

Abstract
Thrombin potently induces endothelial inflammation. One of the responses is upregulation of adhesion molecules such as ICAM-1, resulting in enhanced leukocyte attachment to the endothelium. In this report, we examine the contribution of EphA2 in thrombin-induced expression of ICAM-1 in human umbilical vein endothelial cells (HUVECs). We showed that thrombin transiently induced tyrosine-phosphorylation of EphA2 in a Src-kinase dependent manner. This transactivation was mediated through PAR-1, because a PAR-1 specific agonistic peptide also transactivated EphA2. Expression knockdown of endogenous EphA2 by siRNAs blocked ICAM-1 upregulation and leukocyte/endothelium attachment induced by thrombin. Overexpression of exogenous mouse EphA2 rescued both ICAM-1 expression and leukocyte attachment induced by thrombin in endogenous EphA2-knockdown HUVECs. Mechanistically, we showed EphA2 knockdown suppressed thrombin-induced serine 536 phosphorylation of NFκB, an event critical of ICAM-1 transcriptional upregulation. Collectively, our results strongly suggest EphA2 is a necessary component for thrombin-induced ICAM-1 upregulation.
Keywords: EphA2, thrombin, ICAM-1, endothelial inflammation, receptor tyrosine kinases, PAR-1
Introduction
Thrombin exerts a wide range of cellular and molecular changes in endothelial cells. The principal receptors for thrombin belong to a class of receptors known as the protease-activated receptors (PARs), which initiate a complex network of intracellular signals including protein kinase C, PI3 kinase, Src, MAP kinases, and Rho kinase, to name a few (See reviews, [1-7]).
We hypothesize that thrombin achieves such diverse signaling capability by transactivation of other cell receptors, especially the receptor tyrosine kinases (RTK). In this report, we focused on EphA2 in thrombin-induced endothelial functions.
Eph receptors, the largest family of RTKs, are grouped into two classes based on their ligand specificity [8-11]. Class A Eph receptors bind to Ephrin-A ligands, which are GPI-linked cell surface proteins. Class B Eph receptors bind to Ephrin-B ligands, which are transmembrane cell surface proteins. Eph receptors are structurally similar. At the N-terminus is a globular domain, which contains the ligand binding site. It is followed by a cysteine-rich region, fibronectin-type III repeats, a transmembrane domain, a juxtamembrane region, a kinase domain, a SAM domain, and a PDZ-binding domain [10, 11].
EphA2 appears to play a crucial role in postnatal vascular function. Although EphA2 knockout mice are viable and fertile and show no overt abnormality during embryogenesis [12], endothelial cells isolated from these mice do not undergo vascular assembly in vitro in response to Ephrin-A1 stimulation [12]. Additionally, VEGF-induced endothelial cell migration in vitro and VEGF-induced corneal angiogenesis in mice have been shown to be influenced by the Ephrin-A1/EphA2 system, although the exact mechanism may depend on endothelial cell types. In HUVECs, addition of exogenous soluble EphA2-Fc blocks VEGF-induced angiogenesis, suggesting forward signaling through EphA2 receptor triggered by the interacting Ephrin-A1 ligand mediates the VEGF-induced response [13]. However, since HUVECs also abundantly express Ephrin-A1, it has been argued that the observed reduction of VEGF response may be due to blockade of reverse signaling through Ephrin-A1 [14]. In bovine retinal endothelial cells, where expression of Ephrin-A1 is not detected, stimulation of EphA2 by exogenous Ephrin-A1 appears to inhibit VEGF-induced vascular functions [14].
Importance of EphA2 in endothelial biology is also evidenced in cancer. Both EphA2 and Ephrin-A1 are overexpressed in tumor vasculature [15]. Tumor-induced angiogenesis and metastases are impaired in EphA2-deficient mice [16]. In addition, EphA2 is upregulated in aggressive melanoma cells undergoing vascular mimicry [17, 18]. Transient expression knockdown of EphA2 blocks these tumor cells from forming vascular structures in vitro [17].
Ephrins and Eph receptors have also been implicated in inflammation [9]. Ephrin-A1 was first identified as an immediate-early response gene in endothelial cells that was induced by inflammatory stimuli such as TNF-α, IL-1β, and lipopolysaccharide [19, 20]. Ephrin receptors, including EphA2, are upregulated during inflammation [21]. For example, the EphB/EphrinB system appears to play a role in inflammatory responses in rheumatoid arthritis [22]. Other than attribution of EphA2 as a mediator of TNF-α-induced angiogenesis in micro-pocket corneal assays in mice [23], very little is known about the specific functions of these Eph receptors/Ephrins in endothelial inflammation.
It is well established that thrombin potently induces ICAM-1 upregulation in endothelial cells through an NFκB-dependent pathway [24]. Results from several studies suggest that thrombin triggers a complex network of signals to upregulate ICAM-1 [25]. Examples of such signaling molecules include Gαq, Gβγ PI3 kinase, Akt, PKC-δ, c-Src, actin, ROCK, JNK, and p38 [26-33]. These signals result in activation of NFkB as a p65/p65 homodimer for ICAM-1 transcription [34]. Here, we report that the RTK EphA2 also plays a critical role in thrombin-induced ICAM-1 upregulation through activation of NFκB. Identification of this novel signaling component may provide additional targets for inhibition of endothelial inflammation responses induced by thrombin.
Materials and methods
Cell culture
HUVE cells were obtained from either Cambrex (East Rutherford, NJ) or Cascade Biologics (Portland, OR) and were cultured in EBM2-MV (Cambrex). Cells at passages <8 were used. Phoenix-Ampho cells were obtained from Dr. Nolan's laboratory at Stanford University and cultured in DMEM medium supplemented with 10% fetal bovine serum (Sigma, St. Louis). Human plasma thrombin (citrate-free) was purchased from Calbiochem (San Diego, CA). Soluble human EphA2 and EphrinA1-Fc (a chimeric protein composed of the extracellular domain of mouse Ephrin-A1 (amino acid residues 1 - 182) and the Fc region of human IgG1) were purchased from R&D Systems.
Antibodies
The following antibodies were used: goat polyclonal anti EphA2 (R&D Systems), rabbit polyclonal anti EphA2 (Santa Cruz), anti phosphotyrosine antibody 4G10 (Upstate), mouse monoclonal anti ICAM-1 (Santa Cruz), rabbit polyclonal anti α-actinin (Santa Cruz), and rabbit monoclonal anti phospho NFκB (Cell Signaling).
Phosphorylation of EphA2
To establish a time course of tyrosine phosphorylation of EphA2 by thrombin, a published protocol was used with slight modifications. This protocol was developed to detect tyrosine phosphorylation of VE-cadherin-associated proteins induced by thrombin in endothelial cells [35]. Briefly, confluent HUVECs were stimulated with thrombin (1U/ml) for the desired amount of time. Cells were then treated with pervanadate generated from 1 mM Na3VO4 /0.2 mM H2O2 in PBS at room temperature for 5 mins and lysed in iced-cold triton lysis buffer (50 mM Tris-HCl, pH 8/150 mM NaCl/ 5 mM EDTA/ 5mM EGTA/1%Triton-X100/ 1× complete protease inhibitor (Roche)/ 2 mM sodium orthovanadate/1 mM NaF/2.5 mM β glycerol phosphate/2.5 mM sodium pyrophosphate/1 mM phenylmethylsulphonyl fluoride). After brief sonication and centrifugation, clarified lysates were immunoprecipitated with 2 μg of a polyclonal anti human EphA2 antibody (R&D Systems) and Protein A/G Plus at 4°C overnight. Resins were washed with PBS three times and resuspended in 2× SDS gel loading buffer. Tyrosine phosphorylation was detected by western blot using the 4G10 antibody. The membrane was stripped and reblotted with an rabbit polyclonal antibody against EphA2 (Santa Cruz).
