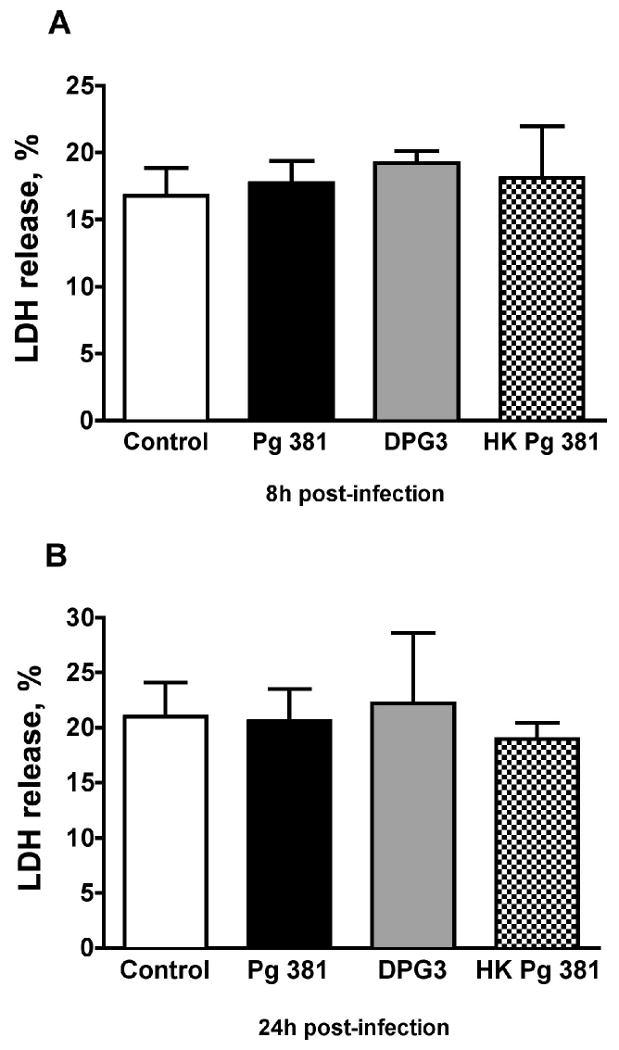
Infection of HASMC with P. gingivalis 381 (Pg 381) at MOI 1:100 did not increase LDH release after 8h (A) compared to non-infected control, non-invasive mutant DPG3, or heat-killed P. gingivalis 381 (HK Pg 381). This was also the case at the 24h time point (B); n=4 for all groups. Mean values are shown and error bars denote standard deviations.
P. gingivalis and TF levels and activity in HASMC
We next determined if P. gingivalis-infected HASMC demonstrated increased TF expression. As shown in Fig. 2, there was no significant upregulation in TF antigen in HASMC extracts at 8h (non-infected 73.14 ±5.19 pg/ml, P. gingivalis 381 75.70 ± 6.70 pg/ml, DPG3 69.46 ± 0.72 pg/ml, and heat-killed P. gingivalis 381 75.01 ± 4.30 pg/ml), or 24h after infection (non-infected 117.81 ± 4.48 pg/ml, P. gingivalis 381 127.45 ± 9.18 pg/ml, DPG3 121.93 ± 6.76 pg/ml, and heat-killed P. gingivalis 381 120.52 ± 6.70 pg/ml). Further, we assessed TF activity, as the extent of TF protein induction in vascular cells does not always correlate well with its activity [23, 24]. P. gingivalis 381-infected HASMC also failed to demonstrate increased TF activity at both times points tested (data not shown).
Figure 2. P. gingivalis and TF antigen levels in HASMC extracts.
