

Abstract
Neurological complications such as inflammation, failure of the blood-brain barrier (BBB), and neuronal death contribute to the mortality and morbidity associated with WNV-induced meningitis. Compromised BBB indicates the ability of the virus to gain entry into the CNS via the BBB, however, the underlying mechanisms, and the specific cell types associated with WNV-CNS trafficking are not well understood. Brain microvascular endothelial cells, main component of the BBB, represent a barrier to virus dissemination into the CNS and could play key role in WNV spread via hematogenous route. To investigate WNV entry into the CNS, we infected primary human brain microvascular endothelial (HBMVE) cells with the neurovirulent strain of WNV (NY99) and examined WNV replication kinetics together with the changes in the expressions of key tight junction proteins (TJP) and cell adhesion molecules (CAM). WNV infection of HBMVE cells was productive as analyzed by plaque assay and qRT-PCR, and did not induce cytopathic effect. Increased mRNA and protein expressions of TJP (claudin-1) and CAM (vascular cell adhesion molecule and E-selectin) were observed at days 2 and 3 after infection, respectively, which coincided with the peak in WNV replication. Further, using an in vitro BBB model comprised of HBMVE cells, we demonstrate that cell-free WNV can cross the BBB, without compromising the BBB integrity. These data suggest that infection of HBMVE cells can facilitate entry of cell-free virus into the CNS without disturbing the BBB, and increased CAM may assist in the trafficking of WNV-infected immune cells into the CNS, via ‘Trojan horse’ mechanism, thereby contributing to WNV dissemination in the CNS and associated pathology.
Introduction
West Nile virus (WNV), an enveloped, single-stranded positive-sense, neurotropic flavivirus, is an important human pathogen that targets neurons to cause potentially lethal encephalitis in 1 to 2% of WNV-infected febrile patients (Diamond et al., 2003; Hayes and Gubler, 2006; Murray et al., 2006). Currently there are no therapeutic agents or vaccines approved for use against WNV infection in humans. The initial target of WNV infection are skin Langerhan's dendritic cells, which after infection migrate to and replicate in regional lymph nodes leading to primary viremia, followed by secondary viremia at significantly higher titers as a result of WNV dissemination and replication in lymphoid and other organs (Davis et al., 2006; Samuel and Diamond, 2006). Further, through un-identified mechanisms, WNV enters the central nervous system (CNS) and causes an array of West Nile neurological disease including meningitis, encephalitis, and acute flaccid paralysis/polio-like myelitis.
WNV-associated encephalitis is characterized by disruption of the blood-brain barrier (BBB), enhanced infiltration of immune cells into the CNS, microglia activation, inflammation and eventual loss of neurons (Cheeran et al., 2005; Glass et al., 2005; Shrestha, Gottlieb, and Diamond, 2003; Sitati et al., 2007). Since high viremia is directly correlated with early WNV entry into the CNS (Samuel and Diamond, 2006), it is suggested that WNV in the periphery enters the CNS by crossing the BBB. Recent studies have documented that inflammatory cytokines, such as TNF-α, macrophage migration inhibitory factor and MMP-9 play an important role in disruption of the BBB in WNV-infected mice (Arjona et al., 2007; Wang et al., 2008; Wang et al., 2004). The BBB, which consists of microvascular endothelial cells, perivascular astrocytes, basement membrane, and pericytes, is a highly regulated interface which separates blood-borne entities from the CNS (Persidsky et al., 2006). The main structural and anatomical basis of BBB integrity is the presence of tight junctions throughout the brain microvascular endothelial cells that regulate paracellular passage of cells, molecules, and ions. Tight junction proteins (TJP) include claudins and occludin that are joined to the cytoskeleton by the cytoplasmic proteins such as zonula occludens (ZO). Regulation of TJP expression and/or sub-cellular distribution plays a key role in the physiology of the BBB (Persidsky et al., 2006).
Infection of BBB endothelial cells and changes in its properties as a result of virus infection has been shown for several viruses, specifically those which manifest acute neuroinflammation and neurodegeneration, such as simian immunodeficiency virus (SIV) (Luabeya et al., 2000; Strelow et al., 1998), measles virus (Cosby and Brankin, 1995), human cytomegalovirus (HCMV) (Lathey et al., 1990) and human T-cell leukemia virus (HTLV). HIV-induced changes in the brain microvascular cells, including alterations in the expressions of TJP and cell adhesion molecules (CAM), are associated with increased influx of infected inflammatory cells as ‘Trojan horse’ and higher viral load in the CNS in both, in vitro and animal models (Eugenin et al., 2006; Kanmogne et al., 2007; Pu et al., 2005).
Since BBB endothelial cells are directly exposed to cell-free virus in the peripheral blood, WNV infection and replication in human brain microvascular endothelial (HBMVE) cells can be one of the possible route by which WNV enters the CNS. Also, cell-free WNV may cross the BBB either by transcytosis mechanism without altering the BBB integrity, or by modulating tight junction properties of the BBB thereby allowing passage of not only cell-free virus, but also cell-associated virus, into the CNS. Leukocyte adhesion and trafficking across endothelial cells depends on sequential activation and expression of cell surface adhesion molecules such as VCAM-1 and ICAM-1 in HIV- and SIV-infected BBB models (Maclean et al., 2004; Nottet et al., 1996). This study characterizes WNV infection in HBMVE cells, an important component of the BBB, specifically the effect on TJP and CAM expressions. Here, we report that WNV infection in confluent HBMVE cells is productive and the peak in WNV infection is correlated to an increase of specific TJP such as claudin-1, and CAM, such as VCAM-1 and E-selectin. Further, using an in vitro human BBB model, we demonstrate that cell-free WNV can cross the BBB without altering its integrity.
Materials and Methods
Cells and virus
Low-passage primary HBMVE cells were propagated as described previously (Chapagain et al., 2007). All experiments were performed with cells between passages 8 to 10. A stock of lineage I WNV strain NY99, originally isolated from a crow in New York and propagated in Vero cells once, was used for all infection experiments. To generate UV-inactivated WNV (UV-WNV), WNV was diluted in 500 μL PBS in a 35-mm culture plate and exposed to UV radiation using a UV-Stratalinker 2400 device (Stratagene) for 10 min. For infection, HBMVE cells grown to 90% confluency in T-25 flasks, 6-well plates or coverslips, were infected with WNV at the multiplicity of infection (MOI) of 1 or 5 as described previously (Verma et al., 2008) and cells, and supernatant were harvested at various time points.
Plaque assay
Complete cell-free supernatants were collected and replaced with fresh media at different time points after WNV infection and production of infectious virus was determined using the plaque assay (Bunning et al., 2002). For plaque assay, 0.1 mL of cell-free supernatants diluted 0 to 10-12 times were added to a confluent monolayer of Vero cells in 6-well plates and incubated for 1 hr at 37°C and 5% CO2. Vero cells were overlaid with 3 mL of M-199 medium containing 1% agarose. Three days later, a second overlay of 3 mL M-199 medium containing 1% agarose and 1% neutral red was added for plaque visualization. The plaques were scored on day 2 after second overlay.
qRT-PCR for host genes expression and intracellular WNV RNA
Cellular RNA from mock-infected, MOI-1 and -5 WNV-infected, and MOI-5 UV-WNV-infected cells at different time points after infection were extracted and one microgram of total RNA was reverse-transcribed into cDNA (Verma et al., 2006). qRT-PCR was performed using 2 to 5 μL of the 1:3 diluted cDNA as template in a iCycler (Bio-Rad). Thermal cycling was initiated with a first denaturation step of 4 min at 95°C followed by 42 cycles of 95°C for 30 s, and annealing/extension for 40 s. Primer sequences, amplicon size and annealing temperatures used for amplification of TJP and CAM are described in Table 1. GAPDH, the house-keeping gene, was used to normalize each gene and the fold-change of expression at each time point was determined compared to corresponding un-infected control as described previously (Verma et al., 2008; Verma et al., 2006). Intracellular WNV RNA was quantitated by using 2 μL of 1:3 diluted cDNA template, 50 pmol/μL each of forward and reverse primers and probe specific for WNV as described previously (Lanciotti et al., 2002). All experiments were performed at least three times in duplicate.