Pharmacological agents PP2 and Y27632 were used to examine the role of Src and ROCK in EphA2 phosphorylation. Confluent HUVECs were pretreated with PP2 (2 μM), Y27632 (5 μM), or DMSO for 1 hour, followed by a 10-min treatment with sodium orthovandate (1mM). Cells were then stimulated for 30 mins with either 1U/ml thrombin or 250ng/ml EphrinA1-Fc. HUVECs were lysed with RIPA buffer supplemented with 5 mM EDTA/ 5mM EGTA/1× complete protease inhibitor (Roche)/ 2 mM sodium orthovanadate/1 mM NaF/2.5 mM β glycerol phosphate/2.5 mM sodium pyrophosphate/1 mM phenylmethylsulphonyl fluoride. After brief sonication and centrifugation, clarified lysates were immunoprecipitated with 2 μg of a polyclonal anti human EphA2 antibody (Santa Cruz) and Protein A/G Plus at 4°C overnight. Resins were washed with the lysis buffer three times and resuspended in 2× SDS gel loading buffer. Tyrosine phosphorylation and EphA2 were detected using the 4G10 and the EphA2 antibody (Santa Cruz), respectively.
To determine the role of PAR-1 in EphA2 transactivation, HUVECs were prepared and stimulated as described above, except that the following peptides at 20 μM were used as stimulants: TFLLR-NH2 (PAR-1), RLLFT-NH2 (negative control for PAR-1), SLIGKV-NH2 (PAR-2), and GYPGKF-NH2 (PAR-4). PAR-1 and its control peptide were purchased from Peptides International. PAR-2 and PAR-4 peptides were synthesized as described [36].
EphA2 knockdown by siRNA, ICAM-1 upregulation, and NFκB phosphorylation
Two validated Stealth siRNA duplexes specific to human EphA2 and a negative control duplexes were purchased from Invitrogen (catalog numbers: 12938-022, 12935-300). A mixture of siRNA (0.2 nmol) and Silentfect (4 μl, BioRad) was prepared in 500 μl serum-free EBM2 basal medium. After incubation at room temperature for 20 mins, the mixture was added to 80%-confluent HUVECs in a 10-cm plate in the presence of 5 ml fresh EBM2-MV medium. Sixteen hours later, cells were split and seeded into 6-well plates. On the next day, the confluent HUVECs were stimulated with thrombin (1U/ml) in 2 ml EBM-2 supplemented with 0.5% FBS for either 1 (for NFκB phosphorylation) or 6 (for ICAM-1 upregulation) hours. Experiments were terminated with lysis of cells using 300μl 2× SDS PAGE loading buffer. NFkB phosphorylation and ICAM-1 protein expression were detected by western blots using an anti phospho-NFκB and an anti ICAM-1 antibodies, respectively.
Stable expression of mouse EphA2 in HUVECs by retrovirus
The coding region of mouse EphA2 was amplified from cDNAs prepared from 4T1 cells and cloned into a pLNCX2-based retroviral vector, in which an IRES-EGFP cassette was inserted downstream of mouse EphA2. To generate retrovirus, either IRES-EGFP-alone or EphA2-IRES-EGFP plasmid was transfected into Phoenix-Ampho cells using Lipofectamine 2000 (Invitrogen). Medium was replaced with serum-free DMEM supplemented with 10ng/ml bFGF and 200ng/ml EGF 16-hours post transfection. After an additional 24 hours, supernatant was collected and used to infect HUVECs in the presence of 40μg/ml protamine sulfate. Transduced cells were selected and expanded in the presence of 0.8mg/ml G418.
U937 attachment assays
A published procedure was followed with modifications [37]. Confluent HUVECs in a 24-well plate were stimulated with thrombin (5U/ml) in 2 ml EBM-2 supplemented with 0.5% FBS for 6 hours. Meanwhile, U937 were labeled with 0.1μM Cell Tracker Red CMTPX (Molecular Probes) in PBS for 10 mins, followed by a 6-hour recovery period in 10%FBS/RPMI. At the end of the 6-hour thrombin stimulation, labeled U937 cells were incubated with HUVECs at room temperature under rotation for 60 mins. Unattached U937 cells were washed off with growth medium. Cells were then fixed in 4% PFA in PBS for 15 mins prior to visualization by fluorescence microscopy.
Results
Thrombin induces tyrosine phosphorylation of EphA2 in HUVECs through PAR-1
Figure 1A illustrates the time course of EphA2 phosphorylation induced by thrombin. Tyrosine phosphorylation of EphA2 was detected as early as 2 minutes and lasted to at least 30 mins post thrombin-stimulation.
Figure 1.
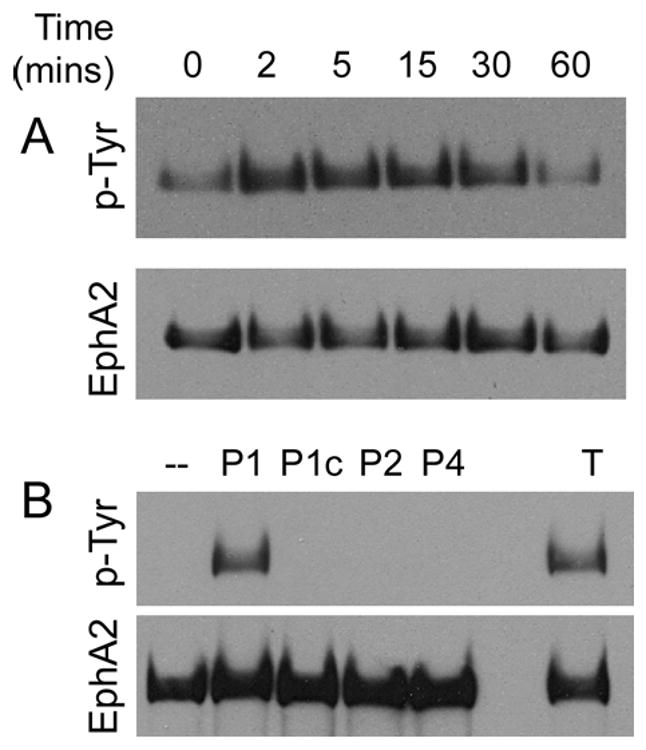
(A) Time course of EphA2 activation by thrombin stimulation in HUVECs. Confluent HUVECs were treated with 1U/ml thrombin for indicated amount of time. EphA2 was immunoprecipitated from clarified cell lysates using an EphA2-polyclonal antibody. Tyrosine phosphorylation was detected by western blot using the 4G10 anti-phosphotyrosine antibody. The blot was stripped and reblotted with the EphA2 antibody for loading. Representative data from three independent experiments are shown. (B) PAR-1 activated EphA2. Confluent HUVECs were stimulated with agonistic peptides TFLLR-NH2 (PAR-1, P1), RLLFT-NH2 (negative control for PAR-1, P1c), SLIGKV-NH2 (PAR-2, P2), GYPGKF-NH2 (PAR-4, P4), and thrombin (T). Tyrosine phosphorylation of EphA2 was determined by western blot. Experiments were done three times. Representative results are shown.
Thrombin signals through a class of cell surface receptors known as protease-activated receptors (PARs). Four PARs have been identified to date. PAR-1, 2, and 3 are expressed on human endothelial cells [38-48]. Although, northern analysis failed to detect PAR-4 expression in HUVEC in vitro [46], PAR-4 is detected in human coronary endothelial cells [49]. Amongst these receptors, thrombin specifically activates PAR-1, PAR-3, and PAR-4 [2, 50]. Based on the mechanism of PAR activation by thrombin, agonistic peptides have been developed to activate PAR-1, -2, and -4 specifically. To date, no agonistic peptide for PAR-3 has been reported (see reviews [1-5, 7]).