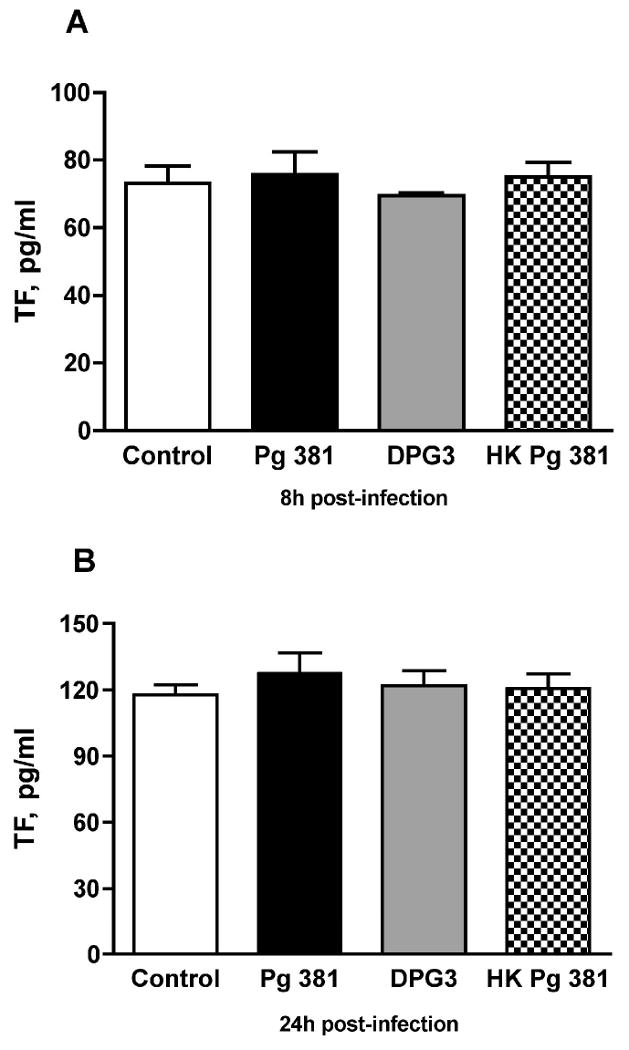
There was no significant upregulation in TF levels in HASMC extracts at 8h (A) or 24h (B) after P. gingivalis 381 infection (Pg 381), compared to non-infected control, DPG3 infection, or heat-killed P. gingivalis 381 (HK Pg 381); n=5 for Pg 381 group and 4 for all other groups. Mean values are shown and error bars denote standard deviations.
P. gingivalis and TFPI antigen and activity in HASMC
Since TFPI, the endogenous inhibitor of TF, is also an important regulator of coagulation, we assessed its expression in HASMC in response to P. gingivalis infection. As shown in Fig. 3A, at 8 hours after infection with P. gingivalis 381 we found a statistically significant 40% decrease in levels of TFPI in HASMC supernatants (0.35 ± 0.05 ng/ml) as compared to the non-infected control group (0.58 ± 0.07 ng/ml; n=4 for both, p=0.0029). Similarly, the levels of TFPI in P. gingivalis 381 infected cells were significantly suppressed compared to both those in DPG3-infceted cells (0.55 ± 0.1 ng/ml; p=0.0107) and in the heat-killed P. gingivalis 381 group (0.68 ± 0.01 ng/ml, p=0.0003; n=4 for all). Further, as shown in Fig. 3B, at 24 hours post-infection P. gingivalis 381 significantly reduced TFPI levels by approximately 70% in HASMC (1.41 ± 0.22 ng/ml), as compared to 5.25 ± 0.44 ng/ml in the non-infected control group (p<0.001), 4.45 ± 0.79 ng/ml in the DPG3-infected group (p=0.0036) and 4.77 ± 0.47 ng/ml in the heat-killed P. gingivalis 381 group (p=0.0001; n=4 for all). At both time points tested, levels of TFPI antigen were similar in control, DPG3- and heat killed P. gingivalis 381-infected cells.
Figure 3. P. gingivalis and TFPI antigen levels in HASMC.
