

Abstract
We report a general method for screening, in solution, the impact of deviations from canonical Watson-Crick composition on the thermodynamic stability of nucleic acid duplexes. We demonstrate how fluorescence resonance energy transfer (FRET) can be used to detect directly free energy differences between an initially formed “reference” duplex (usually a Watson-Crick duplex) and a related “test” duplex containing a lesion/alteration of interest (e.g., a mismatch, a modified, a deleted, or a bulged base, etc.). In one application, one titrates into a solution containing a fluorescently labeled, FRET-active, reference duplex, an unlabeled, single-stranded nucleic acid (test strand), which may or may not compete successfully to form a new duplex. When a new duplex forms by strand displacement, it will not exhibit FRET. The resultant titration curve (normalized fluorescence intensity vs. logarithm of test strand concentration) yields a value for the difference in stability (free energy) between the newly formed, test strand-containing duplex and the initial reference duplex. The use of competitive equilibria in this assay allows the measurement of equilibrium association constants that far exceed the magnitudes accessible by conventional titrimetric techniques. Additionally, because of the sensitivity of fluorescence, the method requires several orders of magnitude less material than most other solution methods. We discuss the advantages of this method for detecting and characterizing any modification that alters duplex stability, including, but not limited to, mutagenic lesions. We underscore the wide range of accessible free energy values that can be defined by this method, the applicability of the method in probing for a myriad of nucleic acid variations, such as single nucleotide polymorphisms, and the potential of the method for high throughput screening.
Thermodynamic studies of nucleic acid duplexes containing base adducts, mismatches, bulges, and other deviations from canonical Watson-Crick pairing/stacking reveal that relatively modest structural defects can be accompanied by profound energetic consequences (1–3). In fact, it has been shown that such defects/lesions can result in significant destablizations of the duplex, with magnitudes that are well beyond that which can be rationalized in terms of local structural perturbations (3). Because the free energy term, ΔGo, quantifies duplex stability, a systematic detection/screening for and comparison of defect-induced duplex destabilization requires a method (preferably a high throughput one) that yields accurate ΔGo determinations.
The free energy change associated with formation of a duplex can be obtained from the equilibrium association constant, K, via the well known relation ΔGo = −RTlnK. Because the values of K for oligonucleotide duplexes typically are beyond the range accessible by standard titration techniques, most thermodynamic studies of DNA have relied on indirect methods for determination of K and ΔGo (4). These methods often make use of the fact that the equilibrium association constant of a duplex can be lowered by increasing the temperature; in other words, the duplex can be melted. Several methods are available to extract K values from the resulting equilibrium melting curves (5). Such data, whether determined by optical methods or by scanning calorimetry, require knowledge of ΔHo so that the measured ΔG can be extrapolated to a desired reference temperature. This extrapolation can significantly reduce the precision with which ΔGo for oligonucleotide duplexes can be determined. A quick and reliable method, therefore, is needed that directly yields a solution value for ΔGo at a desired reference temperature.
We describe in this report a fluorescence-based assay that allows one to measure, in solution, the free energy consequences of any defect in a DNA duplex. The assay uses very little material and takes much less time than the standard panel of spectroscopic and calorimetric experiments necessary to obtain reliable ΔGo values. The assay yields free energies for association of “test” duplexes relative to a “reference” duplex. In one application of the assay, the association free energy of the reference duplex is determined separately, whereas in another application, one need not know ΔGo for the reference duplex. The assay is adaptable to virtually any deviation from Watson-Crick pairing, including, but not limited to, mismatches, chemical modification of single nucleotides (bases, sugars, or phosphate backbone), and DNA folding anomalies/motifs (e.g., loops, hairpins, etc.). If desired, detailed subsequent thermodynamic studies can be directed toward the most interesting defects and sequences that are identified by this screening assay.
MATERIALS AND METHODS
Oligonucleotide Synthesis.