Therefore, we asked whether addition of agonistic peptides of PAR-1, -2, and -4 to HUVECs would recapitulate EphA2 transactivation by thrombin. In this experiment, we pretreated HUVECs with sodium orthovanadate for 10 mins, followed by a 30-min incubation with or without thrombin and PAR peptides. This protocol was designed to increase the detection sensitivity of any tyrosine phosphorylation of EphA2 induced by the agonistic peptide treatment. First we demonstrated that EphA2 transactivation by thrombin was once again detected using this protocol (Figure 1B). Activation of HUVECs with PAR-1 specific agonistic peptide induced EphA2 transactivation seen by thrombin treatment. A control PAR-1 peptide with a reverse sequence, PAR-2, and PAR-4 specific agonist failed to induce EphA2 tyrosine phosphorylation at the same concentration. These results strongly suggest that thrombin transactivates EphA2 through PAR-1 in HUVECs.
Soluble EphA2 fails to block thrombin-induced EphA2 phosphorylation
One possible mechanism of EphA2 activation by thrombin is through interaction of EphA2 and ephrins. To address this possibility, we tested the ability of the soluble extracellular domain of human EphA2 to block EphA2 transactivation by thrombin. This soluble EphA2 was expected to bind to its ligands. As shown in Figure 2, excess soluble EphA2 at 15 μg/ml did not block thrombin-induced phosphorylation of EphA2, while at the same concentration, the excess soluble EphA2 effectively blocked EphA2 phosphorylation induced by EphrinA1-Fc (at 250ng/ml). Therefore, our results strongly suggest transactivation of EphA2 induced by thrombin was independent of EphA2-ephrin interactions.
Figure 2.
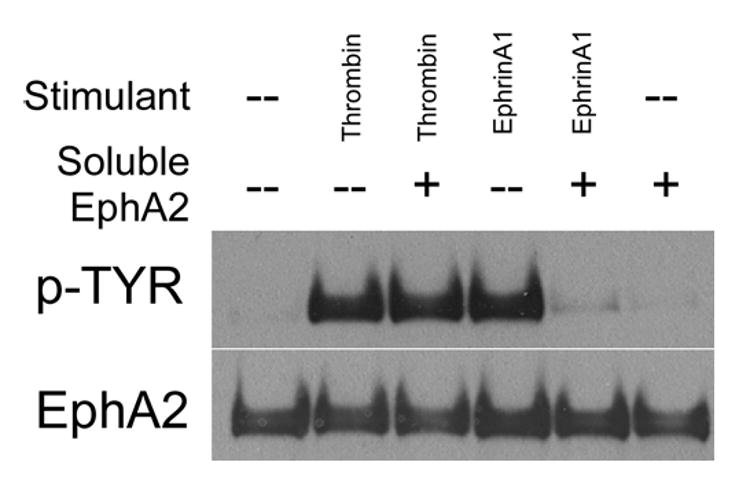
Soluble EphA2 failed to block endogenous EphA2 phosphorylation induced by thrombin. Confluent HUVECs were stimulated with either thrombin (1U/ml) or EphrinA1-Fc (250ng/ml) in the presence or absence of soluble EphA2 (15μg/ml) as in Figure 1B. EphA2 was immunoprecipitated and its tyrosine phosphorylation content determined by western blots using 4G10 antibody. The membrane was stripped and reblotted for EphA2 to ensure equal loading.
Transactivation of EphA2 induced by thrombin is Src-kinase dependent
Next, we investigated the mechanism by which thrombin caused EphA2 phosphorylation. We used pharmacological inhibitors to identify critical component(s) of thrombin signaling pathways in endothelial cells. Y-27632, a specific Rho-associated kinase (ROCK) inhibitor, was chosen because it has been shown to block tyrosine-phosphorylation of kinases down stream of thrombin stimulation in endothelial cells such as focal adhesion kinase (FAK) [51]. PP2, a well-characterized src-family kinase inhibitor, was also tested because it has been shown to mediate thrombin-induced transactivation of EGFR in cardiomyocytes and cardiofibroblasts [52, 53].
As shown in Figure 3A, thrombin-induced EphA2 tyrosine phosphorylation was completely abrogated by PP2, but not Y-27632. Our results suggest that thrombin caused EphA2 activation via a Src-family kinase. Next, we contrasted the activation mechanisms of EphA2 by thrombin and the canonical ligand EphrinA1. As expected, EphrinA1, when presented as a Fc-fusion protein, induced significant activation of EphA2 in endothelial cells (Figure 3B), as did thrombin. However, in contrast to thrombin stimulation, tyrosine phosphorylation of EphA2 by its cognate ligand EphrinA1 was insensitive to PP2 treatment. Therefore thrombin and EphrinA1 induced EphA2 phosphorylation via a Src-dependent and Src-independent pathway, respectively.
Figure 3.
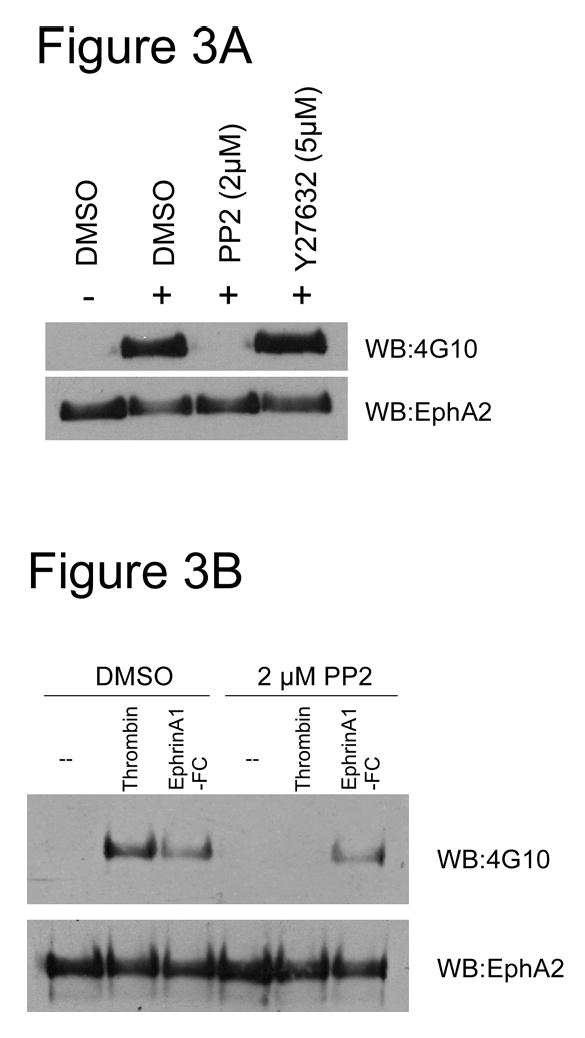
(A) Thrombin-induced tyrosine phosphorylation of EphA2 was dependent on Src kinase but not ROCK activity. Confluent HUVECs were pretreated with either DMSO, PP2 or Y27632 for 10 mins and then stimulated with 1 U/ml thrombin, as indicated by “+”. EphA2 was immunoprecipitated from clarified cell lysates using an EphA2-polyclonal antibody. Tyrosine phosphorylation was detected by western blot using the 4G10 anti-phosphotyrosine antibody. The blot was stripped and reblotted with the EphA2 antibody for loading. Representative data from 2 independent experiments were shown. (B) Mechanisms of activation of EphA2 by thrombin and by EphrinA1 were distinct. Samples were prepared as in (A), except EphrinA1 was also used as a stimulus.