Table 1. Primer sequences used for qRT-PCR.
| Gene GenBank No. |
Primer Sequence (5′-3′) | Amplicon (bp) |
Tm (°C) |
|---|---|---|---|
| Claudin-1 NM_021101 | |||
| Forward | GAA GTG CTT GGA AGA CGA TGA G | 190 | 56 |
| Reverse | GCC CAG CCA GTG AAG AAG G | ||
|
| |||
| Claudin-5 NM_003277 | |||
| Forward | CTC TGC TGG TTC GCC AAC AT | 87 | 57 |
| Reverse | CAG CTC GTA CTT CTG CGA CA | ||
|
| |||
| E-selectin NM_000450 | |||
| Forward | TGA GAC AGA GGC AGC AGT G | 199 | 57 |
| Reverse | CCG TGG AGG TGT TGT AAG AC | ||
| GAPDH BC025925 | |||
| Forward | AGT TAG CCG CAT CTT CTT TTG C | 96 | 57 |
| Reverse | CAA TAC GAC CAA ATC CGT TGA CT | ||
|
| |||
| ICAM-3 NM_002162 | |||
| Forward | AGC GGC AGT TAC CAT GTT AGG G | 146 | 58 |
| Reverse | GGC TTT ATT GGT GCG GAA TCT GAG | ||
|
| |||
| Occludin DQ892423 | |||
| Forward | TCC AGA GTC TTC CTA TAA ATC CAC | 106 | 57 |
| Reverse | ACC ACC GCT GCT GTA ACG | ||
|
| |||
| PECAM NM_000442 | |||
| Forward | GCA GTC TTC ACT CTC AGG ATG | 143 | 57 |
| Reverse | GGC AGG CTC TTC ATG TCA AC | ||
|
| |||
| VCAM-1 BC068490 | |||
| Forward | TGC TGC TCA GAT TGG AGA CTC | 113 | 55 |
| Reverse | TCC TCA CCT TCC CGC TCA G | ||
|
| |||
| ZO1 NM_003257 | |||
| Forward | GAA TGA TGG TTG GTA TGG TGC G | 191 | 60 |
| Reverse | TCA GAA GTG TGT CTA CTG TCC G | ||
|
| |||
| WNV (ENV) NC_009942 | |||
| Forward | CAG ACC ACG CTA CGG CG | 112 | 60 |
| Reverse | CTA GGG CCG CGT GGG | ||
| Probe | TCT GCG GAG AGT GCA GTC TGC GAT | ||
Western immunoblot for TJP and CAM analysis
Total cellular protein was extracted from mock-infected and MOI-5 WNV-infected cells at 12 hr, and from days 1 to 4 after infection (Chapagain et al., 2006; Verma et al., 2008). Total 40-60 μg of cellular protein extract was fractionated on a 4-12% gradient SDS polyacrylamide gel, and then transferred onto 0.2 or 0.45 μm PVDF membranes (Invitrogen). Non-specific binding sites were blocked with 5% BSA in 1× PBS with 0.1% Tween (PBST), and membranes were incubated overnight at 4°C with antibodies against polyclonal rabbit anti-claudin-1 (Zymed, Carlsbad, CA), polyclonal mouse anti-VCAM-1 (Santa Cruz Biotech, CA), and β-actin (Sigma). After three vigorous washings with PBST, the membranes were further incubated with horseradish peroxidase-conjugated secondary antibodies for 2 hr at room temperature and developed using enhanced chemiluminescence (ECL plus) detection kit (Invitrogen). The blots were scanned and quantitated using the Biorad Quantity one Program.
Indirect-immunofluorescence microscopy for detection of WNV antigen and host proteins
HBMVE cell monolayers were grown on coverslips at a confluency of 80-90% in 24-well plates and infected with WNV at MOI-5. At days 2, 3 and 4 after infection, mock-infected control cells and WNV-infected HBMVE cells were fixed in 4% paraformaldehyde (PFA) and immunostained as described previously (Verma et al., 2008), using primary monoclonal human anti-WNV envelope antibody (1:800), or monoclonal mouse anti-ZO-1 (1:100, Zymed) or polyclonal rabbit anti-claudin-1 (1:100), or polyclonal mouse anti-VCAM-1 (1:100) followed by Alexa Fluor 488 conjugated goat anti-mouse or fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit (1:100) (Amersham, Pittsburgh, PA) secondary antibodies (Invitrogen). Fluorescent immunostained cells were examined using a Zeiss confocal Pascal equipped with a Zeiss Axiovert 200 microscope, or a Axiocam MRm camera mounted on a Zeiss Axiovert 200 microscope, both equipped with appropriate fluorescence filters and objectives.
In vitro BBB model and WNV infection
An in-vitro BBB model was constructed using a BioCoat® Cell Environment™ Human Fibronectin PET (polyethylene terephthalate) insert with 3.0 μm pore and 0.33 cm2 in a 24-well plate (BD Bioscience, Bedford, MA) (Mahajan et al., 2008; Mukhtar and Pomerantz, 2000). Briefly, the inserts were rehydrated with pre-warmed medium, 5 × 104 HBMVE cells were seeded on the upper surface of the inserts, and the inserts were incubated at 37°C, 5% CO2 and 100% humidity. The integrity of the in-vitro BBB model was determined every day from day 4 after seeding, by measuring the transendothelial electrical resistance (TEER) and FITC-dextran (4-KDa mol. wt., Sigma) permeability assays. FITC-dextran (150 μL of 100 μg/mL medium) was added in each upper chamber and after 2 hr incubation, 150 μL of medium was removed from the lower chamber to read the fluorescence of transmigrated FITC-dextran on a Victor™ 1420 fluorescence microplate reader (Perkin-Elmer Wallace, Inc.). The FITC-dextran transmigrated across the inserts at each time point was calculated as percentage of the total amount added in the upper well (100 ug/mL). In addition, the BBB model integrity was measured at each time point using TEER (EVOMX, World Precision Instrument) (Mahajan et al., 2008), which was calculated as per the manufacturer's instructions. At day 8 after seeding, the in vitro BBB models were infected with UV-WNV or WNV at MOI-1 as described previously and complete upper (luminal)- and lower (abluminal)-chambers supernatants, UCS and LCS, respectively, were collected at 2, 6 and 12 hr and on days 1 to 3 after infection, and replaced with fresh medium. Viral titers and RNA in both, UCS and LCS were determined by plaque assay and qRT-PCR as described above.
Statistical analyses
All statistical tests, including paired and unpaired students t-test for immunoblotting and for in vitro BBB integrity assays were performed using Graphpad InStat version 5.0 (GraphPad software, San Diego, CA). P values of <0.05 were considered significant.