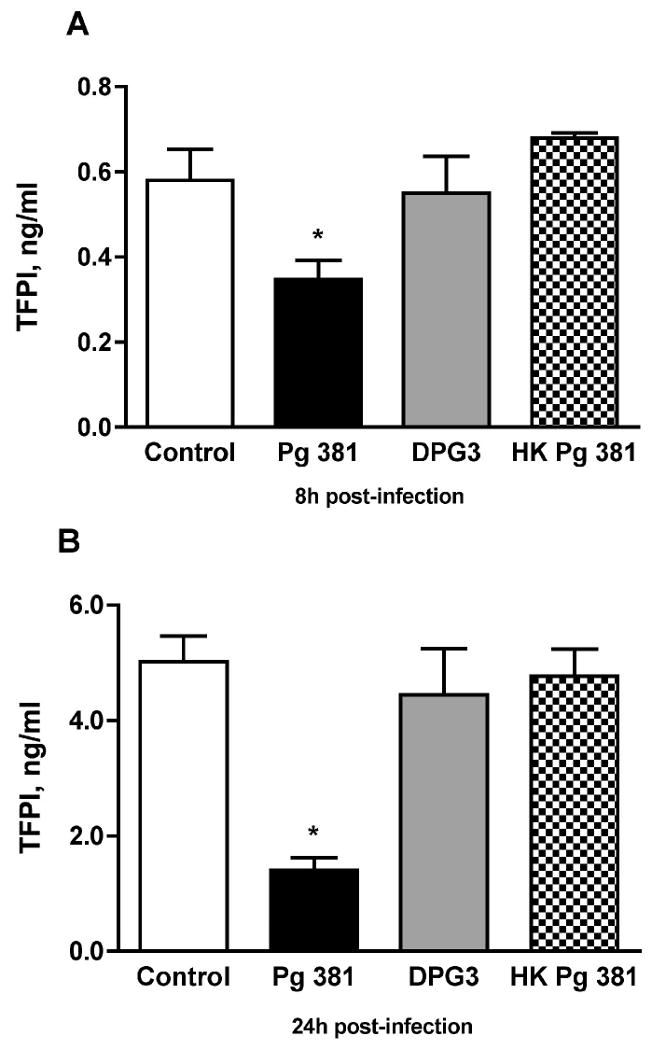
(A) At 8 hours after infection with P. gingivalis 381 (Pg 381), levels of TFPI in HASMC supernatants were significantly decreased as compared to the non-infected control group (n=4 for both, p=0.0029). Similarly, the levels of TFPI in P. gingivalis 381 infected cells were significantly suppressed compared to those in DPG3-treated cells (p=0.0107) and the heat-killed P. gingivalis 381 (HK Pg 381) group (p=0.0003; n=4 for all groups). (B) At 24 hours after infection P. gingivalis 381 significantly reduced TFPI in HASMC, as compared to the non-infected group (p<0.001), the DPG3-treated group (p=0.0036) and the heat killed P. gingivalis 381 group (p=0.0001; n=4 for all). At both time points tested, levels of TFPI antigen were similar in control, DPG3- and heat killed P. gingivalis 381-infected cells. Mean values are shown and error bars denote standard deviations. *indicates statistically significant differences (i.e., p < 0.0125 which under the Bonferroni adjustment for multiple comparisons is necessary for significance at the 5% level) compared to control, DPG3 and HK Pg 381 groups (individual p values for each comparison shown above)
We then assessed TFPI activity, which was below detection at the early time point, but could be quantified at the 24h timepoint. P. gingivalis 381 infection completely suppressed TFPI activity in HASMC (0.001 ± 0.0005 units/ml) compared to the non-infected control group (0.055 ± 0.009 units/ml; p=0.0011), the DPG3-infected group (0.013 ± 0.004 units/ml; p=0.0099) and the heat-killed P. gingivalis 381-treated group (0.047 ± 0.011 units/ml; p=0.0039; n=4 for all groups). TFPI activity in DPG3-infected cells was also significantly suppressed compared to control cells (p=0.0006), but it was still significantly higher than in P. gingivalis 381-infected cells (p=0.0099).