Oligonucleotides used in this study were synthesized in-house on a PerSeptive Biosystems (Framingham, MA) 8909 synthesizer by using standard phosphoramidite chemistry. Oligonucleotides to be fluorescently labeled were synthesized with C6-amino-linker amidites (PerSeptive Biosystems) at the 5′ end. Cleavage and deprotection were achieved by incubation in ammonium hydroxide of the synthesis column support medium (72 h, room temperature). The support subsequently was washed twice with 200 μl of either water or ethanol:acetonitrile:water (3:1:1). Purification of unlabeled oligonucleotides was performed as described (6). Dye coupling and purification of the dye-labeled oligonucleotides are described below.
Fluorescent Modification of Amino-Linker-Terminated Oligonucleotides.
Fluorescent dyes with reactive succinimidyl ester groups were purchased from Molecular Probes. For this study, the succinimidyl ester forms of Oregon Green 514 (OG) and Rhodamine-Red-X (RdRX) were used. The linkage-forming chemistry should be similar, if not identical, for any similarly modified dye. The generic chemistry has been described elsewhere (7). Modifications were developed to optimize the purification of the 13 base oligonucleotides used in this study.
Although the amino-linker monomers terminate with an MMT group, rather than the usual DMT group, standard trityl-on purification via reverse phase HPLC was the first step in our purification, as recommended by the manufacturer. The purified oligonucleotides were dried under vacuum and were resuspended in water. Visible nonsoluble material was removed by pipetting the solution into a fresh tube. Excess salt was removed by ethanol precipitation. The DNA solution was adjusted to a final volume of 300–400 μl in water, was mixed thoroughly with 100 μl of 2 M sodium chloride and 950 μl of ethanol, and was chilled at −20°C for at least 1 h. Precipitation was induced by centrifugation at 16,000 × g in a benchtop centrifuge for 12 min, at a temperature below 20°C. The supernatant was removed by pipette and discarded. The remaining pellet was dried under vacuum.
The terminal monomethoxytrityl group was removed by dissolving the pelleted DNA in 200 μl of 80% acetic acid and incubating for 1 h at room temperature. The sample was evaporated to dryness, or until a glassy material remained. Water was added, and the sample was redried if the first drying step did not achieve dryness/glassiness within 2 h. The dried sample then was ethanol precipitated, as before.
Reverse phase HPLC using a standard “trityl-off” protocol was used to purify the deblocked DNA. Although this DNA has the six-carbon-amino-linker at the 5′ end, the elution profile of the DNA is quite similar to unmodified DNA on our polymeric reverse phase column (Hamilton). The resulting purified DNA was dried and ion-exchanged to sodium via ethanol precipitation.
Labeling of the DNA was performed by using a procedure adapted from the standard protocol recommended by the manufacturer (Molecular Probes). DNA (30–40 OD260 units) was dissolved in 270 μl of water in a sealable tube. Sodium hydrogen carbonate solution (30 μl of 1M, pH 8.3) was added. For each 20 OD of DNA used, ≈1 mg of the succinimidyl ester-modified fluorophore was dissolved in fresh DMSO (80 μl of DMSO/mg of dye). The DMSO solution was added to the reaction mixture, with an additional 20 μl of DMSO resulting from washing of the vial used to dissolve the dye. This solution was mixed thoroughly and allowed to incubate at 37°C overnight. Longer times did not significantly improve the labeling yield. The DNA was separated from unreacted dye by using a PD-10 Sephadex G-25 column (Amersham Pharmacia). The reacted mixture, diluted with water to 1 ml, was loaded onto the column, which first had been washed thoroughly with water, according to the manufacturer’s instructions. To “wash in” the sample, 1.6 ml of water was loaded. Four to six 600-μl elution fractions, resulting from loading 600 μl of water onto the column and collecting the outflow, were collected. Usually, there was a visible separation of the color during migration through the PD-10 column, with early eluting fractions containing the DNA and later fractions containing excess unreacted dye. The volume of the collected fractions was reduced to 300 μl by evaporation. Ethanol precipitation was performed on each fraction independently. Any precipitate was a mixture of unlabeled and labeled DNA, because any remaining unreacted dye would not precipitate.