Results
WNV can infect and replicate in HBMVE cells
Though WNV is a neurotropic virus, its ability to infect and replicate in cells of the BBB is unknown. Therefore, we first determined WNV infection and replication kinetics in primary HBMVE cells by immunostaining, plaque assay and qRT-PCR. By using immunofluorescence microscopy, WNV antigen was detected as early as 12 hr after infection and the number of infected cells gradually increased from day 1 to 3, with a peak of infected cells at day 2 after infection (Fig. 1A and 1B). As observed in figure 1C and 1D, viral antigen was not detected in mock-infected HBMVE cells or in the cells stained with only secondary antibody, respectively. Based on a total of 8,000 cells counted in ten independent fields, approximately 5% of HBMVE cells were infected with WNV at day 2 after infection.
Figure 1. WNV replication kinetics in HBMVE cells.

HBMVE cells grown and fixed on coverslips at day 2 after infection were stained with anti-WNV envelope antibody (red) and counterstained with DAPI (blue). (A and B) WNV-infected HBMVE cells immunostained with WNV-specific antibody demonstrate robust virus replication in the cytoplasm. (C) Mock-infected HBMVE cells stained with primary and secondary antibodies and (D) WNV-infected cells stained with secondary antibody only were used as negative control, which do not show any WNV antigen staining. Scale bar represents 20 μm at 20× magnification. (E) WNV titers in infected HBMVE cells supernatant collected at various time points were determined by plaque assay using Vero cells. Viral titers are expressed as plaque forming units (PFU)/mL of supernatant and are represented as mean ± SD of data obtained from four independent experiments conducted in duplicate. (F) Total RNA extracted from HBMVE cell lysate at different time points after infection were used to determine intracellular WNV RNA copies by qRT-PCR. UV-WNV did not replicate in HBMVE cells, however both, WNV MOI-1 and -5 infected HBMVE cells had a replication kinetic profile similar to that observed in the plaque assay. Data represents mean ± SD of four independent infection experiments in duplicate.
To further confirm the immunofluorescence observations, WNV replication was quantitated using plaque assay in WNV-infected cell supernatants collected at 0, 6, 12 hr and from days 1 to 5 after infection. No significant change was observed in the virus titers between 0 to 12 hr after infection. Productive WNV replication, in the form of release of virions was first detected at day 1 after infection in both MOI- 1 and -5 infected cells. Viral titers peaked at day 2 after infection followed by a sharp decline in the virus titers till day 5 after infection (Fig. 1E). Intracellular WNV replication assessed by qRT-PCR also followed a similar pattern, demonstrating a peak in virus replication in both, MOI-1 and -5 infected cells, at day 2 after infection, after which WNV RNA declined (Fig. 1F). As expected, UV-WNV did not replicate as measured by plaque assay (data not shown) and WNV RNA qRT-PCR (Fig 1F). WNV-infected cells were also examined for cytopathogenic effects (CPE) and cell-death by phase contrast microscopy and CytoTox-One assay (Promega) (Verma et al., 2008), respectively. CPE and elevated levels of LDH, marker for cell death, were not observed at any time points (data not shown).
WNV infection can alter expression of TJP in HBMVE cells
Disruption of the BBB during viral infections is mostly associated with TJP degradation or redistribution. Therefore, the expression of key TJP such as ZO-1, occludin, claudin-1 and -5 were analyzed by qRT-PCR in mock, UV-WNV and WNV-infected HBMVE cells from 6 hr to day 4 after infection. Fig. 2A depicts the fold-change of claudin-1 and -5 in WNV-infected HBMVE cells as compared to the mock-infected controls at the same time point. WNV infection did not affect the transcript level of claudin-5 in infected HBMVE cells at any time point after infection, however the expression of claudin-1 increased from day 2 after infection and remained significantly high at days 3 and 4 after infection. This increase was observed in both MOI-1 and -5 WNV-infected cells but not in cells infected with UV-WNV (Fig. 2A). The expression of ZO-1 and occludin did not change till day 3 after infection, when a marginal 2-fold increase in only ZO-1 gene expression was observed in HBMVE cells infected with WNV at MOI-5 (Fig. 2B). Significant increase in the claudin-1 transcript level at days 3 and 4 after infection was further validated using western blot (WB) and densitometric analysis, and this increase of claudin-1 was statistically significant, p<0.05 (Fig. 2C). However, the marginal increase in ZO-1 mRNA expression was not observed using WB assay, suggesting that the mRNA increase was not significant (data not shown).
Figure 2. WNV differentially modulates tight junction protein expression in HBMVE cells.
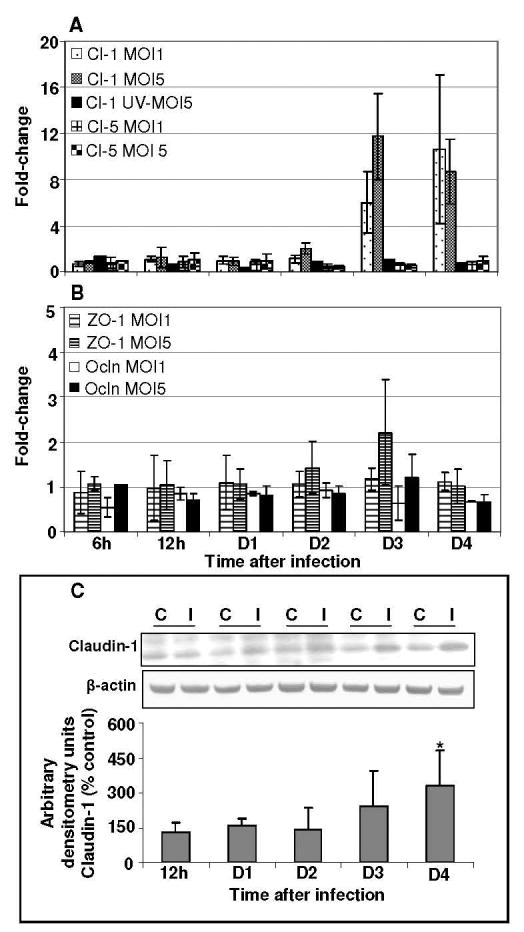
cDNA templates from mock, UV-WNV and WNV-infected HBMVE cells at different time points after infection were used to determine the fold-change of (A) claudin-1, and -5, and (B) ZO-1 and occludin. Changes in the levels of TJP were first normalized to the GAPDH gene and then the fold-change in infected cells as compared to corresponding controls was calculated. Data represents mean of five independent experiments conducted in duplicate. (C) 50 μg of cellular proteins extracted from mock- and WNV-infected HBMVE cells were separated on PAGE, transferred onto PVDF membranes and immunoblotted with antibodies specific to claudin-1 and β-actin. Blots were scanned using BioRad Phosphorimager and analyzed using Quantity one program. Data is represented as percentage of un-infected HBMVE cells at the corresponding time point. Mean comparisons were based upon extrapolated CI for three data points. *p<0.05, compared to mock-infected cells.
BBB integrity is influenced by both TJP expression and its cellular distribution in the HBMVE cells tight junctions. Re-distribution or localization of TJP away from tight junctions can also lead to loss of BBB integrity. Therefore, we assessed the staining pattern of ZO-1 and claudin-1 by immunocytochemistry in WNV MOI-5-infected HBMVE cells. As illustrated in figure 3, ZO-1 staining at day 3 after infection was prominent at cell-cell border in uninfected HBMVE cells and WNV infection did not change its staining pattern and localization. Unlike ZO-1, claudin-1 immunostaining in mock-infected HBMVE cells exhibited an intracellular and junctional staining pattern at days 3 and 4 after infection as seen by confocal microscopy (Fig. 3). However, WNV infection resulted in a marked increase in claudin-1 immunoreactivity at day 3 after infection, which became more intense at day 4 after infection, as compared to the corresponding uninfected controls. Double immunostaining for claudin-1 and WNV antigens revealed that the increase of claudin-1 is a global response similar to that observed in WNV-infected as well as neighboring -uninfected HBMVE cells (Fig. 3). Overall, the immunocytochemistry results support our qRT-PCR and western blot data, which demonstrate increase of only specific TJP, i.e., claudin-1, but not others.
