

Abstract
The therapeutic use of T cell receptor (TCR)-transduced peripheral blood lymphocytes (PBL) targeting tumor-associated antigens is emerging as a promising investigational treatment for patients with cancer. Initial response rates to therapy were low, suggesting the need to improve the function of TCR-transduced PBL. We constructed standard bicistronic retroviral vectors using an internal promoter or internal ribosomal entry site element as well as vectors incorporating coding sequences for 2A linker peptides between coding sequences for α and β chains targeting the cancer-testis (CT) antigen, NY-ESO-1. Incorporation of coding sequences for 2A linker peptides in the bicistronic TCR expression cassette resulted in up to a fourfold increase in TCR expression and a significant improvement in effector function as measured by interferon-gamma release following co-culture with peptide-pulsed targets and NY-ESO-1+ tumors. We also sought to enhance reactivity of TCR-transduced PBL against tumor targets by modulation of tumor antigen expression on target cells. Induction of NY-ESO-1 expression on tumor targets using the demethylating agent 5-aza-2′-deoxycytidine (alone or in combination with the histone deacetylase inhibitor depsipeptide) resulted in enhanced interferon-gamma secretion by the TCR-transduced PBL on culture with treated targets. Taken together, these results indicate that design of TCR-based vectors incorporating 2A linker peptides improves TCR expression and effector function of transduced PBL. Furthermore, induction of CT antigen expression through treatment of tumor targets with chromatin-remodeling agents may augment TCR-based immunotherapy targeting these antigens. These results have relevance for TCR-based gene therapies targeting common epithelial malignancies.
Electronic supplementary material
The online version of this article (doi:10.1007/s00262-008-0562-x) contains supplementary material, which is available to authorized users.
Keywords: NY-ESO-1, TCR, Retroviral vector, Gene therapy, Epigenetics
Introduction
Immunotherapy involving the transfer of genes encoding T cell receptors (TCRs) into peripheral blood lymphocytes (PBL) is based on the foundation of knowledge gained from the study of tumor-infiltrating lymphocytes (TIL). Fundamental advances have been made in the past two decades with regard to adoptive transfer of TIL, resulting in response rates exceeding 50% when autologous reactive TIL were transferred following a lymphodepleting conditioning regimen [1]. Despite these advances, several limitations of this technology exist. This therapy requires a surgical procedure to procure TIL, and mandates that tumor-reactive cells be isolated and expanded ex vivo, which limits the application of this therapy to one-half of eligible patients with metastatic melanoma.
To overcome these limitations, approaches to cancer immunotherapy have been developed based on the genetic modification of normal PBL. We successfully cloned TCRs against several tumor-associated antigens (TAA) found in melanoma, including MART-1 and gp100 [2, 3]. Retroviral vectors incorporating these cloned TCR were used to transduce PBL, resulting in the generation of cells capable of recognizing and destroying both fresh and cultured melanoma cells in vitro [2, 4]. Based on these observations, 15 patients with metastatic melanoma were treated with genetically engineered lymphocytes transduced to express a TCR targeted at MART-1, and durable engraftment at levels exceeding 10% of PBL was observed for at least 2 months [3]. Two of the 15 patients demonstrated high sustained levels of circulating cells at 1 year following infusion, which was associated with objective regression of metastatic lesions [3].
In an effort to extend the scope of immunotherapy toward diverse cancer histologies, we cloned a TCR targeting NY-ESO-1 [5], a cancer-testis (CT) antigen frequently expressed in melanoma as well as a wide range of non-melanoma epithelial malignancies [6, 7]. CT antigens are proteins encoded by genes that are normally expressed only in germ cell tissues, trophoblasts, and certain tumors but not in normal tissues [6, 7]. There has been growing interest in the use of CT antigens as targets for immunotherapy.
NY-ESO-1 was originally discovered by screening the serum of a patient with esophageal cancer using ‘serological identification of antigens by recombinant expression cloning’ [8]. Since that time, its expression has been described in a variety of common tumors (20–80% at the RNA level), including melanoma and cancers of the breast, lung, bladder, liver, prostate, and ovary [6]. Of note, some of these cancer patients develop antibody responses to NY-ESO-1 [6]. NY-ESO-1 is one of the most immunogenic of the CT antigens, and has been a target for immunotherapy in multiple clinical trials [9–12]. NY-ESO-1 offers a potential advantage as a target for TCR-based gene therapy as its expression is limited to neoplastic cells and germ line tissues, thus the risk for autoimmune complications is low. This is in contrast to the melanoma differentiation antigens such as MART-1 and gp100, which are expressed in normal tissues as well as in tumors.
We generated a retroviral vector encoding DNA for TCR genes targeting the HLA-A2-restricted epitope the NY-ESO-1 antigen [5], and demonstrated that T cells engineered with this vector recognize multiple cancer histologies. The original vector design for the NY-ESO-1 TCR vector employed an internal promoter, phosphoglycerate kinase (PGK), to drive expression of the second of the TCR genes (the β chain, in this case). However, use of an internal promoter in retroviral vectors can lead to substantially lower expression of the second gene [13]. Recently, a mechanism to improve expression in multi-cistronic constructs was described utilizing 2A linker peptides [14, 15]. These peptide sequences are derived from viruses such as the porcine teschovirus, foot-and-mouth disease virus, and Thosea asigna virus [16–18]. 2A peptides contain a consensus motif that utilizes a ribosomal skip mechanism to inhibit peptide bond formation between adjacent protein coding sequences. As a result, use of these 2A peptides to link sequences encoding several proteins in the same open reading frame results in near-complete separation and stoichiometric production of the encoded proteins [14]. This technique has been used in other model systems including the production of monoclonal antibodies [15]. We thus incorporated these 2A linker peptides in construction of retroviral vectors targeting the class I-restricted epitope of the CT antigen, NY-ESO-1.
One potential advantage in targeting CT antigens such as NY-ESO-1 is that the expression of these antigens can be up-regulated in tumor cells [19]. We have previously reported that treatment with the demethylating agent 5-aza-2′-deoxycytidine (DAC) can induce expression of NY-ESO-1 in melanoma and non-melanoma cell lines [20]. We have now employed this approach in an effort to sensitize tumor targets to PBL transduced with a TCR targeting the NY-ESO-1 antigen, and report the findings herein.
