

Abstract
Converging evidence suggests that salience-associated modulation of behavior is mediated by the release of monoamines and that monoaminergic activation of D1/D5 receptors is required for normal hippocampal-dependent learning and memory. However, it is not understood how D1/D5 modulation of hippocampal circuits can affect salience-associated learning and memory. We have observed in CA1 pyramidal neurons that D1/D5 receptor activation elicits a bidirectional long-term plasticity of NMDA receptor-mediated synaptic currents with the polarity of plasticity determined by NMDA receptor, NR2A/B subunit composition. This plasticity results in a decrease in the NR2A/NR2B ratio of subunit composition. Synaptic responses mediated by NMDA receptors that include NR2B subunits are potentiated by D1/D5 receptor activation, whereas responses mediated by NMDA receptors that include NR2A subunits are depressed. Furthermore, these bidirectional, subunit-specific effects are mediated by distinctive intracellular signaling mechanisms. Because there is a predominance of NMDA receptors composed of NR2A subunits observed in entorhinal–CA1 inputs and a predominance of NMDA receptors composed of NR2B subunits in CA3–CA1 synapses, potentiation of synaptic NMDA currents predominates in the proximal CA3–CA1 synapses, whereas depression of synaptic NMDA currents predominates in the distal entorhinal–CA1 synapses. Finally, all of these effects are reproduced by the release of endogenous monoamines through activation of D1/D5 receptors. Thus, endogenous D1/D5 activation can (1) decrease the NR2A/NR2B ratio of NMDA receptor subunit composition at glutamatergic synapses, a rejuvenation to a composition similar to developmentally immature synapses, and, (2) in CA1, bias NMDA receptor responsiveness toward the more highly processed trisynaptic CA3–CA1 circuit and away from the direct entorhinal–CA1 input.
Introduction
Salience of environmental cues has been associated with the release of a neuromodulator, dopamine (DA), after activation of DA neurons in the ventral tegmental area (VTA) (Fiorillo et al., 2003). Recent work indicates that the salience-evoked DA release, acting through D1/D5 receptors in the hippocampus, is associated with altered plasticity (Li et al., 2003) and hippocampal-dependent learning and memory (Lemon and Manahan-Vaughan, 2006; O'Carroll et al., 2006). A possible mechanism mediating these effects could involve modulation of NMDA receptors (NMDARs), and, indeed, it has been shown that activation of DA D1/D5 receptors potentates synaptic NMDAR currents in prefrontal cortical neurons (Seamans et al., 2001; Chen et al., 2004) and CA1 pyramidal neurons (Yang, 2000) and inhibits NMDA receptor currents in cultured hippocampal neurons (Lee et al., 2002).
Two pathways provide glutamatergic synaptic input onto CA1 pyramidal neurons in CA1 of the hippocampus. The temporal ammonic (TA) pathway projects directly from the entorhinal cortex (EC) and synapses distally on CA1 apical dendrites in the stratum lacunosum molecular (SLM), whereas the Schaffer collaterals (SCs) project from CA3 via the trisynaptic pathway and synapse proximally on CA1 dendrites in the stratum radiatum. Accordingly, CA1 neuronal firing patterns may reflect drive from either CA3 neurons projecting via the SC input or EC neurons projecting via the TA input.
We examined the effect of D1/D5-mediated modulation of NMDAR-dependent synaptic currents activated from both afferent pathways to CA1 pyramidal cells of the hippocampus to gain a better understanding of how DA can modulate hippocampal CA1 circuit function in a salience-relevant manner.
Materials and Methods
In vitro slice preparation and solutions.
Coronal slices from dorsal hippocampus were obtained from 6- to 8-week-old male C57BL/6 mice (The Jackson Laboratory and Charles River Laboratories). In brief, after deep anesthesia with Isothesia (isoflurane; USP Inc. via Butler Scientific Products), the brain was removed and put in a chilled solution containing the following (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 2.6 NaH2PO4, 2.6 MgCl2, 2.5 CaCl2, and 0.5 kynurenic acid, pH 7.35 (after bubbling with a mixture of 95% O2/CO2, 320 ± 4 mOsm). The brain was then blocked in coronal orientation and sliced (300 μm; Vibratome 1500 sectioning system), and the slices were kept in a chamber with the above solution and continuously oxygenated for at least 1 h before recording.
The slices were transferred to the recording chamber as needed and perfused with a magnesium-free external solution containing the following (in mm): 124 NaCl, 26 NaHCO3, 10 glucose, 3 KCl, 2.6 NaH2PO4, and 2.5 CaCl2. We added the selective AMPA receptor antagonist, 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (10 μm; a disodium salt from Sigma-Aldrich or Tocris Cookson) and the GABAA receptor antagonist picrotoxin (100 μm; from Sigma-Aldrich).