Figure 3. Immunoflourescence analysis of TJP in WNV-infected HBMVE cells.
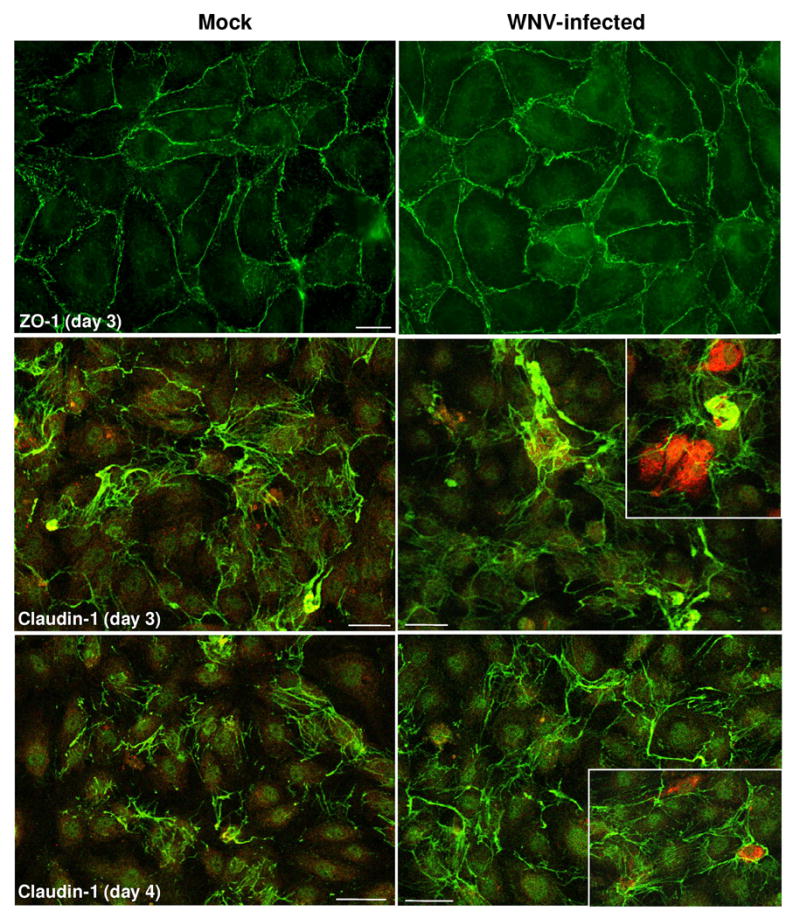
Confluent primary HBMVE cells were infected with WNV at MOI-5 and ZO-1 and claudin-1 expression, were determined using immunofluorescence. ZO-1 staining analyzed by epifluorescence microscopy at day 3 after infection was characterized by a staining pattern primarily confined to cell-cell borders and did not change as a result of WNV infection. Claudin-1 immunoreactivity analyzed by confocal microscopy was characterized by intracellular and junctional pattern in mock-infected HBMVE cells, while WNV-induced a significant increase in the overall immunoreactivity at days 3 and 4 after infection. The increase was evident in both, WNV-infected (inset, red) as well as neighboring un-infected HBMVE cells. The images shown are representative results of three independent experiments. Scale bar represents 10 μm at 63× magnification for ZO1 and 100 μm at 10× magnification for caludin-1.
Effect of WNV infection on cell-matrix adhesion molecules in HBMVE cells
To investigate whether WNV infection of HBMVE cells would potentially affect their adhesive interactions with leukocytes, expression of important CAM such as ICAM-3, VCAM-1, E-selectin and PECAM were evaluated by qRT-PCR at various time points after infection. While the expression levels of ICAM and PECAM did not show any change at any time point after infection, (data not shown), we did observe a dose dependent increase in the transcripts of VCAM-1 and E-selectin from days 2 to 4 after infection (Fig 4A), which corresponded with the peak increase in virus replication. Further, UV-WNV did not elicit any increase in CAM expression, suggesting an important relationship between active virus replication and increase in CAM. WB analysis of VCAM-1 further confirmed our gene expression data and a significant increase in VCAM-1 protein levels (2 to 6-fold increase, p<0.05) were observed from day 2 after infection (Fig. 4B). Similarly, immunocytochemical analysis did not exhibit any VCAM-1 staining in mock-infected HBMVE cells thus confirming that resting endothelial cells do not express VCAM-1 molecules (Fig. 5A). However, a strong signal of VCAM-1 expression was observed at days 2 and 3 after infection in WNV-infected HBMVE cells (Figs. 5B and 5C).
Figure 4. WNV infection induces expression of selective cell adhesion molecules in HBMVE cells.
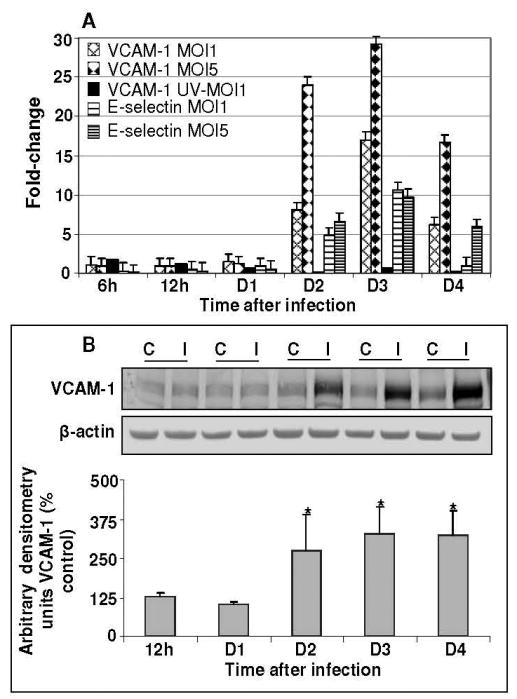
(A) cDNA templates from mock, UV-WNV and WNV-infected HBMVE cells at different time points after infection were used to determine the fold-change of VCAM-1 and E-selectin as compared to mock-infected controls at same time point. Data represents mean of four independent experiments conducted in duplicate. (B) 50-70 μg of cellular proteins extracted from mock- and WNV-infected HBMVE cells were separated on PAGE, transferred onto PVDF membranes and immunoblotted with antibodies specific to VCAM-1 and β-actin. Blots were scanned using BioRad Phosphorimager and analyzed using Quantity one program. Data is represented as percentage of un-infected HBMVE cells at the corresponding time point. Mean comparisons were based upon extrapolated CI for three data points. *p<0.05, compared to corresponding mock-infected cells. C, mock and I, WNV infected.
Figure 5. Up-regulation of VCAM-1 in WNV-infected HBMVE cells.

Mock- and WNV-infected HBMVE cells were fixed and immunostained with VCAM-1 (green) and DAPI at days 2 and 3 after infection. (A) Mock-infected HBMVE cells did not exhibit any VCAM-1 staining, whereas (B) WNV-infected cells exhibited immunoreactivity in the cytoplasm at day 2 after infection, (C) which further increased at day 3 after infection. The images shown are representative results of three independent experiments. Scale bar represents 10 μm at 63× magnification.