This report presents the results of studies indicating that the use of 2A linker peptides in retroviral vectors containing bicistronic TCR expression cassettes leads to significantly improved expression and enhanced effector function of TCR-transduced PBL. This vector design has significant implications for TCR-based therapies targeting melanoma as well as non-melanoma epithelial malignancies. In addition, induction of NY-ESO-1 antigen expression in patients may provide a useful adjunct to adoptive cell transfer in the treatment of malignancy.
Materials and methods
Patient PBL and cell lines
All of the PBL used in this study were from metastatic melanoma patients treated at the Surgery Branch, National Cancer Institute (NCI), NIH, Bethesda, MD, USA. T2 is a lymphoblastoid cell line deficient in TAP function, whose HLA class I proteins can be easily loaded with exogenous peptides [21]. Melanoma lines 1300mel (NY-ESO-1+, HLA-A2+), 624.38mel (NY-ESO-1+, HLA-A2+), 888mel (NY-ESO-1−, HLA-A2−), 526mel (NY-ESO-1−, HLA-A2+), A375 (NY-ESO+, HLA-A2+), and SK23 (NY-ESO−, HLA-A2+) were generated at the Surgery Branch from resected tumor lesions, as previous described [22]. The H2373 pleural mesothelioma cell line (NY-ESO-1−, HLA-A2+), BE-3 esophageal cancer cell line, and H1299 non-small cell lung carcinoma cell line (NY-ESO-1+, HLA-A2−) were kindly provided by Dr David Schrump, National Institutes of Health. The H1299 small cell lung carcinoma line was transduced with retroviral construct to express HLA-A*0201 as previously described [23], and was designated H1299-A2. Cells obtained from other sources include: PG13 gibbon ape leukemia virus-packaging cell line (ATCC CRL 10-686), the human lymphoid cell line SupT1 (ATCC CRL-1942), the human pancreatic cancer cell line Panc-1 (ATCC CRL-1469) (NY-ESO-1−, HLA-A2+), the human glioblastoma cell line LNZTA3WT4 (ATCC CRL-11543) (NY-ESO-1−, HLA-A2+), the human ovarian cancer cell line OVCAR-3 (ATCC HTB-161) (NY-ESO-1−, HLA-A2+) and the human ecotropic packaging cell line, Phoenix Eco (kindly provided by G. Nolan, Stanford University, Stanford, CA, USA). All the cell lines described above were maintained in R10 [RPMI 1640 (Invitrogen, Inc. Rockville, MD, USA) supplemented with 10% FCS (Biofluid, Inc., Gaithersburg, MD, USA)]. Culture medium (CM) for T lymphocytes was AIM V (Invitrogen), 300 IU/ml IL-2 (Chiron Corp., Emeryville, CA, USA), plus 5% human AB serum (Valley Biomedical, Winchester, VA, USA).
Peptide synthesis
Synthetic peptides used in this study were made using a solid phase method on a peptide synthesizer (Gilson Co. Inc., Worthington, OH, USA) at the Surgery Branch of the NCI. The quality of each peptide was evaluated by mass spectrometry (Biosynthesis Inc., Lewisville, TX, USA). The sequences of the peptide used in this study are as follows: NY-ESO-1 p157–165 (SLLMWITQC), gp100 209-217 [3], Mart-1 27-35 [2].
Cysteine-modified TCR generation and cloning
We generated cysteine-modified α and β chains using a polymerase chain reaction (PCR) megaprimer approach. The first fragment was generated by amplifying the chains with a 5′-specific primer and either the α-Cys48rev primer, TAGACCTCATGTCTAGCACGCATTTGTCTGTGATATACACATC or the β-Cys57rev primer, TGAGGGGCTGCGGGTCCGTGCAGACCCCACTGTGCACCTCC for the α and β chains, respectively (mutated bases are underlined). The product of this reaction was then used as a megaprimer to amplify full-length TCR chain with a 3′-specific primer. The full-length chain was subcloned into pGEM vector as previously described [23].
Construction of retroviral vectors
The retroviral vector backbone used in this study, pMSGV1, is a derivative of the vector pMSGV [murine stem cell virus (MSCV)-based splice-gag vector] which utilizes a MSCV long terminal repeat (LTR) [5]. Five vectors were constructed using PCR of four DNA fragments: pMSGV1 (NcoI/NotI), NY-ESO-1 TCR α chain (NcoI/XbaI), PGK promoter (NotI/XhoI), internal ribosomal entry site (IRES) (NotI/XhoI), or one of four different 2A linker peptides—GSGP2A, GSGT2A, T2A, furinSGSGP2A (NotI/XhoI), and NY-ESO-1 TCR β chain (XhoI/EcoRI). The cloned inserts were determined by PCR, restriction enzyme digest, and DNA sequencing.
Production of retroviral supernatants by transient transfection of 293 GP cells
To produce retroviral supernatants for initial testing, 9 μg of vector DNA from each of the constructs was co-transfected with 4 μg of envelope DNA (RD114) into transient viral producer cell lines (293-GP) using the Lipofectamine 2000 reagent (Invitrogen) and Optimem medium (BD Biosciences). Media was changed to DMEM with 10% FBS after 6 h, and viral supernatants were harvested at the 48-h time point. These supernatants were then used to transduce target cells to express a TCR targeting the NY-ESO-1 antigen.
Production of retroviral supernatant from stable producer cell clones
Generation of PG13-packaging cell clones was initiated by first transfecting Phoenix Eco cells with 4 μg of DNA for each construct using the Lipofectamine reagent (Invitrogen). PG13 cells were then transduced with supernatant from Phoenix Eco cells, and PG13 cell clones were obtained by limiting dilution. Clones were expanded, and high titer clones were selected by the dot-blot titration method [24]. Clones were determined to be producing biologically active retroviral vector by transduction of SupT1 cells and analysis of FACS data (using anti-CD3 and NY-ESO-1 tetramer). MSGIN, whose name is derived from MSCV-based splice vector green fluorescent protein (GFP)-internal ribosomal entry site (IRES)-neo, was used as a control vector that expresses GFP.