Two basic types of internal solution were used. For experiments performed at a holding voltage of −60 mV, a potassium-based solution was used (containing 130 mm K-gluconate, 10 mm KCl, 10 mm HEPES, 3 mm MgCl2, 2 mm Mg-ATP, and 1 mm Na-GTP, pH 7.30, 280 ± 4 mOsm). For experiments performed at depolarizing voltages, a cesium-based solution was used (containing 115 mm CsMeSO4, 20 mm CsCl, 10 mm HEPES, 4 mm Mg-ATP, 0.4 mm Na-GTP, and 0.6 mm EGTA, pH 7.30, 285 ± 4 mOsm). For experiments with internal buffering of Ca2+, BAPTA [10 mm (Sigma-Aldrich), tetra-potassium salt, i.e., 40 mm potassium] replaced equimolar K-gluconate in the recording solution. For experiments examining blockade of G-protein-mediated activity, guanosine-5′-O-(2-thiodiphosphate) (GDPβS) (200 μm; Sigma-Aldrich) was added directly to the aliquots of recording solution, without significant change in the pH or osmolality. Finally, all internal solutions had 10 mm lidocaine-N-bromide (QX-314; Sigma-Aldrich or Tocris Cookson), a selective sodium channel blocker.
Electrophysiological recording methods.
CA1 pyramidal neurons were visualized using a Zeiss Axoskop 2A equipped with infrared differential interference contrast optics and a contrast gradient light source. Whole-cell voltage-clamp recordings were obtained using a Multiclamp 700A amplifier (Molecular Devices). Patch-clamp electrodes with resistances of 4–6 MΩ were made from borosilicate glass (outer diameter, 1.5 mm; inner diameter, 0.86 mm; Sutter Instruments) using a horizontal puller (model P-97; Sutter Instruments) Synaptic stimulation of TA and SC inputs was performed using two Iso-Flex stimulus isolators (A.M.P.I.) with stimuli of 100 μs duration. Bipolar stimulating electrodes (homemade, 100 μm.) were placed in both the hippocampal fissure against the SLM and within 100 μm of the stratum pyramidale in the stratum radiatum at opposite ends of CA1. In this way, we selectively activated the TA or the SCs as described previously and validated (Steward and Scoville, 1976; Witter et al., 1988; Amaral and Witter, 1989; Bikson et al., 2004; Judge and Hasselmo, 2004; Ang et al., 2005; Vervaeke et al., 2006; Dudman et al., 2007; Speed and Dobrunz, 2009).
To confirm stimulation of nonoverlapping pathways, we performed paired-pulse stimulation (20 Hz; n = 12 neurons) between both the two pathways. No interaction was ever observed. The amplitude of the stimulation varied between 3 and 30 μA and was selected to evoke a postsynaptic current of <100 pA, reducing voltage escape at the distal dendritic synapses while still evoking EPSCs of sufficient amplitude to allow for assessment of both potentiation and depression. Data was digitized using a Digidata board (Molecular Devices) and pClamp version 9.5 (Molecular Devices) and analyzed using Igor Pro version 5.04β (WaveMetrics). We further tested the specificity of our selective stimulation protocol by perfusion of a specific group II metabotropic glutamate receptor (mGluR) agonist (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine (20 μm; Tocris Cookson), at the end of some of the experiments. Group II mGluR activation causes presynaptic inhibition of TA but not SC inputs. We found that EPSCs evoked from SLM stimulation of TA inputs were inhibited by 89 +/-8% (n = 5) and those evoked by SC stimulation were inhibited by 7 ± 9% (n = 3), suggesting that our stimulation protocol was selective for TA and SC inputs.
An important issue is the control we exert over the membrane potential needed to assess NMDA-EPSCs, especially those from the distal dendrites (i.e., the TA inputs). It can be argued that the synaptic response from the TA pathway having traveled through the dendritic arbor is heavily attenuated attributable to the passive properties of the dendrites. If this is the case, then any observation from the distal synapse is potentially confounded and the observed changes would be an underestimate of the real response. The peak amplitude of the EPSC recorded in the soma is especially affected by dendritic filtering. We measured the total charge transfer of the EPSC because it is less so affected but may nonetheless be attenuated, primarily as a function of the ratio of the dendritic membrane to cytoplasmic resistance.
An estimate of the attenuation factor of the TA input relative to the SC input can be made using the observed reversal potential of the NMDAR currents for each synapse. The relative attenuation was derived from the following equation: α = [Erev, SC − Vrest]/[Erev, SC − Vrest + ΔErevTA] (modified from Häusser and Roth, 1997). Based on our observation that ΔErevTA < 2 mV (n = 5), it was estimated that, at a holding potential of approximately +40 mV, the TA response, assessed by total charge transfer, was attenuated by <10% compared with the SC response. Thus, we consider the clamp control over the TA NMDA-EPSCs sensitive enough to give an accurate relative measure of the potentiation and depression that resulted during our experiments.
All average measurements are expressed as the mean ± SEM. In addition, the coefficient of variance in Figure 2 was calculated as (SD/Mean) × 100 and expressed as percentage for every 20 s time point for the duration of the experiment.
Figure 2.