Cell-free WNV migrates across the in vitro BBB model without altering the BBB permeability
To determine the ability of cell-free WNV to migrate across the HBMVE cells of the BBB, we constructed an in-vitro monolayer BBB model using HBMVE cells and quantitated the WNV released in the UCS and LCS. Since BBB restricts movements of ions, the well documented methods to assess the tightness of the BBB are TEER and transmigration of molecules such as FITC-labeled dextran. The BBB permeability assessed from days 4 to 16 after seeding, demonstrated increased resistance and restricted FITC-dextran permeability, 400-500 Ω/cm2 vs. 70-80 Ω/cm2 and 1.5 to 3% vs. 75-85% in inserts with and without HBMVE cells, respectively. The in vitro BBB model employed in this study and the TEER and FITC-dextran permeability results of the in vitro BBB model are consistent with earlier studies of membranes with 0.3 cm2 surface area (Deli et al., 2005; Jong et al., 2001; Mahajan et al., 2008). At day 8 after seeding, the BBB model was infected with WNV or UV-WNV and virus titers and viral RNA in the media collected from the upper and lower chambers, UCS and LCS, respectively, were determined at various time points after infection. High WNV RNA levels were detected in the UCS at 2 hr after infection, which represents the residual inoculum (Fig. 6A). At the same time point, very little WNV RNA was detected in the LCS (0.0001% of the total inoculum). At 6 and 12 hr after infection, WNV RNA detected in UCS remained low, 0.04 and 0.005% of the total inoculum, respectively, which most likely represents residual WNV in the UCS after two washes. Similarly, very little WNV RNA was detected in the LCS at these time points, and corresponded to 0.001 to 0.003% of the total inoculum. However at day 1 after infection, there was a significant increase in WNV RNA in the UCS, which remained elevated till day 2 after infection, following which the WNV RNA declined at day 3 after infection (Fig.6A). Interestingly, the WNV RNA in the LCS at day 1 after infection did not increase as compared to UCS. However, a significant increase in the WNV RNA was detected in the LCS on days 2 and 3 after infection.
Figure 6. Cell-free WNV crosses the in vitro BBB model without affecting the BBB integrity.
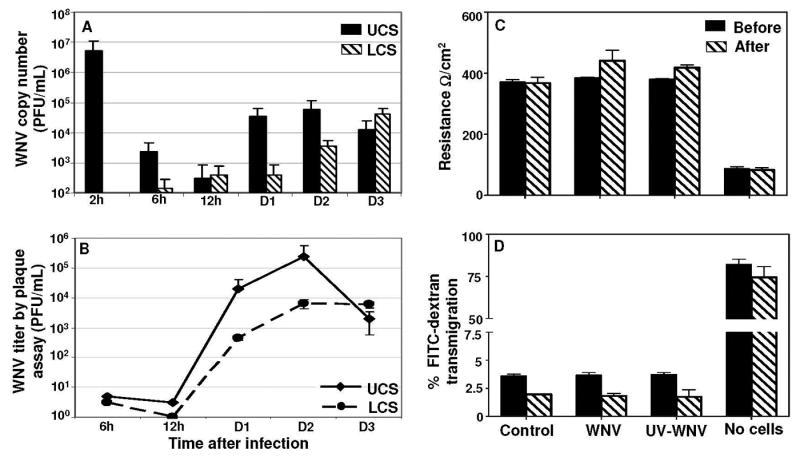
At day 8 post seeding, the upper chamber of the in vitro BBB models were infected with mock, UV-WNV and WNV (MOI1) and UCS and LCS were collected at different time points. (A) WNV copy numbers as determined by qRT-PCR were very low in both UCS and LCS at 6 and 12 hr after infection, however a significant increase in copy numbers was observed in both, UCS and LCS supernatants from days 1 to 3 after infection. Data represents mean ± SD of two independent infection experiments in duplicate. (B) WNV titers in UCS and LCS, determined by plaque assay demonstrate a peak in titer at day 2 after infection, similar to the profile observed using qRT-PCR. WNV titers are expressed as plaque forming units (PFU)/mL of supernatant and are represented as mean ± SD of data obtained from two independent experiments conducted in duplicate. BBB models infected with UV-WNV did not show any plaques, at any time point after infection (data not shown). (C) The integrity of the in vitro BBB models as determined by measuring the BBB TEER before and at day 4 after infection. TEER values are presented as Ω/cm2 and did not change significantly after infection in control, WNV or UV-WNV infected inserts as compared to their respective TEER values before infection. The data is representative of at least two independent experiments. (D) FITC-dextran permeability assay of the BBB models before and at day 4 after infection. The percentage of FITC-dextran that crossed the BBB model was determined based on the total input FITC-dextran and did not vary significantly in WNV or UV-WNV infected inserts as compared to un-infected controls at day 4 after infection. The data is representative of at least two independent experiments.
The viability of WNV released in the UCS and LCS, collected at various time points were determined by plaque assay (Fig. 6B). The profile of virus titers as determined by plaque assay (Fig 6B) and qRT-PCR (Fig. 6A) were very similar. Infectious virus was not detected in the UCS and LCS at 6 and 12 hr after infection; however at day 1 after infection, a sharp increase in virus was observed, approximately log 4 and log 2.5 PFU/mL in UCS and LCS, respectively, which further peaked at day 2 after infection (Fig 6B). The replication kinetics of WNV infection observed in the in vitro BBB model (Fig. 6B) was similar to that observed in HBMVE cells infected with WNV in culture wells/plates (Fig 1E), where approximately 2-3 log increase in virus replication was observed between 12 h to day 1, followed by a peak at day 2 after infection. At the same time in the BBB models infected with UV-WNV, the virus RNA copy number determined in the UCS and LCS was very low; range, 0.1-1 log PFU/mL, and WNV titers analyzed by plaque assay did not show any plaques in UCS and LCS, at any time points after infection (data not shown).
To further assess the effect of increased claudin-1 (Fig. 2) and WNV transmigration (Figs. 6 A and 6B) on the integrity of the in vitro BBB model, BBB permeability was measured before and at day 4 after infection. The TEER values were in the range of 380±10 Ω/cm2 in all the BBB models before infection and did not alter significantly at day 4 after infection in control, WNV- and UV-WNV-infected BBB models, 370±14 Ω/cm2 (p=0.67), 440±34 Ω/cm2 (p=0.117) and 418±9 Ω/cm2 (p=.063), respectively, as compared to their respective TEER values before infection (Fig. 6C). Also, similar to the TEER data, the percent of FITC-dextran which crossed the BBB models before infection was very low, approximately 3.5% as compared to the inserts with no cells with 75% FITC-dextran transmigration. At day 4 after infection, there was slight decrease in the overall FITC-dextran transmigration (range, 1.6 to 2.3%) but as compared to control BBB models, it did not change significantly in WNV (p=0.466) or UV-WNV (p=.671) infected BBB models, further confirming that transmigration of cell-free WNV did not alter the integrity of the BBB model (Fig 6D).
Discussion
Breakdown of the BBB has been previously demonstrated in WNV-infected mice (Wang et al., 2004), and it is believed that during high viremia, one of the potential mechanisms of virus entry into the CNS is through the hematogenous spread, by crossing the BBB. However, there have not been any studies demonstrating the relative contributions of the specific BBB cell types and mechanism(s) underlying this process. Here, we demonstrate that HBMVE cells, the principal barrier against virus invasion into the CNS, can be a target of WNV infection and can be one of the mechanism by which WNV gains entry into the CNS via the transcellular pathway, without compromising the BBB integrity. WNV infection of HBMVE cells is productive, does not induce CPE but induces expression of claudin-1. In addition, up regulation of VCAM-1 and E-selectin by WNV suggests a potential role of HBMVE cells in the transmigration of activated and/or infected immune cells across the BBB, thereby facilitating another route of virus entry into the brain i.e. ‘Trojan horse’ pathway.