Thrombin-induced ICAM-1 upregulation in HUVECs requires EphA2
Next, we sought to identify the function of EphA2 activation by thrombin in endothelial cells by using siRNA knockdown technology. Because thrombin potently upregulates ICAM-1 expression in endothelial cells, we opted to determine whether EphA2 was involved in this regulation. Two siRNA duplexes specific to human EphA2 were tested. As shown in Figure 4, transient transfection of each of these two siRNA duplexes significantly reduced EphA2 protein expression in HUVECs. Transfection of a control siRNA duplex had no effect on EphA2 expression. Thrombin significantly upregulated ICAM-1 expression in HUVECs treated with the control siRNA but failed to induce ICAM-1 expression when EphA2 was knockdown by either EphA2-specific siRNA duplex. To further corroborate the involvement of EphA2 in ICAM-1 upregulation by thrombin, we performed expression rescue experiments. HUVECs were transduced by either GFP- or mouse-EphA2 retrovirus. Infected cells were selected and expanded in the presence of G418. Retrovirus infection and G418 selection had no effects on thrombin-induced, EphA2-mediated ICAM-1 upregulation (Figure 5, left). Expression knockdown of endogenous EphA2 by siRNA again blocked ICAM-1 expression after thrombin stimulation in the GFP-virus infected group. However, in the presence of exogenously expressed mouse EphA2, thrombin-induced ICAM-1 upregulation was significantly restored even when endogenous human EphA2 was knocked down by siRNA (Figure 5, right). Collectively, our results strongly suggest that EphA2 is a downstream effector of thrombin stimulation and is critical in upregulation of ICAM-1 in endothelial cells.
Figure 4.
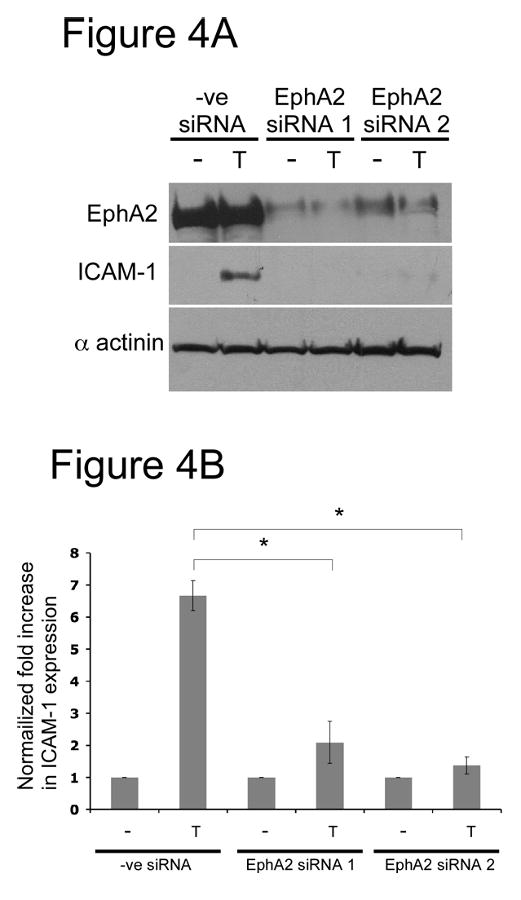
Thrombin-induced ICAM-1 upregulation was dependent on EphA2. HUVECs cells were transfected with either a control siRNA or one of the two EphA2-specific siRNA. Confluent HUVECs were stimulated with 1U/ml thrombin (T) for 6 hours. (A) Total lysates were used for western blots. Effective expression knockdown by both EphA2-specific siRNAs, thrombin-induced ICAM-1 expression, and protein loading were examined by western blot analyses using antibodies specific to EphA2, ICAM-1, and α-actinin, respectively. Representative data from 3 experiments are shown. (B) Densitometry analysis of ICAM-1 upregulation by thrombin in the presence or absence of EphA2. Western blots were scanned and analyzed by using the program ImageJ. Results were reported as fold-increase in ICAM-1 expression relative to unstimulated controls. Data are mean ± s.d. of three experiments; * p<0.002.
Figure 5.
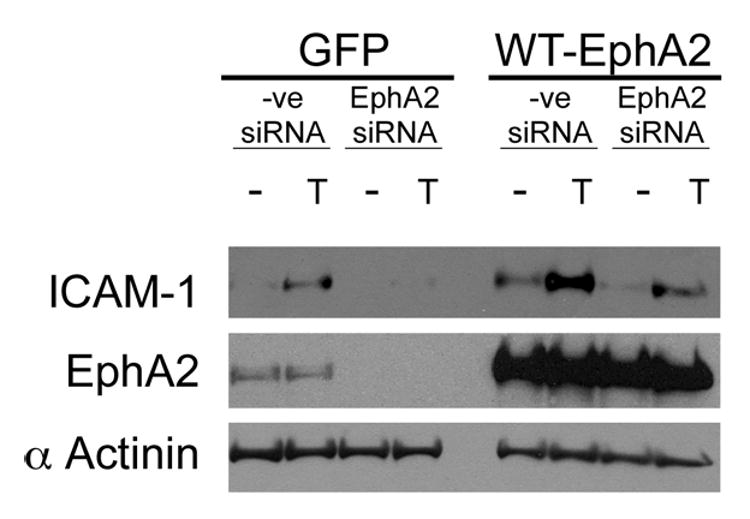
Overexpression of mouse EphA2 rescues thrombin-induced ICAM-1 upregulation in HUVECs with endogenous EphA2 knocked down. Either GFP or mouse EphA2 was stably overexpressed in HUVECs via retroviral infection. GFP or mouse EphA2 overexpressing cells were treated with either a control or a human EphA2 specific siRNA. ICAM-1 upregulation was then induced with 1U/ml thrombin (T) for 6 hours. Expression levels of EphA2, thrombin-induced ICAM-1 expression, and protein loading were examined by western blot analyses using antibodies specific to EphA2, ICAM-1, and α-actinin, respectively. Representative data from 4 experiments are shown.
Soluble EphA2 antibody fails to block thrombin-induced ICAM-1 expression in endothelial cells
We next asked whether addition of soluble EphA2 would block upregulation of ICAM-1 in endothelial cells. Soluble EphA2 was tested at 15μg/ml, because at this concentration soluble EphA2 effectively blocked EphA2 phosphorylation induced by EphrinA1-Fc (Figure 2). As shown in Figure 6A, addition of excess soluble EphA2 failed to inhibit ICAM-1 expression induced by thrombin. This result is consistent with our observation that excess soluble EphA2 was not able to block EphA2 activation induced by thrombin (Figure 2). Therefore, induction of ICAM-1 expression upon thrombin stimulation was not due to EphA2 interactions with ephrins. Indeed, addition of EphrinA1-Fc did not stimulate ICAM-1 expression in endothelial cells (Figure 6B).
Figure 6.