Purification of labeled DNA from unlabeled DNA was performed on a Mono Q HR 5/5 FPLC column (Amersham Pharmacia). The column was initially equilibrated in a buffer (Buffer A) of 15% acetonitrile and 50 mM Tris⋅HCl (pH 7.2), and the elution buffer (Buffer B) was the same, except with 1M sodium chloride. The DNA precipitates were dissolved in water and were loaded onto the column. The typical elution gradient was from 10% Buffer B to 80% Buffer B over 20 min. Outflow was monitored at both 260 nm, detecting all DNA, and at the maximum absorbance of the dye (508 nm for OG or 575 nm for RdRX), thereby allowing discrimination of labeled and unlabeled DNA. The labeled samples were desalted by another round of ethanol precipitation.
Fluorescence Energy Transfer (FRET)-Based Titration Assay.
FRET experiments were performed in 100 mM sodium phosphate buffer at 25°C in a 4- × 4-mm cuvette using a Perkin–Elmer LS50B Luminescence Spectrometer. Relative fluorescence readings were taken with excitation slit at 5 nm and emission slit at either 5 or 15 nm. The wavelengths used were 508 nm for excitation, 528 nm for emission of OG, and 592 nm for emission of RdRX. After each addition of nucleic acid, an annealing step was performed (90°C for 3–5 min) by heating the cuvette in a heat block and cooling at 25°C in the jacketed fluorometer cuvette holder. Oligonucleotide concentrations were determined using extinction coefficients determined previously (6). We have demonstrated (data not shown) that, for the systems studied here, the presence of the fluorophore does not significantly alter the 260 nm absorbance of the DNA. Thus, the extinction coefficients for the unmodified oligonucleotides are still appropriate.
The assay was performed by determining first the relative fluorescence of a sample of the FRET donor-bearing oligonucleotide (D strand, in this case bearing OG). A stoichiometric amount of the acceptor strand (A strand), in this case the RdRX, was added from a concentrated stock solution, and the relative fluorescence was determined. These relative fluorescence values define the full range of observable fluorescence for the “free” D strand (ID) and complexed with acceptor form of the D strand (IAD).
Aliquots of the unlabeled competing strand, X, were added, and the relative fluorescence intensities were measured. The titration continued until the dilution-corrected fluorescence was restored to at least half of the difference between ID and IAD. This strand displacement titration of the reference duplex, DA, by the test strand, X, is shown below in Scheme 1. ![]()
Data Analysis for Determining Relative Free Energy Values for Duplex Association.
Equations analogous to those developed by Linn and Riggs (8) for competitive binding of lac repressor to operator and nonoperator DNA are used to analyze the titration data. Two equilibrium association constants are defined below, with subscripts f and t indicating free and total concentrations, respectively. The AD, AX, D, A, and X expressions represent the donor/acceptor, reference complex (AD), the competitor/acceptor, newly formed complex (AX), the donor strand (D), the acceptor strand (A), and the competitor or test strand (X).
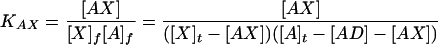 |
1 |
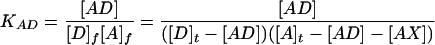 |
2 |
Note that these equations are for the test strand forming a complex with the acceptor strand. The opposite competition, with X binding to D, also can be described by simple substitution of the terms.
These basic equilibrium constant expressions can be combined and rearranged to yield the following equation for the concentration of the AD reference duplex:
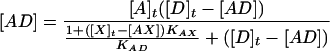 |
3 |
We define a value, θ, which is the fraction of the initially observed fluorescence energy transfer at each point in the titration. The value of θ is related to the ratio of the concentrations of the donor/acceptor complex ([AD]) and the total donor strand, ([D]t).