Transduction of PBL
Peripheral blood lymphocytes were collected by leukapheresis and lymphocytes were resuspended at a concentration of 1 × 106/ml in media supplemented with OKT3 (50 mg/ml) and IL-2 (300 IU/ml). Lymphocytes were plated at 1 × 106/ml in 24-well plates (Costar), and were cultured for 48 h prior to transduction. Lymphocytes were transduced with retroviral vectors by centrifugation (2,000g) with retroviral supernatant and protamine sulfate (10 μg/ml).
Flow cytometry analysis
Cell surface expression of CD3, CD4, CD8 (BD Biosciences), Vβ13.1 (Immunotech), and NY-ESO-1 tetramer (Beckman Coulter) on PBL was measured by immunofluorescence using FITC, PE, PE-Cy7, or APC. HLA class I expression in cultured cancer cells was evaluated by flow cytometric techniques using the W6/32 antibody (Sera-Lab, Sussex, UK) that recognizes only functional class I molecules. For analysis, the relative log fluorescence of live cells was measured using a FACScan flow cytometer (BD Biosciences).
Cytokine release assays
Peripheral blood lymphocyte cultures were tested for reactivity in cytokine release assays using commercially available ELISA kits (IFN-γ; Endogen, Cambridge, MA, USA). T2 cells were pulsed with peptide (1 μg/ml, or as described in figures) in CM for 3 h at 37°C, followed by washing (three times) prior to incubation of co-cultures. For these assays, 1 × 105 effector cells (PBL) and 1 × 105 stimulator cells (T2 cells or tumor cells) were incubated in 0.2 ml culture volume in individual wells of 96-well plates. Effector cells and responder cells were co-cultured for 24 h. Cytokine secretion (interferon-gamma) was measured in culture supernatants diluted so as to be in the linear range of the assay.
DAC-DP Exposure
5-Aza-2′-deoxycytidine was purchased from Sigma Chemical (St Louis, MO, USA). The Developmental Therapeutics Program at the NCI provided depsipeptide (DP). Following optimization of the kinetics of gene induction in preliminary experiments, the effects of DAC, DP, or sequential DAC-DP treatment on NY-ESO-1 expression was determined after exposure to (1) normal media (NM) for 96 h; (2) 0.1 μmol/l DAC for 72 h, followed by NM for 24 h; (3) NM for 72 h, then 25 ng/ml DP for 6 h, followed by NM for 18 h; or (4) sequential DAC-DP, followed by NM for 18 h. For each experiment, day 0 represented the time when cells were initially exposed to either NM or DAC. During the treatment regimen, media were changed every day, and after appropriate drug exposure, cells were washed with Hanks balanced salt solution. Quantitative reverse-transcriptase PCR (RT-PCR) and gel PCR were performed at the 96-h time point.
RNA extraction and real-time quantitative reverse-transcriptase-polymerase chain reaction analysis
Total RNA was isolated from cell lines using reagents and protocols provided in the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Synthesis of cDNA was performed with 2 μg total RNA using a reverse transcription kit (Promega, Madison, WI, USA) and oligo (dT)15 primers. Quantitative RT-PCR (TaqMan) analysis of gene expression was performed in a blinded manner using an ABI prism 7700 Sequence Detection System (Perkin-Elmer, Foster City, CA, USA) [25, 26]. TaqMan probes [labeled at the 5′ end with the reporter dye molecule FAM (6-carboxyfluorescein) and at the 3′ end with the quencher dye TAMRA (6-carboxytetramethylrhodamine)] were purchased from PE Biosystems Oligonucleotide Factory (Foster City, CA, USA). NY-ESO-1, β-actin, and GAPDH primer sequences and methods for quantitative RT-PCR analysis, including generation of cDNA standards, and thermal cycling parameters have been described [27, 28]. cDNA from 586mel was used to generate the relative standard curves for NY-ESO-1, β-actin, and GAPDH. Samples were standardized by dividing the copy number of the target gene by that of the endogenous reference gene (β-actin or GAPDH).
Results
TCR expression of transduced PBL
Using the anti-NY-ESO-1 TCR previously described [29], five retroviral vectors, MSGV1-1G4-APB (APB), MSGV1-1G4-AIB (AIB), MSGV1-1G4-AGSGP2AB (GSGP2A), MSGV1-1G4-AGSGT2AB (GSGT2A), and MSGV1-1G4-AT2AB (T2A), were initially constructed. The APB construct used the retroviral MSCV LTR to drive the expression of α chain with the β chain governed by an internal PGK promoter, while the AIB construct incorporated an IRES between coding sequences for the α and β chains of the TCR (Fig. 1). The remaining three retroviral constructs, GSGP2A, GSGT2A, and T2A used the retroviral MSCV LTR to drive expression of the of α and β chains as a single open reading frame with the incorporation of 2A linker peptides (Fig. 1).
Fig. 1.