Kinetics of WNV replication in HBMVE cells
HBMVE cells are critical players in the maintenance of BBB integrity and regulate influx of molecules and immune cells. In this study we chose to investigate HBMVE cells because they cover the largest surface area of the BBB and therefore allow extensive targeting by neurotropic viruses. Primary HBMVE cells grown in culture retain their structural and functional characteristics and have been successfully used for pathogen transmigration studies using in vitro BBB models (Chaudhuri et al., 2008; Kanmogne et al., 2007; Mahajan et al., 2008; Persidsky et al., 1997).
Both in vitro and in vivo studies establish that neurons are the primary targets of WNV replication, though in vitro, other CNS-based cells such as astrocytes and human oligodendroglial cell lines can also support efficient WNV replication (Cheeran et al., 2005; Jordan et al., 2000). Our study demonstrating intracellular WNV replication by qRT-PCR, WNV antigen by immunocytochemistry and release of infectious virions by plaque assay, indicate a short virus replication cycle with peak virus replication at day 2 after infection, following which the viral titers drop rapidly. This trend of virus replication is unlike other susceptible cells such as neurons or Vero cells where persistently high virus replication is observed for almost 8 to 14 days after infection, accompanied by gross CPE (Cheeran et al., 2005). Further, unlike Vero or neuronal cells, WNV infection in HBMVE cells is not accompanied by gross CPE. However, WNV replication kinetics in HBMVE cells is similar to that observed in human monocytes-derived macrophages where peak viral titers reach within 3-6 days after infection and then decline sharply, also without any noticeable CPE (Rios et al., 2006). Presence of WNV antigen in the brain endothelial cells has not been previously described in brain tissues of humans, mice or horses. One explanation for this can be that the WNV replication cycle in endothelial cells is transient and without CPE, thus the virus is not detected in these cells at later stages of infection, by which time the brain pathology is mostly observed in neurons and glial cells. However, two independent studies have recently documented WNV antigen localization in several brain cells including BMVE cells in owls and American crows (Lopes et al., 2007; Wunschmann et al., 2004). Similarly, though new evidence documents occasional presence of Japanese encephalitis virus (JEV), also a neurotropic flavivirus, in the human brain vascular endothelium (German et al., 2006), these studies do not confirm whether WNV can replicate in these cells.
Role of TJP in WNV infection
In vitro and in vivo infection of brain endothelial cells with neurotropic viruses such as HTLV-1 (Afonso et al., 2007), HIV-1 (Andras et al., 2003; Kanmogne, Primeaux, and Grammas, 2005), SIV (Luabeya et al., 2000), and HCMV (Bentz et al., 2006) has been associated with decrease and/or redistribution of TJP. Up-regulation of any TJP in microvascular endothelial cells has never been reported in viral infections. Therefore our results demonstrating significant up-regulation of claudin-1 by WNV but not by UV-WNV, is quite intriguing. Since the increase of claudin-1 is observed in infected as well as neighboring un-infected exposed cells, it seems likely that it is a response of inflammatory mediators such as cytokines released by WNV-infected HBMVE cells. Recent evidence indicates role of IL-1β in the up-regulation of claudin-1 in HUVEC cells (Williams et al., 2008). However we believe that this increase in claudin-1 is a specific response of HBMVE cells to WNV infection and may only be an in vitro phenomenon, since in vivo several immune and inflammatory mediators such as TNF-α and MMP-9 are known to be released by peripheral WNV-infected cells, which are shown to compromise the BBB integrity in WNV-infected mouse models (Arjona et al., 2007; Wang et al., 2008; Wang et al., 2004)
Migration of WNV across the BBB
In vitro BBB models comprising of both monolayer of HBMVE cells, and bilayer of HBMVE cells and astrocytes, have been routinely used for virus transmigration studies (Chaudhuri et al., 2008; Kanmogne et al., 2007; Mahajan et al., 2008; Persidsky et al., 1997). In this study, we employed the monolayer BBB model to determine whether WNV-induced claudin-1 has any effect on the integrity of BBB model and on the ability of the replicated virus to be released on the abluminal side of HBMVE cells without the influence of astrocytes, which also support WNV infection, and release inflammatory mediators that might compromise the TJP.
Our data demonstrating the release of infectious virus on the abluminal side of the in vitro BBB model indicates the ability of cell-free WNV to migrate across the BBB. As demonstrated in figures 6A and 6B, very little WNV was detected in the abluminal compartment at 2, 6, and 12 hr after infection as compared to the virus detected at later time points. This suggests that the progeny virus is crossing the BBB via the transcellular pathway instead of paracellular pathway, in which case we would expect an increase in WNV RNA at earlier time points, due to passive diffusion. In support of our data, it has been previously demonstrated that cell-free HIV can cross the in vitro BBB model (Liu et al., 2002). Other than retroviruses, paramyxoviruses with neurovirulent potential such as canine distemper virus (CDV), also employs multiple routes to enter CNS and one of the early routes of virus entry into the brain is via infecting the cerebral blood vessels (Rudd, Cattaneo, and von Messling, 2006). In addition, the cell-free transmigration of WNV across the BBB, unlike other neurotropic viruses (Kanmogne, Primeaux, and Grammas, 2005; Luabeya et al., 2000), does not affect the BBB integrity as compared to controls and UV-WNV infected BBB models (Fig. 6C and D). It is not surprising since WNV infection of HBMVE cells is not accompanied by noticeable CPE and loss of any TJP (Figs. 2 and 3). These results indirectly support the recent in vivo studies by Morrey and colleagues, where increased BBB permeability was not found to be correlated with WNV mortality in BALB/c and C57BL/6 mice, suggesting that the first wave of virus enters the CNS without breaching the BBB (Morrey et al., 2008). Similar to these lines of thoughts, we also speculate that BBB failure is more likely an indirect result of inflammatory response induced by infected brain cells, but not due to the infection of BBB endothelial cells.
Role of CAM in WNV infection
Direct infection of HBMVE cells can have multiple implications in WNV-CNS invasion by hematogenous route. One such mechanism of WNV spread can be that, cell-free WNV, could infect the BBB cells and enter BBB via the transcellular pathway as demonstrated in this study. Other mechanism could be that WNV-infected HBMVE cells assist in the entry of virus by ‘Trojan horse’ mechanism, where infected monocytes/macrophages take up residence in the CNS and disseminate virus to neighboring brain cells.
During several viral and bacterial infections, stimulation of endothelial cells results in the induction of CAM that permit migration of monocytes/macrophages through the BBB. CAM in the vascular endothelial surface includes immunoglobulin superfamily (IgSF) such as ICAMs, and VCAM-1, and selectins. Selectins mediate rolling of leukocytes while ICAMs and VCAM-1 mediate more sustained adhesion of immune cells to the endothelium (Nottet et al., 1996; Wolf et al., 2002). Increased expression of CAM on endothelial cells have been a characteristic feature of neuroinflammatory diseases such as HIV encephalitis (Seigneur et al., 1997), HSV infection (Kim et al., 2000), measles (Cosby and Brankin, 1995) and multiple sclerosis (Avolio et al., 2003; Brankin et al., 1995; Muller, 2003; Petri and Bixel, 2006). While inflammatory mediators are considered as the main triggers of CAM expression in most viral infections, in vitro, cell-free SIV and HIV gene products such as Tat is also known to induce CAM in BMVE cells. Nonvirulent strain of WNV (sarafend) has been shown to induce expression of ICAM-1, VCAM-1 and E-selectin but not P-selectin in human umbilical cord vascular endothelial cells, at early stage after infection, 2 to 6 hr, followed by decrease to normal levels by 24 hr after infection (Shen et al., 1997). In contrast, using primary HBMVE cells during early to late infection, i.e., 6 hr to day 4 after infection, we demonstrate increased expression of only specific CAM such as, VCAM-1 and E-selectin, when the virus replication is at peak. The difference in Shen and colleagues studies and our study may be because of the different cell types as well as strain of the WNV (Shen et al., 1997).