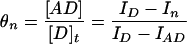 |
4 |
In Eq. 4, In is the dilution-corrected relative fluorescence at each point n; IAD is the fluorescence of the fully formed FRET duplex; and ID is the fluorescence of the donor strand alone. Note that, at the beginning of the titration, [AD] = [D]t. In Eq. 5, shown below, the dilution corrections are added explicitly, where Vo is the initial volume of the D-containing solution; VA is the volume of the added A-containing solution; and vi is the volume of the ith aliquot of the X-containing solution.
 |
5 |
With the reasonable assumption that [X]t ≫ [AX], which is valid when KAX < 0.1⋅KAD, Eqs. 3 and 4 can be combined to yield Eq. 6.
 |
6 |
We define X0.5 as the concentration of competing strand X at which θ = 0.5: in other words, the point at which exactly half of the acceptor/donor duplex, AD, has been disrupted. The value of X0.5 can be interpolated readily from a plot of θ vs. log[X]t. When θ = 0.5, the following simple relation exists between the desired equilibrium constant KAX, the measured value X0.5, and the known values [D]t and KAD:
 |
7 |
Application of the well known relation between ΔGo and K yields Eq. 8:
 |
8 |
As noted above, the value of KAD in the above equations is that which has been determined previously by independent methods, such as differential scanning calorimetry (DSC) and/or UV-absorbance melting (see, for example, refs 2, 3, and 6).
By extension, the impact of the difference between two oligonucleotides (X1 and X2) on duplex stability, ΔΔGo, also can be evaluated in separate experiments by using titrations of the two competitors (X1 and X2) against the same reference AD duplex, at the same AD concentration. In such a “differential” application, the relevant equation further simplifies to
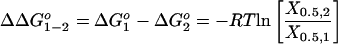 |
9 |
Note that, in Eq. 9, the value for KAD cancels. Therefore, one can evaluate the free energy impact (ΔΔGo) of a defect without independent thermodynamic characterization of the AD reference duplex. In this differential application of the assay, one simply determines the ratio of the concentration values of the two competitor strands required to reduce the signal from the reference duplex to half its original value.
RESULTS
Control Experiments
Before the new method described here can be applied to the measurement of ΔGo, a series of control experiments needs to be performed. First, it must be demonstrated that the DA duplex forms and that fluorescence energy transfer can be observed. It also must be demonstrated that energy transfer does not occur in the absence of an acceptor dye on the complementary strand. Finally, it must be shown that a test strand titrated into a solution of the DA duplex can compete thermodynamically for duplex formation with one of the dye labeled strands, without interference from kinetic effects. We describe below the controls that demonstrate that each of these requirements are met by the system studied here, thereby allowing the equations presented above to be used for calculating ΔGo.
Formation of a FRET-Labeled Duplex with One Dye of the Pair at Each 5′ End.
Although the basis of this assay is competition of an unlabeled test strand for duplex formation with an initially formed FRET reference duplex, it is imperative that formation of the FRET duplex itself first be demonstrated. The upper panel in Fig. 1 shows fluorescence spectra and relative fluorescence intensities collected during the course of a titration of acceptor strand, A, into a solution of donor strand, D (see scheme at top of Fig. 1). As expected, the fluorescence of the donor is reduced as acceptor is added, consistent with energy transfer. Further, a binding curve can be constructed that shows the expected one-to-one stoichiometry for duplex formation (see lower panel in Fig. 1). This curve, with the strands being present at concentrations (≈10 μM) that are considerably higher than the duplex dissociation constant, displays the expected linear dependence on the concentration of the acceptor strand and a clear discontinuity when saturation of the donor strand by the acceptor strand is achieved.
Figure 1.

Formation of the AD duplex. The top schematic shows the two labeled single strands binding to form a FRET-active duplex. The top graph shows the fluorescence emission spectra (excitation at 508 nm) throughout the titration. Note that the donor fluorescence decreases as acceptor is added. The inset shows emission spectra of the acceptor (RdRX), which fluoresces only very weakly. The lower graph shows a Job plot of the relative fluorescence at the emission maximum of the donor versus the mole fraction of acceptor strand. The discontinuity at 0.5 confirms the expected 1:1 stoichiometry of the duplex.