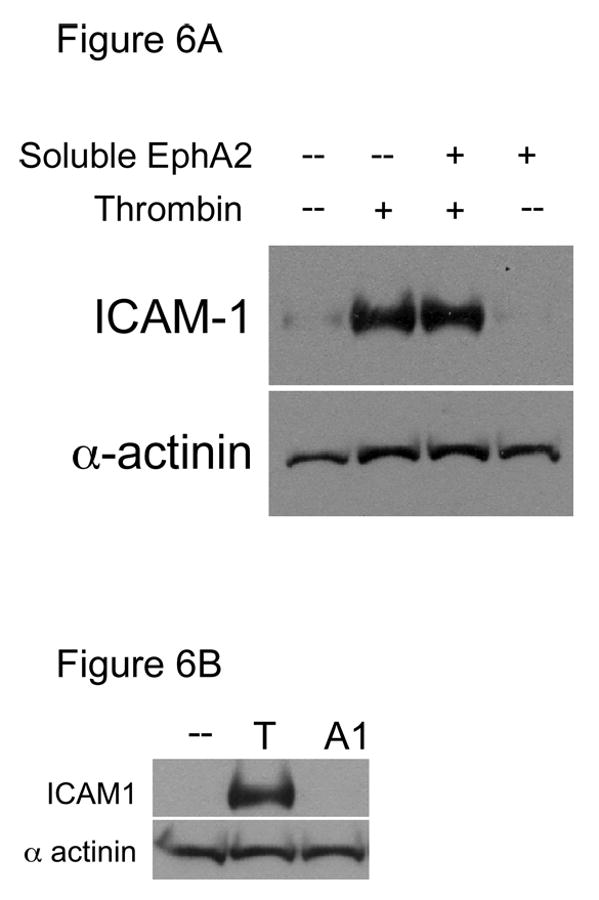
(A) Soluble EphA2 did not block thrombin-induced ICAM-1 expression in HUVECs. Confluent HUVECs were treated with 1U/ml thrombin (T) for 6 hours in the presence or absence of 15μg/ml soluble EphA2. Expression level of thrombin-induced ICAM-1 expression and protein loading was examined by western blot analyses using antibodies specific to ICAM-1 and α-actinin, respectively. Representative results from two experiments are shown. (B) EphrinA1 did not induce ICAM-1 expression. Confluent HUVECs were either untreated, thrombin- (T, 1U/ml), or EphrinA1-treated (A1, 250ng/ml) for 6 hrs. Ephrin A1 did not induce ICAM-1 expression, whereas at the same concentration it activated EphA2 (See Figure 2). In fact, EphrinA1 up to 10 μg/ml did not induce ICAM-1 expression (not shown). Representative results from three experiments are shown.
Thrombin activates NFκB via EphA2 in endothelial cells
It is well established that thrombin upregulates ICAM-1 in endothelial cells through the NFκB pathway [6, 24, 26, 27, 31, 32, 34, 54]. Therefore, we asked whether EphA2 played a role in NFκB activation by thrombin stimulation. As shown in Figure 7, when HUVECs were treated with the control siRNA duplex, thrombin induced serine-phosphorylation of NFκB, a well established marker for NFκB activation. When EphA2 expression was suppressed by either EphA2-specific siRNA duplexes, thrombin-induced serine-phosphoryation of NFκB was reduced. Our data suggest that phosphorylation of NFκB induced by thrombin is dependent on EphA2.
Figure 7.
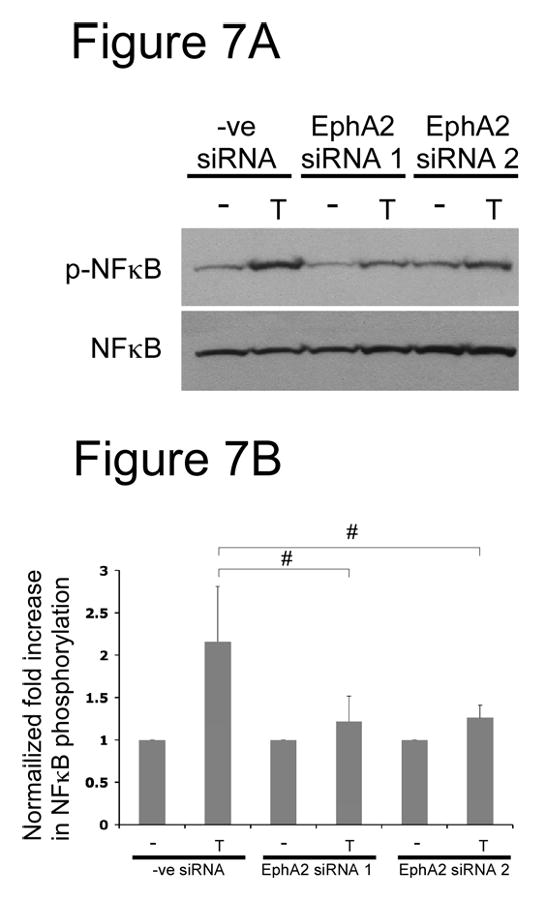
Thrombin-induced NFκB activation was dependent on EphA2 in endothelial cells. HUVECs cells were transfected with either a control siRNA or one of the two EphA2-specific siRNA. Confluent HUVECs were stimulated with 1U/ml thrombin (T) for 1 hours. (A) Total lysates were used for western blots. Thrombin-induced NFκB phosphorylation and protein loading were examined by western blot analyses using antibodies specific to phosphorylated and non-phosphorylated NFκB, respectively. Representative data from 3 experiments were shown. (B) Densitometry analysis of NFκB by thrombin in the presence or absence of EphA2. Western blots were scanned and analyzed by using the program ImageJ. Results were reported as fold-increase in NFκB phosphorylation relative to unstimulated controls. Data are mean ± s.d. of three experiments; # p<0.05.
Suppression of EphA2 blocks monocyte attachment to thrombin-stimulated HUVECs
One of the consequences of thrombin-induced ICAM-1 upregulation is enhanced attachment of leukocytes to the endothelium. Since suppression of EphA2 blocked ICAM-1 upregulation induced by thrombin, we reasoned that it would also block leukocyte attachment to stimulated HUVE monolayer. Therefore, we tested the ability of U937, a monocytic cell line, to attach to thrombin-treated HUVECs in the presence or absence of EphA2. In this experiment, both GFP-expressing and mouse-EphA2-expressing HUVECs were used. As expected, thrombin potently induced U937 attachment to HUVECs when cells were treated with the non-specific control siRNA, both in the GFP- and mouse-EphA2 expressing HUVECs (Figure 8). However, U937 attachment to thrombin-stimulated HUVECs was significantly blocked when endogenous EphA2 was knockdown by siRNA. In contrast, overexpression of mouse EphA2 in HUVECs with endogenous EphA2 suppressed restored thrombin-induced U937 attachment. These results were consistent with our observation that EphA2 was required for ICAM-1 upregulation induced by thrombin.
Figure 8.
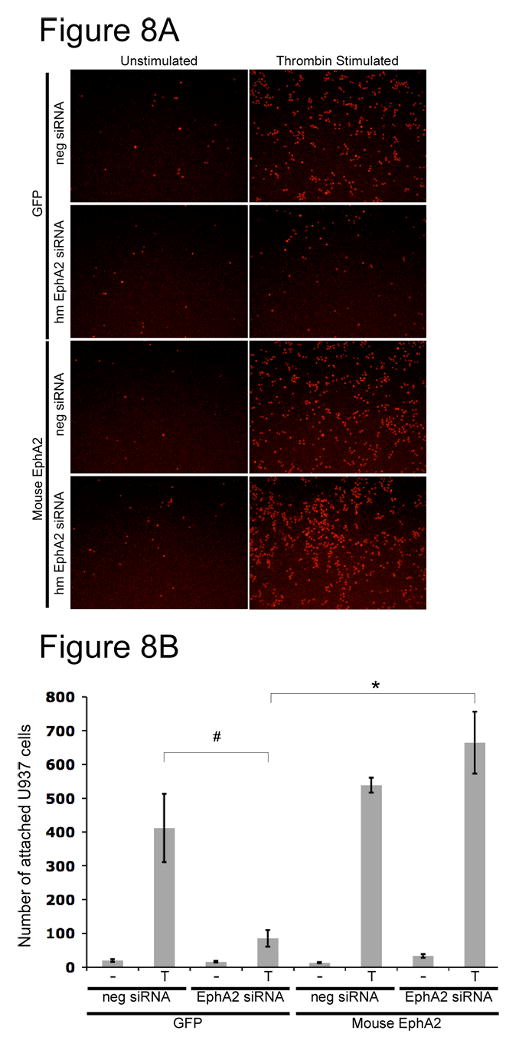
Endothelial EphA2 was crucial in mediating leukocyte attachment to thrombin-stimulated HUVECs. Either GFP or mouse EphA2 was stably overexpressed in HUVECs via retroviral infection. GFP or mouse EphA2 overexpressing cells were treated with either a control or a human EphA2 specific siRNA. Confluent HUVECs were stimulated with 5U/ml thrombin for 6 hours. Fluorescently labeled U937 cells were then added. After one hour of incubation at room temperature, unattached cells were gently aspirated away. Cells were then fixed in 4% PFA. (A) U937 cells were detected by fluorescence microscopy. Experiments were done in triplicate. Each condition was done three times. Representative results are shown. (B) Number of attached U937 cells were counted in 4 randomly chosen fields of each experiments. Three experiments per condition were done. Data are mean ± s.d. of three experiments; * p<0.005; # p<0.02.