Resting brain endothelial cells do not normally express VCAM-1 and E-selectin, which are only induced in activated endothelium. Therefore, the increase of these molecules may have significant impact on the leukocyte transmigration process in WNV-infected brain. In vivo, simultaneous insults by various cytokines and pathogens can result in the breakdown of the BBB and massive infiltration of leukocytes into the brain. Our in vitro study suggests that the CAM induced as a result of cell-free infection of HBMVE cells may play a role in the eventual perivascular infiltration of immune cells via the ‘Trojan horse’ mechanism, thereby contributing to the severe inflammation observed in WNV-associated meningitis. Collectively, these data suggest that WNV infection of HBMVE cells modulates the BBB properties and may assist in the migration of cell-free as well as -associated WNV into the CNS, thereby contributing to WNV dissemination in the CNS and associated pathology.
Acknowledgments
This work was partially supported by grants from the Hawaii Community Foundation (20050405), institutional funds, and Research Centers in Minority Institutions Program (G12RR003061) and Centers of Biomedical Research Excellence (P20RR018727), National Center for Research Resources, and National Institute of Neurological Disorders and Stroke (NS060647), National Institutes of Health. We thank Dr. Duane J. Gubler for the generous gift of the WNV strain NY99 and the monoclonal human anti-WNV envelope antibody. We also thank Dr. Frederick Mercier for constructive comments and Mr. Bruce Cropp and Thomas Bui for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonso PV, Ozden S, Prevost MC, Schmitt C, Seilhean D, Weksler B, Couraud PO, Gessain A, Romero IA, Ceccaldi PE. Human blood-brain barrier disruption by retroviral-infected lymphocytes: role of myosin light chain kinase in endothelial tight-junction disorganization. J Immunol. 2007;179(4):2576–83. doi: 10.4049/jimmunol.179.4.2576. [DOI] [PubMed] [Google Scholar]
- Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toborek M. HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res. 2003;74(2):255–65. doi: 10.1002/jnr.10762. [DOI] [PubMed] [Google Scholar]
- Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, Wong SJ, Montgomery RR, Fikrig E, Bucala R. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. 2007;117(10):3059–66. doi: 10.1172/JCI32218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avolio C, Giuliani F, Liuzzi GM, Ruggieri M, Paolicelli D, Riccio P, Livrea P, Trojano M. Adhesion molecules and matrix metalloproteinases in Multiple Sclerosis: effects induced by Interferon-beta. Brain Res Bull. 2003;61(3):357–64. doi: 10.1016/s0361-9230(03)00098-4. [DOI] [PubMed] [Google Scholar]
- Bentz GL, Jarquin-Pardo M, Chan G, Smith MS, Sinzger C, Yurochko AD. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J Virol. 2006;80(23):11539–55. doi: 10.1128/JVI.01016-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankin B, Hart MN, Cosby SL, Fabry Z, Allen IV. Adhesion molecule expression and lymphocyte adhesion to cerebral endothelium: effects of measles virus and herpes simplex 1 virus. J Neuroimmunol. 1995;56(1):1–8. doi: 10.1016/0165-5728(94)00110-a. [DOI] [PubMed] [Google Scholar]
- Bunning ML, Bowen RA, Cropp CB, Sullivan KG, Davis BS, Komar N, Godsey MS, Baker D, Hettler DL, Holmes DA, Biggerstaff BJ, Mitchell CJ. Experimental infection of horses with West Nile virus. Emerg Infect Dis. 2002;8(4):380–6. doi: 10.3201/eid0804.010239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapagain ML, Nguyen T, Bui T, Verma S, Nerurkar VR. Comparison of real-time PCR and hemagglutination assay for quantitation of human polyomavirus JC. Virol J. 2006;3:3. doi: 10.1186/1743-422X-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapagain ML, Verma S, Mercier F, Yanagihara R, Nerurkar VR. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology. 2007;364(1):55–63. doi: 10.1016/j.virol.2007.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A, Yang B, Gendelman HE, Persidsky Y, Kanmogne GD. STAT1 signaling modulates HIV-1-induced inflammatory responses and leukocyte transmigration across the blood-brain barrier. Blood. 2008;111(4):2062–72. doi: 10.1182/blood-2007-05-091207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeran MC, Hu S, Sheng WS, Rashid A, Peterson PK, Lokensgard JR. Differential responses of human brain cells to West Nile virus infection. J Neurovirol. 2005;11(6):512–24. doi: 10.1080/13550280500384982. [DOI] [PubMed] [Google Scholar]
- Cosby SL, Brankin B. Measles virus infection of cerebral endothelial cells and effect on their adhesive properties. Vet Microbiol. 1995;44(24):135–9. doi: 10.1016/0378-1135(95)00006-v. [DOI] [PubMed] [Google Scholar]
- Davis LE, DeBiasi R, Goade DE, Haaland KY, Harrington JA, Harnar JB, Pergam SA, King MK, DeMasters BK, Tyler KL. West Nile virus neuroinvasive disease. Ann Neurol. 2006;60(3):286–300. doi: 10.1002/ana.20959. [DOI] [PubMed] [Google Scholar]
- Deli MA, Abraham CS, Kataoka Y, Niwa M. Permeability studies on in vitro blood-brain barrier models: physiology, pathology, and pharmacology. Cell Mol Neurobiol. 2005;25(1):59–127. doi: 10.1007/s10571-004-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003;16(3):259–78. doi: 10.1089/088282403322396082. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW. CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci. 2006;26(4):1098–106. doi: 10.1523/JNEUROSCI.3863-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German AC, Myint KS, Mai NT, Pomeroy I, Phu NH, Tzartos J, Winter P, Collett J, Farrar J, Barrett A, Kipar A, Esiri MM, Solomon T. A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model. Trans R Soc Trop Med Hyg. 2006;100(12):1135–45. doi: 10.1016/j.trstmh.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. 2005;202(8):1087–98. doi: 10.1084/jem.20042530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes EB, Gubler DJ. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu Rev Med. 2006;57:181–94. doi: 10.1146/annurev.med.57.121304.131418. [DOI] [PubMed] [Google Scholar]
- Jong AY, Stins MF, Huang SH, Chen SH, Kim KS. Traversal of Candida albicans across human blood-brain barrier in vitro. Infect Immun. 2001;69(7):4536–44. doi: 10.1128/IAI.69.7.4536-4544.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan I, Briese T, Fischer N, Lau JY, Lipkin WI. Ribavirin inhibits West Nile virus replication and cytopathic effect in neural cells. J Infect Dis. 2000;182(4):1214–7. doi: 10.1086/315847. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol. 2005;64(6):498–505. doi: 10.1093/jnen/64.6.498. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Schall K, Leibhart J, Knipe B, Gendelman HE, Persidsky Y. HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab. 2007;27(1):123–34. doi: 10.1038/sj.jcbfm.9600330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Bang D, Lee S, Lee KH. The effect of herpesvirus infection on the expression of cell adhesion molecules on cultured human dermal microvascular endothelial cells. J Dermatol Sci. 2000;24(1):38–47. doi: 10.1016/s0923-1811(00)00080-3. [DOI] [PubMed] [Google Scholar]