The lack of an increase in acceptor fluorescence seen in the upper panel of Fig. 1 is a departure from ideal energy transfer behavior. The RdRX dye has intrinsic fluorescence maxima at 572 nm for excitation and 592 nm for emission. The direct intrinsic fluorescence of the dye, albeit much weaker than the Oregon Green fluorescence, is observed for both the single strand and in duplexes (data not shown). Weak intrinsic fluorescence of dyes linked to oligonucleotides often is attributed to interaction of the dye with the DNA or undefined sequence effects at the point of attachment of the dye. To help evaluate these potential effects, we attached the dye to the opposite strand, thereby changing the nearest base from a G to a C. This exercise did not restore the intrinsic fluorescence (data not shown). We conclude that the low intrinsic fluorescence is caused by quenching of the excited state of the fluorophore because energy transfer is being monitored from the decrease in donor emission intensity.
An additional control is needed to prove that the decrease in donor fluorescence is indeed caused by energy transfer and not merely a fluorescence-altering artifact attributable to interaction of the donor with the newly formed duplex. This control is important because of the very low intrinsic fluorescence of the acceptor. An increase in acceptor fluorescence would provide conclusive proof of energy transfer; however, its absence does not mean FRET is not occurring. Titration of a donor-labeled strand with a complementary unlabeled strand, instead of a complementary acceptor-labeled strand, does not alter the OG intrinsic fluorescence (data not shown). The only difference between the two titrants is the presence of the acceptor dye. We therefore conclude that duplex formation can be monitored by fluorescence energy transfer, although, in this case, we are limited to monitoring a decrease in the donor intensity. The donor intensity decrease has complementary information content to the ideally expected increase in acceptor emission intensity (9).
An Annealing Step Is Required for Formation of the FRET Duplex.
For the sequences studied here, we find that aliquot addition must be followed by a heating/cooling cycle to rapidly achieve the expected duplex association. This annealing requirement is because both the OG- and RdRX-bearing single strands self-associate in solution. This self-association is reflected in optical melting transitions reminiscent of complex dissociation (data not shown). The stoichiometry and structures of the self-association complexes will be the subject of future studies. The following observations, however, suggest that these complexes are not thermodynamically significant when competing with the equilibrium that leads to complementary duplex formation. The observation of 1:1 stoichiometry for formation of FRET-labeled complexes is consistent with formation of duplex structures when the two strands are mixed in solution. This stoichiometry argues strongly for formation of duplexes, rather than reassociation into same-strand structures. Furthermore, if not heated and cooled, the donor fluorescence decreases over a period of minutes to hours, asymptotically approaching the intensity observed after an annealing cycle. Thus, the self-association complexes of the single strands are kinetically trapped. Heating to facilitate dissociation allows the proper duplexes to be formed directly. Because there is no change in fluorescence in the control experiment in which the OG strand is bound to an unlabeled complementary strand, we conclude that the self-associated states of the single strands do not interfere with OG fluorescence and therefore do not compromise the assay or the data analysis.
Unlabeled Oligonucleotides Can Compete for Duplex Formation, Disrupting the FRET Complex.
As shown in the upper panel of Fig. 2, we find that addition to a solution containing a donor-acceptor duplex (DA) of an unlabeled competitor or test strand (X), which is complementary to either the donor or acceptor strands of the DA duplex, causes an increase in donor emission. This observation of an increase in donor emission (or a decrease in FRET) is consistent with the unlabeled strand being able to compete for duplex formation by disruption of the FRET complex. As cartooned at the top of Fig. 2, formation of a duplex containing the unlabeled competitor strand results in displacement from the reference duplex of one of the dye-labeled strands. The loss of energy transfer because of separation of the donor/acceptor pair results in an increase in the donor intensity. Recall that duplex formation between the donor strand and unlabeled DNA does not alter the donor fluorescence. Therefore, the addition of the test strand must induce disruption of the initially formed FRET reference duplex.
Figure 2.

Titration of the unlabeled competitor strand, X, disrupts the AD duplex. The top schematic represents the competitive equilibria described in the text. The top graph shows the fluorescence emission spectra throughout the titration. Note that the donor fluorescence increases as competitor strand is added. In this case, the competitor strand (5′CGCATGCGTACGC3′) has the same sequence as the donor strand, so fluorescence of the donor can be nearly completely restored. The lower graph shows a plot of θ versus the logarithm of competitor concentration for two different competitors: the unlabeled donor strand as a control (which forms a Watson-Crick duplex designated as C), and a strand with a tetrahydrofuranyl abasic site analogue (5′CGCATGFGTACGC3′), which is known to form a duplex (designated as F) with lower free energy (6). Note that both curves exhibit the expected sigmoidal shape. Note also that the poor competitor strand, with the abasic site, must be added in considerably higher concentration to achieve a comparable reduction in θ.