Design of NY-ESO-1 TCR retroviral vectors. Schematic representation of six retroviral vectors used to transfer and express the TCR genes targeting the HLA-A2-restricted immundominant epitope of NY-ESO-1. Vector MSGV1-1G4-AIB is a bicistronic vector design where the LTR promoter drives the expression of both TCR α and β genes linked by an IRES. In vector MSGV1-1G4-APB (APB), the expression of the α chain is mediated by the vector LTR gene promoter, whereas β chain expression is driven by an internal PGK promoter. In each of the vectors incorporating 2A linker peptides, the expression of both the α and β chains is mediated by the vector LTR gene promoter through an open reading frame, and ‘cleavage’ of the α- and β chains is accomplished at the protein level, where incomplete peptide bond formation between two components in the linker peptide design results in two separate proteins. MSGV1-1G4-AGSGP2AB (GSGP2A) incorporates coding sequences generated from the porcine teschovirus-1 and includes a GSG (glycine-serine-glycine) ‘spacer’ sequence. MSGV1-1G4-AGSGT2AB (GSGT2A) incorporates coding sequences generated from the Thosea asigna virus and includes a GSG ‘spacer’ sequence. MSGV1-1G4-AT2AB (T2A) is essentially identical to MSGV1-1G4-AGSGT2AB with the exception that it lacks a GSG ‘spacer’ sequence. The MSGV1-Cys1G4-AfrnSGSGP2AB (CysfrnSGSGP2A) vector incorporates α and β chains of the 1G4 NY-ESO-1 TCR in which we replaced a Thr48 on the α chain and a Ser57 on the β chain with cysteines to facilitate the creation of an additional disulfide bond between the TCR constant regions in an effort to enhance TCR stability
To assess vector function prior to generation and cloning of stable producer cell clones, anti-NY-ESO-1 TCR vectors were produced by transient transfection as described (see Sect. ‘Materials and methods’). Lymphocytes were transduced on day 2 post-stimulation with supernatant from these cultures. Five days following transduction, cells were harvested and stained for CD8, and NY-ESO-1 tetramer. Representative PBL transductions for the APB, AIB, GSGP2A, GSGT2A, and T2A vectors were as shown in Fig. 2. Transduction with constructs incorporating 2A peptides resulted in significantly higher expression of the TCR measured by staining with the NY-ESO-1 tetramer (Fig. 2a). No appreciable staining of NY-ESO-1 tetramer was noted following transduction with supernatant from the IRES construct, thus this construct was not pursued further.
Fig. 2.
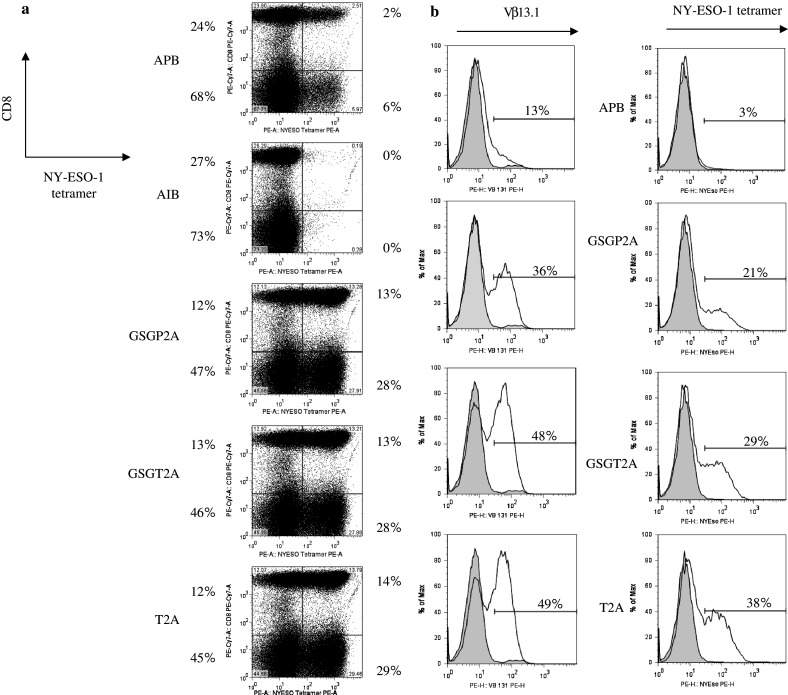
T cell receptor expression of transduced PBL. a Retroviral supernatants were produced by transient transfection of 293-GP cells with vector DNA from the APB, AIB, GSGP2A, GSGT2A, and T2A constructs. OKT3-stimulated PBL were transduced with supernatant from each of the constructs to evaluate transduction efficiency. Five days post transduction, PBL were stained for CD8, and NY-ESO-1 tetramer and expression was analyzed via flow cytometry. The percentage of positive cells for CD8 and NY-ESO-1 are indicated on the dot-plot. b Following generation of stable producer cell clones, OKT3-stimulated PBL were transduced with retroviral supernatant from packaging cell clones APB 38, GSGP2A 3, GSGT2A 36, and T2A 24. Five days post transduction, PBL were stained for Vβ13.1 and NY-ESO-1 tetramer and expression was analyzed via flow cytometry. The percentage of positive cells for Vβ13.1 and NY-ESO-1 are indicated on the histogram
Stable producer cell lines for the APB, GSGP2A, GSGT2A, and T2A constructs were generated as previously described (see Sect. ’Materials and methods’). Six packaging cell clones from each construct with the highest physical titer were used to transduce the human T cell line SupT1, and CD3 expression was assayed by FACS analysis (Supplementary Table 1). SupT1 is defective in endogenous TCR α chain biosynthesis and will not mobilize the CD3 protein complex to the cell surface unless supplied with a substitute α chain. CD3 expression ranged from 6 to 96%. Clones APB 38, GSGP2A 3, GSGT2A 36, and T2A 24 exhibited both high physical titer and high transduction efficiency, and these four constructs were chosen for additional study (Supplementary Table 1).
To test the hypothesis that use of 2A linker peptides would result in improved TCR expression, PBL were transduced with each of the anti-NY-ESO-1 TCR vector constructs on day 2 post-stimulation. Five days following transduction, cells were harvested and stained for Vβ13.1, and NY-ESO-1 tetramer. Examples of transductions using the high tier producer clones of APB, GSGP2A, GSGT2A, and T2A were as shown in Fig. 2b. Transduction with constructs incorporating 2A peptides resulted in significantly higher expression of the TCR, both by Vβ13.1 staining and NY-ESO-1 tetramer (Fig. 2b). These findings were consistent in four independent experiments. There was little variability in TCR expression following transduction of PBL with each of the 2A constructs, yielding equivalent expression and significantly higher TCR expression when compared to the PGK promoter-containing vector.
Functional studies of the transduced PBL
To determine whether the increased TCR expression in the 2A constructs was associated with increased release of effector cytokines by TCR-transduced PBL, lymphocytes were transduced with supernatants from the top producer clone for each of the constructs. The transduced PBL were then co-cultured with T2 cells pulsed with cognate (NY-ESO-1) or irrelevant influenza peptide. Antigen-specific IFN-γ release was detected in co-cultures of peptide-pulsed T2 cells for all constructs, with a higher level of cytokine secretion for PBL transduced with the constructs incorporating the 2A linker peptides (Fig. 3a). These findings were consistent in four independent experiments.