As P. gingivalis is a Gram (-) pathogen, in additional experiments we tried to elucidate the effect of its LPS (in various concentrations) on TFPI production. As seen in Fig. 4A, P. gingivalis 381 LPS did not induce any effect on HASMC TFPI levels at 8h (0.25 μg/ml: 0.59 ± 0.08 ng/ml, 1 μg/ml: 0.55 ± 0.03 ng/ml, 5 μg/ml: 0.50 ± 0.01 ng/ml) as compared to control untreated cells (0.48 ± 0.08 ng/ml; n=3 for all). Similarly at 24h (Fig. 4B), TFPI levels in LPS-treated cells were not significantly affected (0.25 μg/ml: 5.55 ± 0.72 ng/ml, 1 μg/ml: 4.31 ± 0.24 ng/ml, 5 μg/ml: 4.30 ± 0.38 ng/ml, control: 4.80 ± 0.33 ng/ml; n=3 for all). These data suggest that the observed P. gingivalis 381 procoagulant effect cannot be attributed to its LPS, but requires the invasion of HASMC by viable bacterial cells.
Figure 4. Effect of P. gingivalis 381 LPS on HASMC TFPI production.
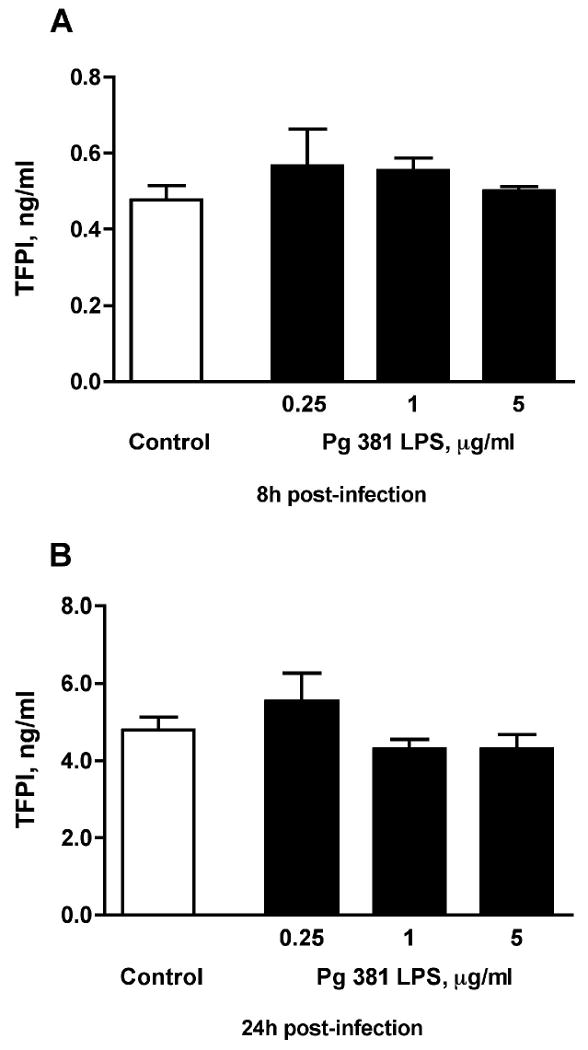
(A) P. gingivalis 381 LPS at 3 different concentrations (0.25 μg/ml, 1 μg/ml, or 5 μg/ml) did not induce any effect on HASMC TFPI levels at 8h as compared to control untreated cells. Similarly at 24h (B), TFPI levels in LPS-treated cells were not significantly affected; n=3 for all groups. Mean values are shown and error bars denote standard deviations.
Discussion
Our data demonstrate that P. gingivalis 381 has the ability to adhere to and invade HASMC and does not have a deleterious effect on smooth muscle cell survival within 24 hours following infection at MOI 1:100. Moreover, infection of HASMC with live whole P. gingivalis 381 does not affect levels or activity of TF, but leads to a significant decrease in TFPI levels and activity. Interestingly, this cannot be attributed to a sole LPS effect, but requires the invasion of smooth muscle cells by viable bacteria.