Discussion
Thrombin is a serine protease that exhibits a wide array of biological activities. In addition to cleaving fibrinogen to fibrin for clot formation, thrombin also activates endothelial cells through cleavages of its canonical receptors PARs to induce endothelial inflammation. Inhibition of thrombin at the level of its protease activity therefore blocks both processes.
EphA2 in endothelial cells can be transactivated by stimulation with VEGF [13] or TNFα [55]. However, in both cases, it was determined that EphA2 transactivation was due to upregulation of EphrinA1 [13, 55]. In this report, we provide evidence that EphA2 is also transactivated in endothelial cells upon thrombin or PAR-1 agonist activation, but through a mechanistically distinct pathway. First, phosphorylation of EphA2 induced by thrombin was rapid and could be detected within minutes. Second, EphA2 activation by thrombin was Src-dependent, whereas that induced by EphrinA1 was independent of Src. Third, excess soluble EphA2 failed to block EphA2 transactivation induced by thrombin. Therefore, EphA2 activation by thrombin in our system was not mediated by EphrinA1 or other ephrins. We believe that the transactivation circuitry lies intracellularly via a Src kinase.
Using two siRNA constructs that targeted different coding regions of the EphA2 mRNA, we showed that suppression of EphA2 expression concomitantly blocked ICAM-1 expression induced by thrombin. Similarly, thrombin-induced leukocyte attachment to HUVECs was also blocked by the EphA2-specific siRNAs. Overexpression of mouse EphA2 rescued thrombin-induced ICAM-1 expression and U937 attachment when endogenous EphA2 was knocked down. Therefore EphA2 is critical in ICAM-1 expression induced by thrombin in HUVECs. However, EphA2-mediated ICAM-1 expression is dependent on the stimulus, because treatment of endothelial cells with Ephrin-A1 did not result in ICAM-1 expression. One possible explanation is that signals from EphA2 are necessary but not sufficient to induce ICAM-1 expression. It is conceivable that EphA2's signals need to be coupled with other thrombin-induced signals to upregulate ICAM-1. Therefore, a single set of signals from EphA2 induced by Ephrin-A1 is not sufficient. Another possibility is that thrombin and Ephrin-A1 trigger different signals from EphA2. For example, the sites of phosphorylation on EphA2 induced by thrombin may be different from that induced by EphrinA1. Indeed, we show evidence that Ephrin-A1 and thrombin appear to use distinct pathways to activate EphA2. Further studies will be needed to map out the differences in signals triggered by Ephrin-A1 and thrombin through EphA2.
Collectively, our results point to a new role of EphA2 in endothelial biology as a mediator of inflammation induction by thrombin. To further understand how EphA2 is linked to ICAM-1 upregulation, we examined the NFκB pathway. NFκB activation is key to ICAM-1 transcription in endothelial cells stimulated with thrombin. NFκB is sequestered in the cytoplasm as a complex with IkB in quiescent cells. Upon activation, IκB is degraded, and NFκB is released and transported to the nucleus. In endothelial cells, it appears that two parallel signaling pathways downstream of PAR-1 activation are needed to activate NFκB to achieve ICAM-1 expression. Gβγ and Gαq subunits of the G-protein coupled to PAR-1 activate PI3 kinase and PKC-δ, respectively. These two signals converge to activate AKT, which leads to NFκB activation [33]. Using phosphorylation of serine 536 of NFκB as a marker for activation, we showed that the role of EphA2 in ICAM-1 expression lies upstream of NFκB activation. At this moment, it is not known whether EphA2 transactivation is upstream or downstream of PI3K/PKC-δ. However, since it is known that tyrosine phosphorylation of EphA2 provides docking site for the SH2 domains of PI3 kinase [55], it is likely that thrombin induced EphA2 phosphorylation precedes PI3 kinase activation. Further experiments will be needed to address this question.
Figure 9 depicts our current model in how transactivation of EphA2 mediates thrombin-induced ICAM-1 upregulation. Thrombin activates the PAR1 receptor, which in turn activates a Src-family kinase to cause tyrosine phosphorylation of EphA2. Transactivation of EphA2 leads to activation of NFκB and subsequent ICAM-1 expression. Increased expression of ICAM-1 promotes attachment of leukoctyes to the endothelium.
Figure 9.
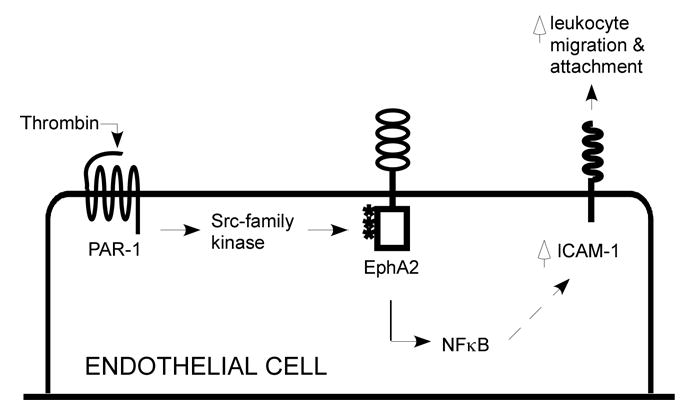
Working model of how EphA2 mediates thrombin-induced ICAM-1 upregulation. See text for details.
In summary, the present study provides evidence that EphA2 plays a critical role in thrombin-induced endothelial inflammation. Expression knockdown of EphA2 inhibits ICAM-1 upregulation in endothelial cells and leukocyte-endothelium attachment induced by thrombin. Our data strongly suggest that EphA2 is required for ICAM-1 expression induced by thrombin in endothelial cells. Therefore, strategies targeting EphA2 may provide a specific method to inhibit thrombin-induced endothelial activation without affecting clot formation in the host.
Acknowledgments
B.C. was supported in part by the Ruth L. Kirschstein National Research Service Award (NRSA) Institutional Research Training Grants from the National Institutes of Health. This work was partially supported by a research grant from Gambro/DaVita and seed funds from Beth Israel Deaconess Medical Center.