Fig. 3.

Cytokine production of TCR-transduced PBLs following co-culture with cells expressing the cognate antigen. a PBL transduced with supernatant from packaging cell clones APB 38, GSGP2A 3, GSGT2A 36, and T2A 24 were co-cultured with T2 cells pulsed with NY-ESO-1 peptide (p157-165v) or influenza-specific peptide. Antigen-specific IFN-γ secretion by retroviral vector-transduced PBLs was determined by ELISA. Data are the mean values (picograms per milliliter) of duplicate samples, with standard deviation (error bars). b PBL were transduced with supernatant from retroviral constructs for GFP (control virus) or from APB, GSGP2A, GSGT2A, and T2A and were then co-cultured with HLA-A2+, NYESO-1+ melanoma lines (624.38mel, 1300mel, A375mel) or HLA-A2+, NY-ESO-1- melanoma line (SK23). Transduced PBL were also incubated with T2 cells alone, or T2 cells pulsed with NY-ESO-1 peptide (p157-165v) or gp100 peptide. Antigen-specific IFN-γ secretion by retroviral vector-transduced PBLs was determined by ELISA. Data are the mean values (picograms per milliliter) of samples, with standard deviation (error bars)
NY-ESO-1 is expressed in a wide range of malignancies, and in over a third of melanoma tumors [11]. We have previously shown that PBL transduced with our original TCR vectors targeting the NY-ESO-1 antigen are able to secrete IFN-γ and lyse HLA-A2+, NY-ESO-1+ melanoma targets in an antigen-specific manner [5]. To determine whether the increased effector cytokine production of PBL transduced with the 2A constructs with peptide-pulse T2 cells translated into increased effector function against melanoma targets, we next measured IFN-γ release following cultured of PBL with melanoma tumor cell lines. Antigen-specific IFN-γ release was detected in co-cultures of HLA-A2+, NY-ESO+ melanoma tumors with all TCR constructs, with a higher level of cytokine secretion for PBL transduced with the constructs incorporating the 2A linker peptides (Fig. 3b). These findings were consistent in four independent experiments.
TCR expression and effector function of PBL transduced with modified TCR vector
We previously demonstrated that supplement of an additional disulfide bond in the constant regions of the TCR results in improve pairing of α and β chains in the introduced TCR with increased surface expression and improved effector function of PBL electroporated with the cysteine-modified TCR [30]. To test the hypothesis that incorporation of this cysteine-modified TCR into our vector constructs would enhance TCR expression beyond that seen with 2A linker peptides alone, we created a vector construct incorporating α and β chains for the 1G4 NY-ESO TCR in which we replaced Thr48 on the α chain and Ser57 on the β chain with cysteines to facilitate the creation of an additional interchain disulfide bond between the TCR constant regions, as had originally been described by Boulter et al. [31].
A retroviral vector, MSGV1-Cys1G4-AfrnSGSGP2AB (CysfrnSGSGP2A), was constructed incorporating genes encoding the cysteine-modified α and β chains joined by a coding sequence for a 2A linker peptide (Fig. 1). In this case, we incorporated a furin cleavage site at the 5′ end of the linker to facilitate cleavage of the residual 2A peptide from the α chain following translation. PBL were transduced with either the GSGP2A construct or the CysfrnSGSGP2A construct on day 2 post-stimulation. Five days following transduction, cells were harvested and stained for Vβ13.1, and NY-ESO-1 tetramer. Examples of transductions for GSGP2A and CysfrnSGSGP2A are shown in Fig. 4. Transduction with constructs incorporating the cysteine-modified 1G4 TCR demonstrated a modest increase in expression of the TCR, both by Vβ13.1 staining and NY-ESO-1 tetramer (Fig. 4a). There was also a slight increase in recognition of tumor targets in CysfrnSGSGP2A-transduced PBL that was consistent with our previous results (Fig. 4b).
Fig. 4.
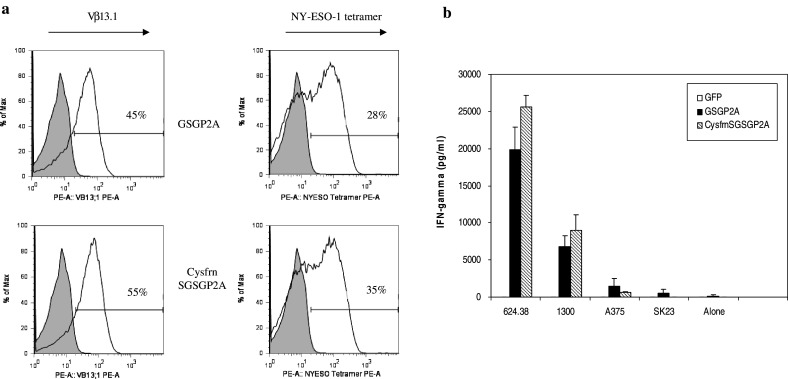
T cell receptor expression and effector function of PBL transduced with modified TCR vector. a A retroviral construct incorporating cysteine-modified α and β chains was constructed as previously described. OKT3-stimulated PBL were transduced with viral supernatant from the GSGP2A and CysfrnSGSGP2A constructs. Five days post transduction, PBL were stained for Vβ13.1 and NY-ESO-1 tetramer and expression was analyzed via flow cytometry. The percentage of positive cells for Vβ13.1 and NY-ESO-1 are indicated on the histogram. b PBL were transduced with supernatant from retroviral constructs for GFP (control virus) or from GSGP2A and CysfrnSGSGP2A and were then co-cultured with HLA-A2+, NYESO-1+ melanoma lines (624.38mel, 1300mel, A375mel) or HLA-A2+, NY-ESO-1− melanoma line (SK23). Antigen-specific IFN-γ secretion by retroviral vector-transduced PBLs was determined by ELISA. Data are the mean values (picograms per milliliter) of samples, with standard deviation (error bars)
Induction of NY-ESO-1 expression by epigenetic modulation of target cells
We have previously shown that sequential treatment of tumor cells with the demethylating agent DAC and the histone deacetylase inhibitor DP resulted in induction of NY-ESO-1 expression in melanoma, lung, and esophageal cancer cell lines [23]. To test the extent to which NY-ESO-1 expression can be induced, a wider range of non-melanoma epithelial malignancies were treated with DAC, DP, or sequential DAC/DP. Each of the cell lines, with the exception of H1299, demonstrated little to no detectable expression of NY-ESO-1 in the basal state in culture with NM (Fig. 5). Minimal induction of NY-ESO-1 expression was observed after 6-h exposure to DP at 25 ng/ml (a concentration approximately 1 log less than that observed in plasma at the maximum tolerated dose for this agent). Treatment of tumor lines with either DAC alone (0.1 μmol/l), or sequential DAC (0.1 μmol/l)—DP (25 ng/ml) resulted in a significant induction of NY-ESO-1 expression (Fig. 5).