Viability of bacteria is necessary for invasion and heat-killed bacteria cannot invade. Indeed, in this study, heat killed P. gingivalis 381 had no effect on HASMC. In an analogous fashion, P. gingivalis 381 LPS had no effect on TFPI production by HASMC. In agreement with our present findings, prior evidence indicates that LPS of P. gingivalis exhibits unique properties distinct from that of other bacterial species [21, 25].
The exact mechanisms by which P. gingivalis may enter human cells are still elusive, but the use of lipid rafts as a portal of entry has been suggested [26]. The precise intracellular fate of internalized bacteria and the exact nature of the processes by which they modulate cell functions are also not completely clarified. However, in coronary endothelial cells as well as epithelial cells, internalized P. gingivalis cells have been shown to multiply and persist in vivo [27, 28]. This feature is common in bacteria that cause mucosal infections and could enable P. gingivalis to avoid host defense mechanisms.
Intracellular prokaryotic pathogens are well known to be able to induce cell death, by either apoptosis or necrosis, in the infected host cells. To ensure that any observed effects were not due to cell death, we addressed the question of smooth muscle cell survival following P. gingivalis infection in our study. At a MOI of 1:100 P. gingivalis 381 did not lead to increased smooth muscle cell death, as confirmed by phase contrast microscopy and LDH release, which is in line with our recent findings in human aortic endothelial cells [29].
Tissue factor is a key initiator of the coagulation cascade. TF-mediated activation can lead to occlusive thrombosis after plaque disruption, which is the immediate cause of most acute coronary symptoms. TF is also involved in promoting atherosclerotic plaque growth, by inducing migration and proliferation of vascular smooth muscle cells [30, 31]. In addition, it plays an important role at the crossroad of coagulation and inflammation. Previous studies have reported that mediators like TNF-α, CD40 ligand or endotoxin can induce TF expression on vascular endothelial cells and smooth muscle cells and thereby commence pathways that lead to thrombin generation [31]. We have previously demonstrated that endothelial cell infection with P. gingivalis 381 lead to a significant upregulation of TF levels and activity [20]. Interestingly, it appears that P. gingivalis infection effects are cell-specific, as in our current studies, we could not demonstrate an influence on levels or activity of TF in HASMC, although P. gingivalis 381 diminished TFPI production and activity in this cell type.
TFPI is a Kunitz-type serine protease inhibitor, which inhibits the initial reactions of the blood coagulation cascade, by specifically blocking TF. TFPI inhibits the FVIIa-TF complex and FXa with its first and second Kunitz domains, respectively [32, 33]. TFPI expression in vascular endothelial and smooth muscle cells is comparable, and its expression and co-localization with TF in atherosclerotic plaques suggests a significant role for TFPI in the regulation of TF activity [34, 35]. Moreover, in animal models reduced endogenous TFPI levels lead to enhanced thrombus formation [36], whereas overexpression was protective after vascular injury and resulted in reduced atherosclerosis [37, 38]. In addition to the anticoagulant effects, recent findings indicate that TFPI has an inhibitory effect on vascular cells [39] and it is able to attenuate the inflammatory response of the vascular wall to TF [32, 40]. We recently demonstrated that live P. gingivalis 381 can adhere to and invade HAEC, and that it is its invasive capacity that is involved in modulating properties of endothelial cells linked to prothrombotic pathways, including suppression of TFPI production [20, 21]. Based on the present findings, it appears that P. gingivalis 381 may also exert a prothrombotic effect in HASMC, by suppressing TFPI levels, and its ability to invade these cells appears to play a vital role in this. Although the in vivo significance of TFPI activity is not as well understood, we found that P. gingivalis 381 also significantly reduced TFPI activity, compared to control or DPG3-infected cells. Interestingly, despite the fact that the non-invasive mutant strain failed to affect TF levels and activity or TFPI production by HASMC, it did reduce TFPI activity. The latter effect was significant when compared to control cells, but was also significantly smaller that the effect of P. gingivalis 381. This suggests that bacterial properties beyond the pathogen's invasive capacity may also contribute in this setting. Previous studies have also shown that DPG3 is unable to invade vascular cells [16] or to accelerate atherosclerosis in hypercholesterolemic apoE-null mice [18], although its LPS is not distinguishable from that of the wild type strain [22].