Abbreviations
- HUVEC
human umbilical vein endothelial cell
- PAR-1
protease activated receptor-1
- DMSO
dimethyl sulfoxide
- ROCK
Rho-associated kinase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bohm SK, McConalogue K, Kong W, Bunnett NW. Proteinase-Activated Receptors: New Functions for Old Enzymes. News Physiol Sci. 1998;13:231–40. doi: 10.1152/physiologyonline.1998.13.5.231. [DOI] [PubMed] [Google Scholar]
- 2.Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci USA. 1999;96:11023–7. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dery O, Corvera CU, Steinhoff M, Bunnett NW. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol. 1998;274:C1429–52. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- 4.Grand RJ, Turnell AS, Grabham PW. Cellular consequences of thrombin-receptor activation. Biochem J. 1996;313:353–68. doi: 10.1042/bj3130353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–82. [PubMed] [Google Scholar]
- 6.Minami T, Sugiyama A, Wu SQ, Abid R, Kodama T, Aird WC. Thrombin and phenotypic modulation of the endothelium. Arterioscler Thromb Vasc Biol. 2004;24:41–53. doi: 10.1161/01.ATV.0000099880.09014.7D. [DOI] [PubMed] [Google Scholar]
- 7.O'Brien PJ, Molino M, Kahn M, Brass LF. Protease activated receptors: theme and variations. Oncogene. 2001;20:1570–81. doi: 10.1038/sj.onc.1204194. [DOI] [PubMed] [Google Scholar]
- 8.Himanen JP, Saha N, Nikolov DB. Cell-cell signaling via Eph receptors and ephrins. Curr Opin Cell Biol. 2007;19:534–42. doi: 10.1016/j.ceb.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanov AI, Romanovsky AA. Putative dual role of ephrin-Eph receptor interactions in inflammation. IUBMB life. 2006;58:389–94. doi: 10.1080/15216540600756004. [DOI] [PubMed] [Google Scholar]
- 10.Kalo MS, Pasquale EB. Signal transfer by Eph receptors. Cell Tissue Res. 1999;298:1–9. [PubMed] [Google Scholar]
- 11.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nature reviews. 2002;3:475–86. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 12.Brantley-Sieders DM, Caughron J, Hicks D, Pozzi A, Ruiz JC, Chen J. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J Cell Sci. 2004;117:2037–49. doi: 10.1242/jcs.01061. [DOI] [PubMed] [Google Scholar]
- 13.Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N, Yancopoulos G, Cerretti DP, Daniel TO, Chen J. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol Cancer Res. 2002;1:2–11. [PubMed] [Google Scholar]
- 14.Ojima T, Takagi H, Suzuma K, Oh H, Suzuma I, Ohashi H, Watanabe D, Suganami E, Murakami T, Kurimoto M, Honda Y, Yoshimura N. EphrinA1 inhibits vascular endothelial growth factor-induced intracellular signaling and suppresses retinal neovascularization and blood-retinal barrier breakdown. Am J Pathol. 2006;168:331–9. doi: 10.2353/ajpath.2006.050435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–52. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- 16.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. Faseb J. 2005;19:1884–6. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]
- 17.Hess AR, Seftor EA, Gardner LM, Carles-Kinch K, Schneider GB, Seftor RE, Kinch MS, Hendrix MJ. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2) Cancer research. 2001;61:3250–5. [PubMed] [Google Scholar]
- 18.Hess AR, Seftor EA, Gruman LM, Kinch MS, Seftor RE, Hendrix MJ. VE-cadherin regulates EphA2 in aggressive melanoma cells through a novel signaling pathway: implications for vasculogenic mimicry. Cancer biology & therapy. 2006;5:228–33. doi: 10.4161/cbt.5.2.2510. [DOI] [PubMed] [Google Scholar]
- 19.Dixit VM, Green S, Sarma V, Holzman LB, Wolf FW, O'Rourke K, Ward PA, Prochownik EV, Marks RM. Tumor necrosis factor-alpha induction of novel gene products in human endothelial cells including a macrophage-specific chemotaxin. J Biol Chem. 1990;265:2973–8. [PubMed] [Google Scholar]
- 20.Holzman LB, Marks RM, Dixit VM. A novel immediate-early response gene of endothelium is induced by cytokines and encodes a secreted protein. Mol cell biol. 1990;10:5830–8. doi: 10.1128/mcb.10.11.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivanov AI, Steiner AA, Scheck AC, Romanovsky AA. Expression of Eph receptors and their ligands, ephrins, during lipopolysaccharide fever in rats. Physiol genomics. 2005;21:152–60. doi: 10.1152/physiolgenomics.00043.2004. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura T, Kabuyama Y, Kamataki A, Homma MK, Kobayashi H, Aota S, Kikuchi SI, Homma Y. Enhancement of lymphocyte migration and cytokine production by ehprinB1 system in rheumatoid arthritis. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00314.2007. [DOI] [PubMed] [Google Scholar]
- 23.Pandey A, Shao H, Marks RM, Polverini PJ, Dixit VM. Role of B61, the ligand for the Eck receptor tyrosine kinase, in TNF-alpha-induced angiogenesis. Science (New York, NY. 1995;268:567–9. doi: 10.1126/science.7536959. [DOI] [PubMed] [Google Scholar]
- 24.Denk A, Goebeler M, Schmid S, Berberich I, Ritz O, Lindemann D, Ludwig S, Wirth T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J Biol Chem. 2001;276:28451–8. doi: 10.1074/jbc.M102698200. [DOI] [PubMed] [Google Scholar]
- 25.Minami T, Abid MR, Zhang J, King G, Kodama T, Aird WC. Thrombin stimulation of vascular adhesion molecule-1 in endothelial cells is mediated by protein kinase C (PKC)-delta-NF-kappa B and PKC-zeta-GATA signaling pathways. J Biol Chem. 2003;278:6976–84. doi: 10.1074/jbc.M208974200. [DOI] [PubMed] [Google Scholar]
- 26.Anwar KN, Fazal F, Malik AB, Rahman A. RhoA/Rho-associated kinase pathway selectively regulates thrombin-induced intercellular adhesion molecule-1 expression in endothelial cells via activation of I kappa B kinase beta and phosphorylation of RelA/p65. J Immunol. 2004;173:6965–72. doi: 10.4049/jimmunol.173.11.6965. [DOI] [PubMed] [Google Scholar]
- 27.Bijli KM, Minhajuddin M, Fazal F, O'Reilly MA, Platanias LC, Rahman A. c-Src interacts with and phosphorylates RelA/p65 to promote thrombin-induced ICAM-1 expression in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;292:L396–404. doi: 10.1152/ajplung.00163.2006. [DOI] [PubMed] [Google Scholar]
- 28.Fazal F, Minhajuddin M, Bijli KM, McGrath JL, Rahman A. Evidence for actin cytoskeleton-dependent and -independent pathways for RelA/p65 nuclear translocation in endothelial cells. J Biol Chem. 2007;282:3940–50. doi: 10.1074/jbc.M608074200. [DOI] [PubMed] [Google Scholar]
- 29.Kaur J, Woodman RC, Kubes P. P38 MAPK: critical molecule in thrombin-induced NF-kappa B-dependent leukocyte recruitment. Am J Physiol. 2003;284:H1095–103. doi: 10.1152/ajpheart.00016.2002. [DOI] [PubMed] [Google Scholar]
- 30.Miho N, Ishida T, Kuwaba N, Ishida M, Shimote-Abe K, Tabuchi K, Oshima T, Yoshizumi M, Chayama K. Role of the JNK pathway in thrombin-induced ICAM-1 expression in endothelial cells. Cardiovasc Res. 2005;68:289–98. doi: 10.1016/j.cardiores.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 31.Rahman A, Anwar KN, Minhajuddin M, Bijli KM, Javaid K, True AL, Malik AB. cAMP targeting of p38 MAP kinase inhibits thrombin-induced NF-kappaB activation and ICAM-1 expression in endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1017–24. doi: 10.1152/ajplung.00072.2004. [DOI] [PubMed] [Google Scholar]