Fig. 5.

Induction of NY-ESO-1 expression following treatment of cells with DAC-DP. RT-PCR and quantitative RT-PCR analysis were performed for NY-ESO-1 mRNA expression in the H1299 small cell lung carcinoma cell line (HLA-A2 transfected), the BE-3 esophageal carcinoma cell line, the H2373 pleural mesothelioma cell line, the human pancreatic cancer cell line Panc-1, the human glioblastoma cell line LNZTA3WT4, and the human ovarian cancer cell line OVCAR-3 after exposure to normal media (NM), 5-aza-2′-deoxycytidine (DAC), depsipeptide (DP), or sequential 5-aza-2′-deoxycytidine and depsipeptide (DAC-DP). A representative image of the RT-PCR products following agarose gel electrophoresis is shown. GAPDH or β-actin served as a control. Data below each band indicates the NY-ESO-1 copy number per 104 β-actin determined by quantitative RT-PCR analysis. Data are representative of three independent experiments
Recognition of tumor cell lines following epigenetic modulation
In our prior studies we demonstrated that the induction of NY-ESO-1 conferred recognition of treated tumor cell lines, but not normal human bronchial epithelial cells, by HLA-A2-restricted NY-ESO-1-specific CTLs [20]. The CTLs in our prior studies were generated by in vitro peptide stimulation of lymphocytes obtained from a patient with HLA-A2+ melanoma who had a high serum antibody titer against NY-ESO-1 [20]. We next sought to determine if the increased NY-ESO-1 expression in the treated tumor cell lines conferred recognition by NY-ESO-1 TCR-transduced PBL. Treated tumor lines were co-cultured with PBL transduced with the CysfrnSGSGP2A construct. Transduction efficiency was 60% by Vβ13.1 and 50% by NY-ESO-1 tetramer staining (data not shown).
All of the HLA-A2+ tumor cell lines treated with either DAC or sequential DAC-DP were recognized by the TCR-transduced PBL (Fig. 6). To test whether the increased sensitivity observed following treatment of tumors with DAC or DAC-DP was due to up-regulation of MHC class I molecules, we assessed HLA class I expression in cultured cancer cells by FACS analysis of the W6/32 antibody that recognizes only functional class I molecules. We observed no significant difference in expression of HLA class I by FACS analysis on tumor cells undergoing no treatment (NM), treatment with DAC, DP, or sequential DAC-DP (data not shown). The magnitude of the cytokine release corresponded with the mRNA copy number in the treated cells, which was consistent with findings from our prior studies [20].
Fig. 6.

Cytokine production of TCR-transduced PBLs following co-culture with NY-ESO-1+, HLA-A2+ and − cell lines. PBL were transduced with supernatant from the CysfrnSGSGP2A retroviral construct and were then co-cultured with the H1299 non-small cell lung carcinoma line (HLA-A2-transfected) (NY-ESO-1+, HLA-A2+), the (BE-3 esophageal carcinoma cell line (NY-ESO-1−, HLA-A2+), the H2373 pleural mesothelioma cell line (NY-ESO-1−, HLA-A2+),), the human pancreatic cancer cell line Panc-1 (NY-ESO-1−, HLA-A2+), the human osteosarcoma cell line LNZTA3WT4 (NY-ESO-1−, HLA-A2+), and the human ovarian cancer cell line OVCAR-3 (NY-ESO-1−, HLA-A2+) after exposure to normal media (NM), 5-aza-2′-deoxycytidine (DAC), depsipeptide (DP), or sequential 5-aza-2′-deoxycytidine and depsipeptide (DAC-DP). Antigen-specific IFN-γ secretion by retroviral vector-transduced PBLs was determined by ELISA. Data are the mean values (picograms per milliliter) of duplicate samples, with standard deviation (error bars)
Discussion
T cell receptor-based genetic immunotherapy for cancer involves the transfer of genes encoding TCRs into PBL with the goal of conferring reactivity against tumor targets expressing the cognate antigen. This therapy is based on knowledge gained through the study of the adoptive transfer of TIL, which can result in objective tumor regression when transferred following a lymphodepleting conditioning regimen in patients with metastatic melanoma [1]. However, TIL therapy has several limitations. First, a tumor must be removed via a surgical procedure to procure TIL. Depending on the location of the tumor, there may be significant associated morbidity and a prolonged recovery. Second, tumor-reactive cells must be isolated and expanded ex vivo, which is generally only successful in half of patients. Third, the process of ex vivo expansion takes up to 2 months, thus treatment may be delayed if reactive TIL cannot be generated. Furthermore, reactive TIL are difficult to generate in histologies other than melanoma, thus cancer immunotherapy using this strategy may not be applicable in to other malignancies.
In TCR-based genetic immunotherapy, TCRs targeting TAA are cloned and inserted into viral vectors. These vectors can then be used to transduce PBL to license them for antigen recognition and tumor destruction. We have recently developed retroviral vectors with TCR targeting several TAA, including those targeting the HLA-A2-restricted immunodominant epitopes of the MART-1 and gp100 antigens [2, 3]. We have employed TCR-transduced PBL in on-going clinical trials to treat patients with metastatic melanoma. Our early experience with this form of therapy yielded a low response rate (13% by RECIST criteria) [4], however we have since made modifications to the TCR and vector constructs which has resulted in improved TCR avidity and tumor reactivity of transduced PBL in vitro [30, 32, 33].