Taken together, the present study makes the novel observation that P. gingivalis elicits a prothrombotic response in HASMC, by modulating TFPI production. Our findings add to the understanding of this pathogen's role in pathways linked to plaque progression and instability, and further help to explain the link between periodontal infections and atherosclerosis-related vascular disease.
Acknowledgments
This work was supported by NIH grant DE14575. We are grateful to Ms. Romanita Celenti for her technical assistance.
Abbreviations
- VSMC
vascular smooth muscle cells
- HAEC
human aortic endothelial cells
- HASMC
human aortic smooth muscle cells
- TF
tissue factor
- TFPI
tissue factor pathway inhibitor
- LPS
lipopolysacharide
- LDH
lactate dehydrogenase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hedin U, Roy J, Tran PK. Control of smooth muscle cell proliferation in vascular disease. Curr Opin Lipidol. 2004;15:559–65. doi: 10.1097/00041433-200410000-00010. [DOI] [PubMed] [Google Scholar]
- 2.Levi M, van der Poll T, Buller HR. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109:2698–704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- 3.Elkind MS, Cole JW. Do common infections cause stroke? Semin Neurol. 2006;26:88–99. doi: 10.1055/s-2006-933312. [DOI] [PubMed] [Google Scholar]
- 4.Papapanou PN. Periodontal diseases: epidemiology. Ann Periodontol. 1996;1:1–36. doi: 10.1902/annals.1996.1.1.1. [DOI] [PubMed] [Google Scholar]
- 5.Beck JD, Offenbacher S. The association between periodontal diseases and cardiovascular diseases: a state-of-the-science review. Ann Periodontol. 2001;6:9–15. doi: 10.1902/annals.2001.6.1.9. [DOI] [PubMed] [Google Scholar]
- 6.Desvarieux M, Demmer RT, Rundek T, Boden-Albala B, Jacobs DR, Jr, Sacco RL, et al. Periodontal microbiota and carotid intima-media thickness: the Oral Infections and Vascular Disease Epidemiology Study (INVEST) Circulation. 2005;111:576–82. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seinost G, Wimmer G, Skerget M, Thaller E, Brodmann M, Gasser R, et al. Periodontal treatment improves endothelial dysfunction in patients with severe periodontitis. Am Heart J. 2005;149:1050–4. doi: 10.1016/j.ahj.2004.09.059. [DOI] [PubMed] [Google Scholar]
- 8.Behle JH, Papapanou PN. Periodontal infections and atherosclerotic vascular disease: an update. Int Dent J. 2006;56:256–62. doi: 10.1111/j.1875-595x.2006.tb00110.x. [DOI] [PubMed] [Google Scholar]
- 9.Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res. 2002;90:2–4. [PubMed] [Google Scholar]
- 10.Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of periodontitis and endothelial function. N Engl J Med. 2007;356:911–20. doi: 10.1056/NEJMoa063186. [DOI] [PubMed] [Google Scholar]
- 11.Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000;71:1554–60. doi: 10.1902/jop.2000.71.10.1554. [DOI] [PubMed] [Google Scholar]
- 12.Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Jr, Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol. 2005;25:e17–8. doi: 10.1161/01.ATV.0000155018.67835.1a. [DOI] [PubMed] [Google Scholar]
- 13.Kinane DF, Riggio MP, Walker KF, MacKenzie D, Shearer B. Bacteraemia following periodontal procedures. J Clin Periodontol. 2005;32:708–13. doi: 10.1111/j.1600-051X.2005.00741.x. [DOI] [PubMed] [Google Scholar]