Application of the Method
Comparison of Two Different Unlabeled Oligonucleotides Competing for the Same Starting FRET Duplex.
To determine the impact of a defect on duplex stability (ΔΔGo), two titrations are required. First, a titration in which the test strand (X) forms a standard Watson-Crick duplex is performed, and X0.5 is determined. Second, a defect-containing duplex is formed by titration of the second test strand (X′), and X′0.5 is determined. The two unlabeled competing strands used include one strand with a sequence identical to the OG-containing strand, representing an exactly matching control, and a second strand with an almost identical sequence except bearing at the central position a tetrahydrofuran (F) abasic site (10). Recent results from our lab have shown reductions in enthalpy and free energy for these same oligonucleotide sequences with F in the central position (6). Indeed, as shown in the lower panel of Fig. 2, our assay reveals that a much higher concentration of the defect-containing F strand is required to reduce the FRET intensity compared with the concentration required when the unmodified “native” strand is used. This observation demonstrates the lower affinity of the F-containing strand for its complement while validating the method.
Versatility of the Assay Over a Range of Concentrations.
Conventional titrations are limited to concentration regimes that depend on the value of the equilibrium association constant. The shape of a titration curve depends on the value of K. The curve then is fit to extract a K value. For the shape of the curve to be useful in determining K, there must be a significant amount of free titrant present in solution throughout the titration. When K is large, as in the case of oligonucleotide duplex equilibria, very low concentrations must be used to ensure reasonable values of free titrant. The requirement for concentrations on the order of 100/K means that detection of such binding/association events becomes problematic. By contrast, because the assay described here is based on quantifying competing equilibria, a wide range of DNA concentrations can be used. Rearrangement of Eq. 8 to derive Eq. 10 shows that ΔGoAX depends on the free energy of association of the AD duplex, ΔGoAD, and a concentration-dependent term.
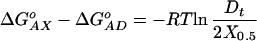 |
10 |
Note that the last term in Eq. 10 depends on the ratio of Dt to X0.5. A change in Dt results in a corresponding change in X0.5. We have demonstrated the expected sigmoidal shape in experiments in which the AD duplex is present initially at 10 μM or at 100 nM (data not shown); as expected, the higher initial duplex concentration requires a higher concentration of competitor to effect strand displacement. Because the concentration dependence of ΔGo is negligible, Dt can be selected to optimize the spectroscopic measurements.
Ability To Resolve Duplexes with Very Similar Free Energies.
Further examination of Eq. 10 indicates that the assay measures (ΔGoAX − ΔGoAD). This feature has significant advantages because the range of accessible ΔGoAX values is determined by ΔGoAD and not by Dt, as is the case with conventional titration experiments. Consequently, the choice of the AD reference duplex allows one to tune the assay for measurements over a very broad range of ΔGo values. A practical limit of Dt/X0.5 is 0.001, which corresponds to a (ΔGoAX − ΔGoAD) value of 3.7 kcal/mol. Note that this ΔΔGo value corresponds to a factor of ≈0.002 in association constant. Values of KAD (and ΔGoAD) can be modulated over a wide range by inclusion of mismatches or modified bases. Thus, the assay has both a broad range and high resolution for screening stability differences between duplexes (such as are imparted by nearly all “lesions” studied to date). We demonstrate below the high quality of the free energy data one can obtain using this method by comparing assay-derived ΔGo values with the corresponding values determined by extensive spectroscopic and calorimetric measurements.
Free Energies Calculated from This FRET Assay Are in Excellent Agreement with Those Measured by Extensive Thermodynamic Studies on Individual Duplexes.