In addition to developing TCRs targeting melanoma antigens, we have generated TCRs targeting TAA expressed in a wide range of non-melanoma epithelial malignancies [5, 34, 35]. One of these TCR targets NY-ESO-1, a CT antigen that is expressed in approximately one-third of melanoma tumors as well as a wide range of other malignancies [6, 7]. Since development of the original anti-NY-ESO-1 TCR vector in 2005 [5], we have made modifications to the TCR to increase its affinity and antigen-specific recognition [29]. Furthermore, modifications utilizing targeted mutations in the CDR2 or CDR3 regions of TCR can significantly enhance antigen-specific recognition, and confer this recognition to both CD4 and CD8+ T lymphocytes [36].
Having isolated a high affinity NY-ESO-1 specific TCR, we sought to optimize gene transfer vector design. The first two TCR vector designs analyzed used either an IRES or internal promoter to express both TCR chains. While these designs had been highly successful with previous TCRs [2, 35], the IRES proved to be ineffective in mediating TCR expression. While we have observed a lack of IRES vector activity with other TCRs, this is not a common finding.
The most active retroviral constructs used in this report incorporated coding sequences for 2A linker peptides between the α and β chains of the NY-ESO-1 TCR. T cells transduced with these vectors demonstrated significantly enhanced TCR expression by FACS analysis and improved effector function as measured by cytokine release following co-culture with peptide-pulsed target cells or tumor cells expressing the NY-ESO-1 antigen (Figs. 2, 3). The improved TCR expression and effector function following TCR transduction of PBL with these vector constructs has direct implications for future vector design for immunotherapy as well as other gene therapy trials. This design strategy may also allow incorporation of coding sequences for additional genes that may further enhance effector function, such as cytokines and co-stimulatory molecules. We also confirmed our previous findings that addition of cysteine modifications to TCRs results in improved expression in PBL, perhaps by facilitating pairing of the introduced α and β [30].
In addition to improving TCR avidity and improved TCR expression through vector design, we sought to enhance reactivity against tumor targets by increasing their level of antigen expression. This is based on evidence that widespread changes in DNA methylation status and chromatin structure occur during the process of carcinogenesis leading to changes in gene expression [37], which include increased expression of the CT antigens [20]. We have demonstrated that expression of antigens such as NY-ESO-1 can be selectively up-regulated in tumor cells by treatment with demethylating agents and histone deacetylase inhibitors, and can sensitize them to recognition by CTLs specific for the antigen [20]. In a phase I clinical trial, we used these agents to treat patients with advanced malignancies, and were able to demonstrate up-regulation of NY-ESO-1, MAGE, or p16 in approximately one-third of evaluable patients [38]. In this trial, all of the 35 patients enrolled were evaluable for toxicity. The most common toxicities were hematologic, with grade 3 leukopenia occurring in 20 of 35, grade 3 thrombocytopenia in 2 of 35, and grade 3 anemia in 3 of 35 patients [38]. We also reported that treatment with these agents with resultant increased expression of NY-ESO-1 in human melanoma and non-melanoma cell lines and can sensitize them to recognition by CTLs specific for the antigen [20]. We have applied these findings in animal studies, in which systemic treatment with DAC resulted in improved responses to adoptive transfer of specific CTL targeting the P1A CT antigen in this model [39], providing the basis for studying the cooperative effects of treatment with these agents in conjunction with adoptive immunotherapy.
Taken together, these data demonstrate that treatment of target cells with demethylating agents alone or in combination with a histone deacetylase inhibitor causes up-regulation of CT antigen expression which confers recognition of these tumor targets to HLA-restricted, TCR-transduced PBL. These findings have significant implications as we move forward with TCR-based genetic immunotherapy for melanoma as well as non-melanoma epithelial malignancies, and suggest that a combination of immunotherapy and epigenetic modulation may be a useful clinical strategy. Incorporation of pre-treatment with chromatin-remodeling agents followed by adoptive transfer of TCR-transduced PBL targeting CT antigens may provide a therapeutic benefit over TCR-based immunotherapy alone.
Electronic supplementary material
Below is the link to the electronic supplementary material.
MOESM1 [INSERT CAPTION HERE] (PPT 43 kb)
Acknowledgments
We thank FACS lab and TIL lab in Surgery Branch for providing technical support and maintenance of tumor cells from patients. This work is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- 1.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, Rogers LJ, Gracia GJ, Jones SA, Mangiameli DP, Pelletier MM, Gea-Banacloche J, Robinson MR, Berman DM, Filie AC, Abati A, Rosenberg SA. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, Morgan RA. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, Zheng Z, Nahvi A, de Vries CR, Rogers-Freezer LJ, Mavroukakis SA, Rosenberg SA. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065X.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 7.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 8.Chen YT, Scanlan MJ, Sahin U, Tureci O, Gure AO, Tsang S, Williamson B, Stockert E, Pfreundschuh M, Old LJ. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci USA. 1997;94:1914–1918. doi: 10.1073/pnas.94.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jager E, Gnjatic S, Nagata Y, Stockert E, Jager D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, Hoffman E, Arand M, Old LJ, Knuth A. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci USA. 2000;97:12198–12203. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shackleton M, Davis ID, Hopkins W, Jackson H, Dimopoulos N, Tai T, Chen Q, Parente P, Jefford M, Masterman KA, Caron D, Chen W, Maraskovsky E, Cebon J. The impact of imiquimod, a Toll-like receptor-7 ligand (TLR7L), on the immunogenicity of melanoma peptide vaccination with adjuvant Flt3 ligand. Cancer Immun. 2004;4:9. [PubMed] [Google Scholar]