- 14.Giacona MB, Papapanou PN, Lamster IB, Rong LL, D'Agati VD, Schmidt AM, et al. Porphyromonas gingivalis induces its uptake by human macrophages and promotes foam cell formation in vitro. FEMS Microbiol Lett. 2004;241:95–101. doi: 10.1016/j.femsle.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 15.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Invasion of human coronary artery cells by periodontal pathogens. Infect Immun. 1999;67:5792–8. doi: 10.1128/iai.67.11.5792-5798.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deshpande RG, Khan MB, Genco CA. Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect Immun. 1998;66:5337–43. doi: 10.1128/iai.66.11.5337-5343.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, et al. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003;23:1405–11. doi: 10.1161/01.ATV.0000082462.26258.FE. [DOI] [PubMed] [Google Scholar]
- 18.Gibson FC, 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–6. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 19.Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, Beck J, et al. Porphyromonas gingivalis bacteremia induces coronary and aortic atherosclerosis in normocholesterolemic and hypercholesterolemic pigs. Arterioscler Thromb Vasc Biol. 2005;25:1446–51. doi: 10.1161/01.ATV.0000167525.69400.9c. [DOI] [PubMed] [Google Scholar]
- 20.Roth GA, Moser B, Huang SJ, Brandt JS, Huang Y, Papapanou PN, et al. Infection with a periodontal pathogen induces procoagulant effects in human aortic endothelial cells. J Thromb Haemost. 2006;4:2256–61. doi: 10.1111/j.1538-7836.2006.02128.x. [DOI] [PubMed] [Google Scholar]
- 21.Roth GA, Moser B, Roth-Walter F, Giacona MB, Harja E, Papapanou PN, et al. Infection with a periodontal pathogen increases mononuclear cell adhesion to human aortic endothelial cells. Atherosclerosis. 2007;190:271–81. doi: 10.1016/j.atherosclerosis.2006.03.018. [DOI] [PubMed] [Google Scholar]
- 22.Malek R, Fisher JG, Caleca A, Stinson M, van Oss CJ, Lee JY, et al. Inactivation of the Porphyromonas gingivalis fimA gene blocks periodontal damage in gnotobiotic rats. J Bacteriol. 1994;176:1052–9. doi: 10.1128/jb.176.4.1052-1059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camera M, Giesen PL, Fallon J, Aufiero BM, Taubman M, Tremoli E, et al. Cooperation between VEGF and TNF-alpha is necessary for exposure of active tissue factor on the surface of human endothelial cells. Arterioscler Thromb Vasc Biol. 1999;19:531–7. doi: 10.1161/01.atv.19.3.531. [DOI] [PubMed] [Google Scholar]
- 24.Steffel J, Akhmedov A, Greutert H, Luscher TF, Tanner FC. Histamine induces tissue factor expression: implications for acute coronary syndromes. Circulation. 2005;112:341–9. doi: 10.1161/CIRCULATIONAHA.105.553735. [DOI] [PubMed] [Google Scholar]
- 25.Darveau RP, Arbabi S, Garcia I, Bainbridge B, Maier RV. Porphyromonas gingivalis lipopolysaccharide is both agonist and antagonist for p38 mitogen-activated protein kinase activation. Infect Immun. 2002;70:1867–73. doi: 10.1128/IAI.70.4.1867-1873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belanger M, Rodrigues PH, Dunn WA, Jr, Progulske-Fox A. Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy. 2006;2:165–70. doi: 10.4161/auto.2828. [DOI] [PubMed] [Google Scholar]