We have conducted two titrations, at the same Dt concentration, for two starting Watson-Crick FRET duplexes, designated A⋅T (5′CGCATGAGTACGC3′⋅3′GCGTACTCATGCG5′) and T⋅A (5′CGCATGTGTACGC3′⋅3′GCGTACACATGCG5′), which differ only by the central base pair of the 13 pairs in the duplex. Competition on each duplex is from nearly the same single strand as present in the FRET duplexes, except this single strand is unlabeled and has a tetrahydrofuranyl abasic lesion site (F) at the central base pair. We designate this titrant single strand as F (5′CGCATGFGTACGC3′), and we designate as F⋅T and F⋅A the two new duplexes that can form via strand displacement by F within each of the two target, Watson-Crick, FRET duplexes (A⋅T and T⋅A). We find that the free energy values measured by our method compare quite favorably to those we have measured by extensive differential scanning calorimetry and UV absorbance melting experiments on these 13-mer duplexes containing a single tetrahydrofuranyl abasic site (F) in the central position (6). Specifically, using the FRET assay, we measured a value of −14.5 ± 0.1 kcal/mole for formation of the FT duplex, compared with a value of −15.1 ± 0.6 kcal/mole determined by using DSC/UV melts. Similarly, using the FRET assay, we measured a value of −16.2 ± 0.1 kcal/mole for formation of the F⋅A duplex compared with a value of −16.0 ± 0.4 kcal/mole by DSC/UV melts. These results correspond to ΔΔGo values of −1.7 ± 0.2 kcal/mole from FRET and −0.9 ± 1.0 kcal/mole from DSC/UV melting studies for substitution of an A residue for a T residue opposite the abasic site. We consider the good ΔGo and ΔΔGo agreements noted above as quantitative validation of the method described here.
DISCUSSION
Advantages of Combining FRET and Competing Equilibria To Determine Free Energy Differences Between Two Different Nucleic Acid Duplexes.
The assay we have described has several advantageous features that, when combined, provide a powerful and rapid method to assess the energetic consequences of nearly any local or global perturbation to a nucleic acid structure. We elaborate below on these features.
Nucleic acid duplexes (even damage-bearing duplexes) exhibit high stabilities and therefore low dissociation (high association) constants. As a result, traditional titrations of one strand into another must be carried out at very low nucleic acid concentrations for one to be able to monitor the progress of duplex formation. These concentrations generally are too low to be detected calorimetrically and often are too low to be detected even by standard spectroscopic means. By contrast, a competition experiment for duplex formation, such as the competing equilibria in the assay described here, can be performed over essentially any concentration range. As a result, the concentrations can be tailored to virtually any method of detection. We describe below the advantage of the FRET method of detection used in our competitive assay.
Competition assays, such as the one described here, require a means of discrimination between the complexes that are in competitive equilibria; in this assay, the AD and AX duplexes. Traditionally, the displaced strand would be labeled, and discrimination would be based on physical separation of the displaced strand from the duplexes by gel electrophoresis, filter binding, or other techniques. In our assay, we discriminate between duplexes with fluorescence energy transfer, which has the advantage of being a direct, in solution, spectroscopic technique that eliminates the need for further manipulations of the sample. FRET also has additional advantages over other common spectroscopic methods for monitoring duplex formation (11–13). UV absorbance can measure hyperchromicity on duplex formation and is typically monitored in “melting” experiments. However, the extinction coefficients of duplexes of similar length are relatively insensitive to the small differences in DNA composition, thereby precluding use of UV absorbance to monitor competitive equilibria. Similarly, circular dichroism, although sensitive to changes in duplex composition and structure, does not provide easy spectral deconvolution of the components of the competitive equilibrium. By contrast, FRET provides an extremely sensitive and essentially binary means of discrimination. Only a duplex with both the donor and acceptor dyes will have the spectroscopic signature of energy transfer.
Because fluorescence is much more sensitive than many other optical techniques, there is a tremendous saving in material needed (possibly by a factor of 1,000,000× or more). Further, although there have been significant advances in the sensitivity of differential scanning calorimeters, the material saved by the fluorescence assay is even more significant than traditional optical methods. Such savings can be particularly important for samples of specifically damaged DNA that may either be extremely difficult to synthesize and/or purify or which may be thermally unstable.