- 32.Rahman A, Anwar KN, Uddin S, Xu N, Ye RD, Platanias LC, Malik AB. Protein kinase C-delta regulates thrombin-induced ICAM-1 gene expression in endothelial cells via activation of p38 mitogen-activated protein kinase. Mol Cell Biol. 2001;21:5554–65. doi: 10.1128/MCB.21.16.5554-5565.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahman A, True AL, Anwar KN, Ye RD, Voyno-Yasenetskaya TA, Malik AB. Galpha(q) and Gbetagamma regulate PAR-1 signaling of thrombin-induced NF-kappaB activation and ICAM-1 transcription in endothelial cells. Circ Res. 2002;91:398–405. doi: 10.1161/01.res.0000033520.95242.a2. [DOI] [PubMed] [Google Scholar]
- 34.Rahman A, Anwar KN, True AL, Malik AB. Thrombin-induced p65 homodimer binding to downstream NF-kappa B site of the promoter mediates endothelial ICAM-1 expression and neutrophil adhesion. J Immunol. 1999;162:5466–76. [PubMed] [Google Scholar]
- 35.Ukropec JA, Hollinger MK, Salva SM, Woolkalis MJ. SHP2 association with VE-cadherin complexes in human endothelial cells is regulated by thrombin. J Biol Chem. 2000;275:5983–6. doi: 10.1074/jbc.275.8.5983. [DOI] [PubMed] [Google Scholar]
- 36.Chan B, Merchan JR, Kale S, Sukhatme VP. Antiangiogenic property of human thrombin. Microvasc Res. 2003;66:1–14. doi: 10.1016/s0026-2862(03)00037-2. [DOI] [PubMed] [Google Scholar]
- 37.Luscinskas FW, Kansas GS, Ding H, Pizcueta P, Schleiffenbaum BE, Tedder TF, Gimbrone MA., Jr Monocyte rolling, arrest and spreading on IL-4-activated vascular endothelium under flow is mediated via sequential action of L-selectin, beta 1-integrins, and beta 2-integrins. J Cell Biol. 1994;125:1417–27. doi: 10.1083/jcb.125.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woolkalis MJ, DeMelfi TM, Jr, Blanchard N, Hoxie JA, Brass LF. Regulation of thrombin receptors on human umbilical vein endothelial cells. J Biol Chem. 1995;270:9868–75. doi: 10.1074/jbc.270.17.9868. [DOI] [PubMed] [Google Scholar]
- 39.Nelken NA, Soifer SJ, O'Keefe J, Vu TK, Charo IF, Coughlin SR. Thrombin receptor expression in normal and atherosclerotic human arteries. J Clin Invest. 1992;90:1614–21. doi: 10.1172/JCI116031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molino M, Woolkalis MJ, Reavey-Cantwell J, Pratico D, Andrade-Gordon P, Barnathan ES, Brass LF. Endothelial cell thrombin receptors and PAR-2. Two protease-activated receptors located in a single cellular environment. J Biol Chem. 1997;272:11133–41. doi: 10.1074/jbc.272.17.11133. [DOI] [PubMed] [Google Scholar]
- 41.D'Andrea MR, Derian CK, Leturcq D, Baker SM, Brunmark A, Ling P, Darrow AL, Santulli RJ, Brass LF, Andrade-Gordon P. Characterization of protease-activated receptor-2 immunoreactivity in normal human tissues. J Histochem Cytochem. 1998;46:157–64. doi: 10.1177/002215549804600204. [DOI] [PubMed] [Google Scholar]
- 42.Bahou WF, Coller BS, Potter CL, Norton KJ, Kutok JL, Goligorsky MS. The thrombin receptor extracellular domain contains sites crucial for peptide ligand-induced activation. J Clin Invest. 1993;91:1405–13. doi: 10.1172/JCI116344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirza H, Yatsula V, Bahou WF. The proteinase activated receptor-2 (PAR-2) mediates mitogenic responses in human vascular endothelial cells. J Clin Invest. 1996;97:1705–14. doi: 10.1172/JCI118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nystedt S, Ramakrishnan V, Sundelin J. The proteinase-activated receptor 2 is induced by inflammatory mediators in human endothelial cells. Comparison with the thrombin receptor. J Biol Chem. 1996;271:14910–5. doi: 10.1074/jbc.271.25.14910. [DOI] [PubMed] [Google Scholar]
- 45.Ngaiza JR, Manley G, Grulich-Henn J, Krstenansky JL, Jaffe EA. The fibrinogen anion-binding exosite of thrombin is necessary for induction of rises in intracellular calcium and prostacyclin production in endothelial cells. J Cell Physiol. 1992;151:190–6. doi: 10.1002/jcp.1041510124. [DOI] [PubMed] [Google Scholar]
- 46.O'Brien PJ, Prevost N, Molino M, Hollinger MK, Woolkalis MJ, Woulfe DS, Brass LF. Thrombin responses in human endothelial cells. Contributions from receptors other than PAR1 include the transactivation of PAR2 by thrombin-cleaved PAR1. J Biol Chem. 2000;275:13502–9. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- 47.Lafleur MA, Hollenberg MD, Atkinson SJ, Knauper V, Murphy G, Edwards DR. Activation of pro-(matrix metalloproteinase-2) (pro-MMP-2) by thrombin is membrane-type-MMP-dependent in human umbilical vein endothelial cells and generates a distinct 63 kDa active species. Biochem J. 2001;357:107–15. doi: 10.1042/0264-6021:3570107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmidt VA, Nierman WC, Maglott DR, Cupit LD, Moskowitz KA, Wainer JA, Bahou WF. The human proteinase-activated receptor-3 (PAR-3) gene. Identification within a Par gene cluster and characterization in vascular endothelial cells and platelets. J Biol Chem. 1998;273:15061–8. doi: 10.1074/jbc.273.24.15061. [DOI] [PubMed] [Google Scholar]
- 49.Hamilton JR, Frauman AG, Cocks TM. Increased expression of protease-activated receptor-2 (PAR2) and PAR4 in human coronary artery by inflammatory stimuli unveils endothelium-dependent relaxations to PAR2 and PAR4 agonists. Circ Res. 2001;89:92–8. doi: 10.1161/hh1301.092661. [DOI] [PubMed] [Google Scholar]
- 50.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–87. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sinnett-Smith J, Lunn JA, Leopoldt D, Rozengurt E. Y-27632, an inhibitor of Rho-associated kinases, prevents tyrosine phosphorylation of focal adhesion kinase and paxillin induced by bombesin: dissociation from tyrosine phosphorylation of p130(CAS) Exp Cell Res. 2001;266:292–302. doi: 10.1006/excr.2001.5219. [DOI] [PubMed] [Google Scholar]
- 52.Sabri A, Guo J, Elouardighi H, Darrow AL, Andrade-Gordon P, Steinberg SF. Mechanisms of protease-activated receptor-4 actions in cardiomyocytes. Role of Src tyrosine kinase. J Biol Chem. 2003;278:11714–20. doi: 10.1074/jbc.M213091200. [DOI] [PubMed] [Google Scholar]
- 53.Sabri A, Short J, Guo J, Steinberg SF. Protease-activated receptor-1-mediated DNA synthesis in cardiac fibroblast is via epidermal growth factor receptor transactivation: distinct PAR-1 signaling pathways in cardiac fibroblasts and cardiomyocytes. Circ Res. 2002;91:532–9. doi: 10.1161/01.res.0000035242.96310.45. [DOI] [PubMed] [Google Scholar]
- 54.Minhajuddin M, Fazal F, Bijli KM, Amin MR, Rahman A. Inhibition of mammalian target of rapamycin potentiates thrombin-induced intercellular adhesion molecule-1 expression by accelerating and stabilizing NF-kappa B activation in endothelial cells. J Immunol. 2005;174:5823–9. doi: 10.4049/jimmunol.174.9.5823. [DOI] [PubMed] [Google Scholar]
- 55.Pandey A, Lazar DF, Saltiel AR, Dixit VM. Activation of the Eck receptor protein tyrosine kinase stimulates phosphatidylinositol 3-kinase activity. J Biol Chem. 1994;269:30154–7. [PubMed] [Google Scholar]