- Lanciotti RS, Ebel GD, Deubel V, Kerst AJ, Murri S, Meyer R, Bowen M, McKinney N, Morrill WE, Crabtree MB, Kramer LD, Roehrig JT. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology. 2002;298(1):96–105. doi: 10.1006/viro.2002.1449. [DOI] [PubMed] [Google Scholar]
- Lathey JL, Wiley CA, Verity MA, Nelson JA. Cultured human brain capillary endothelial cells are permissive for infection by human cytomegalovirus. Virology. 1990;176(1):266–73. doi: 10.1016/0042-6822(90)90252-m. [DOI] [PubMed] [Google Scholar]
- Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol. 2002;76(13):6689–700. doi: 10.1128/JVI.76.13.6689-6700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes H, Redig P, Glaser A, Armien A, Wunschmann A. Clinical findings, lesions, and viral antigen distribution in great gray owls (Strix nebulosa) and barred owls (Strix varia) with spontaneous West Nile virus infection. Avian Dis. 2007;51(1):140–5. doi: 10.1637/0005-2086(2007)051[0140:CFLAVA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Luabeya MK, Dallasta LM, Achim CL, Pauza CD, Hamilton RL. Blood-brain barrier disruption in simian immunodeficiency virus encephalitis. Neuropathol Appl Neurobiol. 2000;26(5):454–62. doi: 10.1046/j.1365-2990.2000.00275.x. [DOI] [PubMed] [Google Scholar]
- Maclean AG, Rasmussen TA, Bieniemy DN, Alvarez X, Lackner AA. SIV-induced activation of the blood-brain barrier requires cell-associated virus and is not restricted to endothelial cell activation. J Med Primatol. 2004;33(56):236–42. doi: 10.1111/j.1600-0684.2004.00077.x. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Sykes DE, Reynolds JL, Bindukumar B, Adal A, Qi M, Toh J, Xu G, Prasad PN, Schwartz SA. Methamphetamine alters blood brain barrier permeability via the modulation of tight junction expression: Implication for HIV-1 neuropathogenesis in the context of drug abuse. Brain Res. 2008;1203:133–48. doi: 10.1016/j.brainres.2008.01.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrey JD, Olsen AL, Siddharthan V, Motter NE, Wang H, Taro BS, Chen D, Ruffner D, Hall JO. Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J Gen Virol. 2008;89(Pt 2):467–73. doi: 10.1099/vir.0.83345-0. [DOI] [PubMed] [Google Scholar]
- Mukhtar M, Pomerantz RJ. Development of an in vitro blood-brain barrier model to study molecular neuropathogenesis and neurovirologic disorders induced by human immunodeficiency virus type 1 infection. J Hum Virol. 2000;3(6):324–34. [PubMed] [Google Scholar]
- Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24(6):327–34. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- Murray K, Baraniuk S, Resnick M, Arafat R, Kilborn C, Cain K, Shallenberger R, York TL, Martinez D, Hellums JS, Hellums D, Malkoff M, Elgawley N, McNeely W, Khuwaja SA, Tesh RB. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol Infect. 2006;134(6):1325–32. doi: 10.1017/S0950268806006339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, Sharer LR, McComb RD, Swindells S, Soderland C, Gendelman HE. Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. J Immunol. 1996;156(3):1284–95. [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1(3):223–36. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J Immunol. 1997;158(7):3499–510. [PubMed] [Google Scholar]
- Petri B, Bixel MG. Molecular events during leukocyte diapedesis. Febs J. 2006;273(19):4399–407. doi: 10.1111/j.1742-4658.2006.05439.x. [DOI] [PubMed] [Google Scholar]
- Pu H, Tian J, Andras IE, Hayashi K, Flora G, Hennig B, Toborek M. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J Cereb Blood Flow Metab. 2005;25(10):1325–35. doi: 10.1038/sj.jcbfm.9600125. [DOI] [PubMed] [Google Scholar]
- Rios M, Zhang MJ, Grinev A, Srinivasan K, Daniel S, Wood O, Hewlett IK, Dayton AI. Monocytes-macrophages are a potential target in human infection with West Nile virus through blood transfusion. Transfusion. 2006;46(4):659–67. doi: 10.1111/j.1537-2995.2006.00769.x. [DOI] [PubMed] [Google Scholar]
- Rudd PA, Cattaneo R, von Messling V. Canine distemper virus uses both the anterograde and the hematogenous pathway for neuroinvasion. J Virol. 2006;80(19):9361–70. doi: 10.1128/JVI.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MA, Diamond MS. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80(19):9349–60. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seigneur M, Constans J, Blann A, Renard M, Pellegrin JL, Amiral J, Boisseau M, Conri C. Soluble adhesion molecules and endothelial cell damage in HIV infected patients. Thromb Haemost. 1997;77(4):646–9. [PubMed] [Google Scholar]
- Shen J, TT SS, Schrieber L, King NJ. Early E-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J Virol. 1997;71(12):9323–32. doi: 10.1128/jvi.71.12.9323-9332.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrestha B, Gottlieb D, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J Virol. 2003;77(24):13203–13. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitati E, McCandless EE, Klein RS, Diamond MS. CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol. 2007;81(18):9801–11. doi: 10.1128/JVI.00941-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow LI, Watry DD, Fox HS, Nelson JA. Efficient infection of brain microvascular endothelial cells by an in vivo-selected neuroinvasive SIVmac variant. J Neurovirol. 1998;4(3):269–80. doi: 10.3109/13550289809114528. [DOI] [PubMed] [Google Scholar]
- Verma S, Molina Y, Lo YY, Cropp B, Nakano C, Yanagihara R, Nerurkar VR. In vitro effects of selenium deficiency on West Nile virus replication and cytopathogenicity. Virol J. 2008;5:66. doi: 10.1186/1743-422X-5-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, Ziegler K, Ananthula P, Co JK, Frisque RJ, Yanagihara R, Nerurkar VR. JC virus induces altered patterns of cellular gene expression: interferon-inducible genes as major transcriptional targets. Virology. 2006;345(2):457–67. doi: 10.1016/j.virol.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Wang P, Dai J, Bai F, Kong KF, Wong SJ, Montgomery RR, Madri JA, Fikrig E. Matrix metalloproteinase 9 facilitates west nile virus entry into the brain. J Virol. 2008;82(18):8978–85. doi: 10.1128/JVI.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10(12):1366–73. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Williams MR, Kataoka N, Sakurai Y, Powers CM, Eskin SG, McIntire LV. Gene expression of endothelial cells due to interleukin-1 beta stimulation and neutrophil transmigration. Endothelium. 2008;15(1):73–165. doi: 10.1080/10623320802092443. [DOI] [PubMed] [Google Scholar]
- Wolf K, Tsakiris DA, Weber R, Erb P, Battegay M. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis. 2002;185(4):456–62. doi: 10.1086/338572. [DOI] [PubMed] [Google Scholar]
- Wunschmann A, Shivers J, Bender J, Carroll L, Fuller S, Saggese M, van Wettere A, Redig P. Pathologic findings in red-tailed hawks (Buteo jamaicensis) and Cooper's hawks (Accipiter cooper) naturally infected with West Nile virus. Avian Dis. 2004;48(3):570–80. doi: 10.1637/7170-022004R. [DOI] [PubMed] [Google Scholar]