Applicability of the Assay to a Broad Scope of Variations in Nucleic Acids.
One of the key features of this method is the flexibility in the nature of the nucleic acid components that can be evaluated. To date, we have studied a simple system using 13-base oligodeoxynucleotides, with single defects incorporated in the competitor strand. However, application of the method is far more general, encompassing virtually any variation in the nature of the duplex and test strand competitor. For example, there is no specific requirement for any of the three strands to be DNA in chemical composition. RNA, peptido-nucleic acids, and other oligomers incorporating modified phosphates (e.g., methyl phosphonates, phosphorothioates, etc.) or modified sugar moieties (carbocyclics, etc.) that can form duplexes are applicable. Similarly, any of the vast number of base variations can be incorporated into any of the components, including intrastrand crosslinks, abasic sites, naturally occurring or synthetic base variants, base mimetics, and base adducts. Furthermore, although the system currently uses three independent strands of the same length, it could be adapted to address other structures such as large bulging/unpaired regions, competing internal loops and/or hairpins, or other deviations from simple duplex formation. The only requirements are that the FRET donor and acceptor be sufficiently close in the starting complex to provide energy transfer and that formation of the competing complex prevents energy transfer by displacement of either the donor or acceptor strand. For the systems studied here, we have demonstrated that these requirements can be readily met and that the method, in fact, can rapidly and conveniently yield data consistent with those obtained by using far more labor-intensive techniques. Furthermore, our technique requires far less material, has high throughput, and probes a duplex energy range inaccessible to other methods. The potential applications of this assay to a wide range of nucleic acid based diagnostics are clear.
Acknowledgments
This research was supported by National Institutes of Health Grants GM-23509, GM-34469, and CA-47795 to K.J.B.
ABBREVIATIONS
- OG
Oregon Green 514
- RdRX
Rhodamine-Red-X
- FRET
fluorescence resonance energy transfer
References
- 1.Plum G E, Breslauer K J. Ann NY Acad Sci. 1994;726:45–56. doi: 10.1111/j.1749-6632.1994.tb52796.x. [DOI] [PubMed] [Google Scholar]
- 2.Plum G E, Grollman A P, Johnson F, Breslauer K J. Biochemistry. 1995;34:16148–16160. doi: 10.1021/bi00049a030. [DOI] [PubMed] [Google Scholar]
- 3.Gelfand C A, Plum G E, Grollman A P, Johnson F, Breslauer K J. Biochemistry. 1998;37:12507–12512. doi: 10.1021/bi981090b. [DOI] [PubMed] [Google Scholar]
- 4.Bloomfield V A, Crothers D M, Tinoco I. Physical Chemistry of Nucleic Acids. New York: Harper & Row; 1974. , Chapter 6. [Google Scholar]
- 5.Breslauer K J. Methods Enzymol. 1995;259:221–242. doi: 10.1016/0076-6879(95)59046-3. (1995). [DOI] [PubMed] [Google Scholar]
- 6.Gelfand C A, Plum G E, Grollman A P, Johnson F, Breslauer K J. Biochemistry. 1998b;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- 7.Eis P S, Millar D P. Biochemistry. 1993;31:9227–9236. [Google Scholar]
- 8.Lin S Y, Riggs A D. J Mol Biol. 1972;72:671–690. doi: 10.1016/0022-2836(72)90184-2. [DOI] [PubMed] [Google Scholar]
- 9.Yang M, Millar D P. Methods Enzymol. 1997;278:417–444. doi: 10.1016/s0076-6879(97)78022-4. [DOI] [PubMed] [Google Scholar]
- 10.Takeshita M, Chang C N, Johnson F, Will S, Grollman A P. J Biol Chem. 1987;262:10171–10179. [PubMed] [Google Scholar]
- 11.Cooper J P, Hagerman P J. Biochemistry. 1990;29:9261–9268. doi: 10.1021/bi00491a022. [DOI] [PubMed] [Google Scholar]
- 12.Clegg R M. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 13.Vamosi G, Clegg R M. Biochemistry. 1998;37:14300–14316. doi: 10.1021/bi9727601. [DOI] [PubMed] [Google Scholar]