- 11.Chen Q, Jackson H, Shackleton M, Parente P, Hopkins W, Sturrock S, MacGregor D, Maraskovsky E, Tai TY, Dimopoulos N, Masterman KA, Luke T, Davis ID, Chen W, Cebon J. Characterization of antigen-specific CD8+ T lymphocyte responses in skin and peripheral blood following intradermal peptide vaccination. Cancer Immun. 2005;5:5. [PubMed] [Google Scholar]
- 12.Davis ID, Chen W, Jackson H, Parente P, Shackleton M, Hopkins W, Chen Q, Dimopoulos N, Luke T, Murphy R, Scott AM, Maraskovsky E, McArthur G, MacGregor D, Sturrock S, Tai TY, Green S, Cuthbertson A, Maher D, Miloradovic L, Mitchell SV, Ritter G, Jungbluth AA, Chen YT, Gnjatic S, Hoffman EW, Old LJ, Cebon JS. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci USA. 2004;101:10697–10702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1:376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 14.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–417. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 15.Fang J, Yi S, Simmons A, Tu GH, Nguyen M, Harding TC, VanRoey M, Jooss K. An antibody delivery system for regulated expression of therapeutic levels of monoclonal antibodies in vivo. Mol Ther. 2007;15:1153–1159. doi: 10.1038/sj.mt.6300142. [DOI] [PubMed] [Google Scholar]
- 16.Arnold PY, Burton AR, Vignali DA. Diabetes incidence is unaltered in glutamate decarboxylase 65-specific TCR retrogenic nonobese diabetic mice: generation by retroviral-mediated stem cell gene transfer. J Immunol. 2004;173:3103–3111. doi: 10.4049/jimmunol.173.5.3103. [DOI] [PubMed] [Google Scholar]
- 17.Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 18.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol. 2001;82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 19.Lee L, Wang RF, Wang X, Mixon A, Johnson BE, Rosenberg SA, Schrump DS. NY-ESO-1 may be a potential target for lung cancer immunotherapy. Cancer J Sci Am. 1999;5:20–25. [PubMed] [Google Scholar]
- 20.Weiser TS, Guo ZS, Ohnmacht GA, Parkhurst ML, Tong-On P, Marincola FM, Fischette MR, Yu X, Chen GA, Hong JA, Stewart JH, Nguyen DM, Rosenberg SA, Schrump DS. Sequential 5-aza-2′-deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY-ESO-1. J Immunother. 2001;24:151–161. doi: 10.1097/00002371-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 21.Salter RD, Howell DN, Cresswell P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 1985;21:235–246. doi: 10.1007/BF00375376. [DOI] [PubMed] [Google Scholar]
- 22.Topalian SL, Solomon D, Rosenberg SA. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J Immunol. 1989;142:3714–3725. [PubMed] [Google Scholar]
- 23.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J Virol. 1996;70:5701–5705. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onodera M, Yachie A, Nelson DM, Welchlin H, Morgan RA, Blaese RM. A simple and reliable method for screening retroviral producer clones without selectable markers. Hum Gene Ther. 1997;8:1189–1194. doi: 10.1089/hum.1997.8.10-1189. [DOI] [PubMed] [Google Scholar]
- 25.Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986–994. doi: 10.1101/gr.6.10.986. [DOI] [PubMed] [Google Scholar]
- 26.Kruse N, Pette M, Toyka K, Rieckmann P. Quantification of cytokine mRNA expression by RT PCR in samples of previously frozen blood. J Immunol Methods. 1997;210:195–203. doi: 10.1016/S0022-1759(97)00188-9. [DOI] [PubMed] [Google Scholar]
- 27.Riker AI, Kammula US, Panelli MC, Wang E, Ohnmacht GA, Steinberg SM, Rosenberg SA, Marincola FM. Threshold levels of gene expression of the melanoma antigen gp100 correlate with tumor cell recognition by cytotoxic T lymphocytes. Int J Cancer. 2000;86:818–826. doi: 10.1002/(SICI)1097-0215(20000615)86:6<818::AID-IJC10>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 28.Kammula US, Lee KH, Riker AI, Wang E, Ohnmacht GA, Rosenberg SA, Marincola FM. Functional analysis of antigen-specific T lymphocytes by serial measurement of gene expression in peripheral blood mononuclear cells and tumor specimens. J Immunol. 1999;163:6867–6875. [PubMed] [Google Scholar]
- 29.Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, Li Y, Molloy PE, Dunn SM, Jakobsen BK, Rosenberg SA, Morgan RA. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol. 2007;179:5845–5854. doi: 10.4049/jimmunol.179.9.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. doi: 10.1093/protein/gzg087. [DOI] [PubMed] [Google Scholar]
- 32.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine–human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson LA, Heemskerk B, Powell DJ, Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, Morgan RA. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol Ther. 2006;13:151–159. doi: 10.1016/j.ymthe.2005.07.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, Morgan RA. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbins PF, Li YF, El-Gamil M, Zheng Z, Xu H, Zhao Y, Wargo J, Jakobsen BK, Bennett AD, Morgan RA, Feldman S, Rosenberg SA Identification of amino acid substitutions in T cell receptor complementarity determining regions that specifically enhance functional avidity. J Immunol (in press)
- 37.Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, Caligiuri MA, Yates A, Nishikawa R, Su Huang H, Petrelli NJ, Zhang X, O’Dorisio MS, Held WA, Cavenee WK, Plass C. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 38.Schrump DS, Fischette MR, Nguyen DM, Zhao M, Li X, Kunst TF, Hancox A, Hong JA, Chen GA, Pishchik V, Figg WD, Murgo AJ, Steinberg SM. Phase I study of decitabine-mediated gene expression in patients with cancers involving the lungs, esophagus, or pleura. Clin Cancer Res. 2006;12:5777–5785. doi: 10.1158/1078-0432.CCR-06-0669. [DOI] [PubMed] [Google Scholar]
- 39.Guo ZS, Hong JA, Irvine KR, Chen GA, Spiess PJ, Liu Y, Zeng G, Wunderlich JR, Nguyen DM, Restifo NP, Schrump DS. De novo induction of a cancer/testis antigen by 5-aza-2′-deoxycytidine augments adoptive immunotherapy in a murine tumor model. Cancer Res. 2006;66:1105–1113. doi: 10.1158/0008-5472.CAN-05-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MOESM1 [INSERT CAPTION HERE] (PPT 43 kb)