- 27.Madianos PN, Papapanou PN, Nannmark U, Dahlen G, Sandros J. Porphyromonas gingivalis FDC381 multiplies and persists within human oral epithelial cells in vitro. Infect Immun. 1996;64:660–4. doi: 10.1128/iai.64.2.660-664.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorn BR, Dunn WA, Jr, Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun. 2001;69:5698–708. doi: 10.1128/IAI.69.9.5698-5708.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth GA, Ankersmit HJ, Brown VB, Papapanou PN, Schmidt AM, Lalla E. Porphyromonas gingivalis infection and cell death in human aortic endothelial cells. FEMS Microbiol Lett. 2007;272:106–13. doi: 10.1111/j.1574-6968.2007.00736.x. [DOI] [PubMed] [Google Scholar]
- 30.Viles-Gonzalez JF, Fuster V, Badimon JJ. Atherothrombosis: a widespread disease with unpredictable and life-threatening consequences. Eur Heart J. 2004;25:1197–207. doi: 10.1016/j.ehj.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 31.Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. 2006;113:722–31. doi: 10.1161/CIRCULATIONAHA.105.567297. [DOI] [PubMed] [Google Scholar]
- 32.Kato H. Regulation of functions of vascular wall cells by tissue factor pathway inhibitor: basic and clinical aspects. Arterioscler Thromb Vasc Biol. 2002;22:539–48. doi: 10.1161/01.atv.0000013904.40673.cc. [DOI] [PubMed] [Google Scholar]
- 33.Broze GJ, Jr, Warren LA, Novotny WF, Higuchi DA, Girard JJ, Miletich JP. The lipoprotein-associated coagulation inhibitor that inhibits the factor VII-tissue factor complex also inhibits factor Xa: insight into its possible mechanism of action. Blood. 1988;71:335–43. [PubMed] [Google Scholar]
- 34.Crawley J, Lupu F, Westmuckett AD, Severs NJ, Kakkar VV, Lupu C. Expression, localization, and activity of tissue factor pathway inhibitor in normal and atherosclerotic human vessels. Arterioscler Thromb Vasc Biol. 2000;20:1362–73. doi: 10.1161/01.atv.20.5.1362. [DOI] [PubMed] [Google Scholar]
- 35.Caplice NM, Mueske CS, Kleppe LS, Peterson TE, Broze GJ, Jr, Simari RD. Expression of tissue factor pathway inhibitor in vascular smooth muscle cells and its regulation by growth factors. Circ Res. 1998;83:1264–70. doi: 10.1161/01.res.83.12.1264. [DOI] [PubMed] [Google Scholar]
- 36.Ragni M, Golino P, Cirillo P, Scognamiglio A, Piro O, Esposito N, et al. Endogenous tissue factor pathway inhibitor modulates thrombus formation in an in vivo model of rabbit carotid artery stenosis and endothelial injury. Circulation. 2000;102:113–7. doi: 10.1161/01.cir.102.1.113. [DOI] [PubMed] [Google Scholar]
- 37.Nishida T, Ueno H, Atsuchi N, Kawano R, Asada Y, Nakahara Y, et al. Adenovirus-mediated local expression of human tissue factor pathway inhibitor eliminates shear stress-induced recurrent thrombosis in the injured carotid artery of the rabbit. Circ Res. 1999;84:1446–52. doi: 10.1161/01.res.84.12.1446. [DOI] [PubMed] [Google Scholar]
- 38.Zoldhelyi P, Chen ZQ, Shelat HS, McNatt JM, Willerson JT. Local gene transfer of tissue factor pathway inhibitor regulates intimal hyperplasia in atherosclerotic arteries. Proc Natl Acad Sci U S A. 2001;98:4078–83. doi: 10.1073/pnas.061004098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamuro T, Kamikubo Y, Nakahara Y, Miyamoto S, Funatsu A. Human recombinant tissue factor pathway inhibitor induces apoptosis in cultured human endothelial cells. FEBS Lett. 1998;421:197–202. doi: 10.1016/s0014-5793(97)01559-7. [DOI] [PubMed] [Google Scholar]
- 40.Esmon CT. Role of coagulation inhibitors in inflammation. Thromb Haemost. 2001;86:51–6. [PubMed] [Google Scholar]