

There is considerable debate about the value of personal genome sequencing (1). In addition to the five individuals whose genomes have been sequenced in their entirety, 68 patients have been evaluated for tumor-specific mutations in all exons of protein coding genes (exomic sequencing). This coincidentally yielded information about germline sequence variations in these individuals (2-4). To explore the utility of such information, we evaluated a pancreatic cancer patient (Pa10) whose tumor DNA had been sequenced in (4). This patient had familial pancreatic cancer, as defined by the fact that his sister also had developed the disease.
Among the 20, 661 coding genes analyzed, we identified 15,461 germline variants in Pa10 not found in the reference human genome. Of these, 7318 were synonymous, 7721 were missense, 64 were nonsense, 108 were at splice sites, and 250 were small deletions or insertions (54% in-frame). Past studies have shown that tumors arising in patients with a hereditary predisposition harbor no normal alleles of the responsible gene: one allele is inherited in mutant form, often producing a stop codon, and the other (wild type) allele is inactivated by somatic mutation during tumorigenesis. In Pa10, only three genes met these criteria: SERPINB12, RAGE and PALB2. Of these, we considered PALB2 to be the best candidate because germline stop codons in SERPINB12 and RAGE, but not in PALB2, are relatively common in healthy individuals and because germline PALB2 mutations have previously been associated with breast cancer predisposition and Fanconi anemia(5) although its function is not well understood. Pa10 harbored a germline deletion of 4 bp (TTGT at c.172-175) producing a frameshift at codon 58; the pancreatic cancer that developed in Pa10 had somatically acquired a transition mutation (C to T) at a canonical splice site for exon 10 (IVS10+2).
To determine whether PALB2 mutations occur in other patients with familial pancreatic cancer, we sequenced this gene in a cohort of 96 familial pancreatic cancer patients, 90 of which were of Caucasian ancestry. Sixteen of these patients had one first degree relative with pancreatic cancer and 80 had at least two additional relatives, at least one of which was first degree, with the disease. Truncating mutations were identified in three of the 96 patients, each producing a different stop codon (Fig. 1). The average age-of-onset of pancreatic cancer in these families was 66.7 years, similar to the mean age of onset of 65.3 years in the families without PALB2 mutations. We determined the germ-line sequence of an affected brother in one of these kindreds, and he harbored the same stop codon. Truncating mutations in PALB2 are rare in individuals without cancer; none have been reported among 1,084 normal individuals in a previous study using a cohort of similar ethnicity to ours (6). While some families we identified with a PALB2 stop mutation had a history of both breast and pancreatic cancer, breast cancer was not observed in all families (pedigrees in Supplementary Online Material). From these data, PALB2 appears to be the second most commonly mutated gene for hereditary pancreatic cancer. Interestingly, the most commonly mutated gene is BRCA2 (7), whose protein product is a binding partner for the PALB2 protein (8).
Fig. 1.
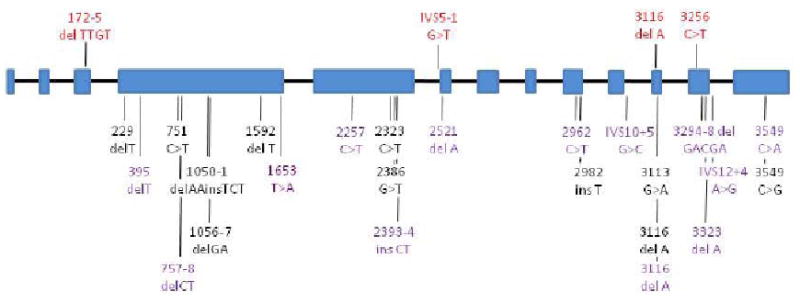
Location of mutations in the PALB2 gene. Exons are represented as blue boxes and introns as black lines (not to scale). Mutations previously identified in patients with familial breast cancer or Fanconi Anemia are shown in black or purple, respectively. Germline mutations identified in patients with familial pancreatic cancer are shown above the gene in red.
In summary, through complete, unbiased sequencing of protein–coding genes, we have discovered a gene responsible for a hereditary disease. We note that this approach is independent of classical methods for gene discovery, such as linkage analysis, which can be challenging in the absence of large families with monogenic diseases. We predict that variations of the approach described here will soon become a standard tool for the discovery of disease-related genes.
Acknowledgments
9. Supported by the The Sol Goldman Pancreatic Cancer Research Center, The Lustgarten Foundation for Pancreatic Cancer Research, The Cancer Foundation. Virginia and D.K. Ludwig Fund for Cancer Research, NIH grants CA62924, CA123483 and RO1CA97075, and the Michael Rolfe Pancreatic Cancer Foundation.
References
- 1.McGuire AL, Cho MK, McGuire SE, Caulfield T. Science. 2007;317:1687. doi: 10.1126/science.1147475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wood LD, et al. Science. 2007;318:1108. [Google Scholar]
- 3.Parsons DW, et al. Science. 2008;321:1807. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones S, et al. Science. 2008;321:1801. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turnbull C, Rahman N. Annu Rev Genomics Hum Genet. 2008;9:321. doi: 10.1146/annurev.genom.9.081307.164339. [DOI] [PubMed] [Google Scholar]
- 6.Rahman N, et al. Nat Genet. 2007;39:165. doi: 10.1038/ng1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maitra A, Hruban RH. Annu Rev Pathol. 2008;3:157. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia B, et al. Mol Cell. 2006;22:719. doi: 10.1016/j.molcel.2006.05.022. [DOI] [PubMed] [Google Scholar]