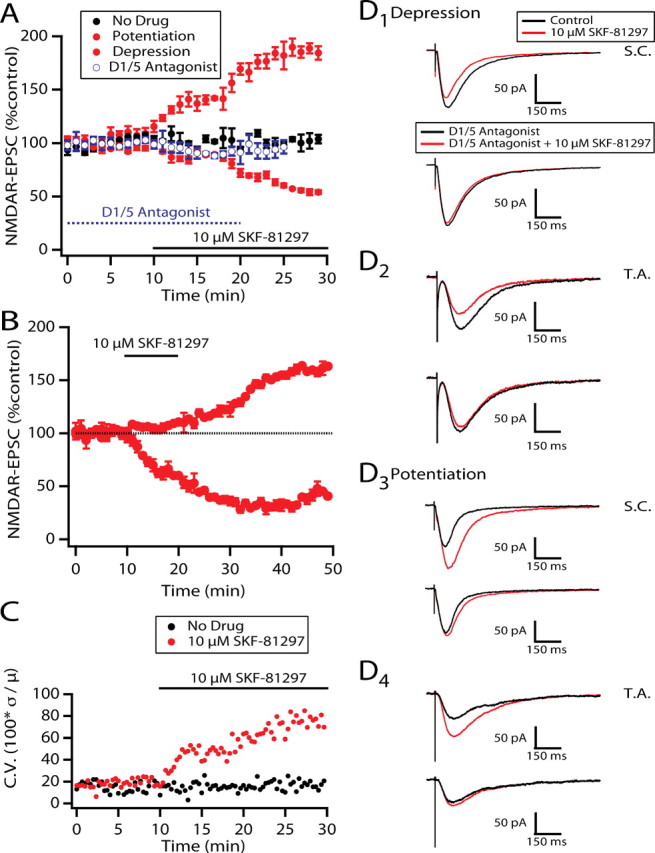
Both polarities of plastic D1/D5 effects are blocked by selective D1/D5 antagonists. A is a graph showing onset of D1/D5 effects under control conditions or with preexposure to the D1/D5 antagonists (results were pooled from both reagents because their antagonistic actions were indistinguishable, as described in Results). B is a similar graph but with a short exposure to the agonist showing a plastic effect that outlasts removal of the agonist by >20 min. C illustrates the change in coefficient of variance in the presence of D1/D5 agonist compared with “no drug” for all recorded NMDAR-EPSCs. D1–D4 are representative averages of current traces (n = 15/trace) evoked by stimulation of SC or TA inputs and recorded from cells in A at time t = 25 min to illustrate the blockade of NMDAR-EPSC responses to SKF-81297 by D1/D5 antagonists.
Unless otherwise indicated, sample NMDAR-EPSC traces are averages recorded in the last 3 min of baseline (7–10 min) for control or recorded after 27–30 min drug applications for experimental.
Results
D1/D5 modulation of NMDAR-EPSCs in CA1 includes both depression and potentiation
Whole-cell patch recordings from hippocampal CA1 pyramidal neurons in vitro were used to assess the effect of D1/D5 receptor activation on the isolated NMDAR component of EPSCs evoked from either the TA or SC inputs. The TA stimulating electrode was placed in the stratum lacunosum moleculare and the SC electrode in the stratum radiatum to obtain selective activation of the direct entorhinal inputs and the CA3 inputs, respectively (see Materials and Methods). Selective activation of each pathway was confirmed by ruling out paired stimulation interaction between electrodes. The effects produced by bath application of the D1/D5 agonist 6-chloro-2,3,4,5-tetrahydro-1-phenyl-1H-3-benzazepine hydrobromide (SKF-81297; 10 μm continuous perfusion for 30 min) on NMDAR-EPSCs (n = 64 EPSCs from n = 43 neurons) were either depression (Fig. 1A1,A2,C1,C2) or potentiation (Fig. 1B1,B2,D1,D2). The D1/D5-induced changes in the NMDAR-mediated component of the evoked EPSCs were quantified as the percentage change in total charge transfer of the EPSC, before and during >10 min of continuous drug exposure, averaged for all NMDAR-EPSCs, for all SC and for all TA NMDAR-EPSCs (Fig. 2A, Table 1). Note that the mean for the percentage change in total charge transfer for all recorded EPSCs during drug exposure (both potentiated and depressed responses) was close to zero, because the induced enhancements and depressions cancelled each other out (Table 1, “Total” column). The coefficient of variance [(SD/mean) × 100] was, nevertheless, increased during drug exposure (n = 64 EPSCs) (Fig. 2C). During sham perfusion (artificial SCF containing no drug; n = 14 NMDAR-EPSCs), the change was 2 ± 5%; the coefficient of variance did not change over time, and it was similar to experimental baseline conditions (n = 14 EPSCs) (Fig. 2C). It was, however, different from the coefficient of variance at 30 min of drug exposure (p < 10−7, two-tailed t test).
Figure 1.

Effect of the D1/D5 agonist SKF-81297 on isolated NMDAR-EPSCs from CA1 pyramidal cells. Each trace is an average of 10 stimulated EPSCs recorded before D1 agonist (10 μm SKF-81297) application (black) or during the last 3 min of D1/D5 agonist application (red). For both SC (A1, B1) and TA (C1, D1) pathway stimulation, both inhibitory (A1, C1) and excitatory (B1, D1) modulation of NMDAR-EPSCs are apparent at holding potentials of either −60 mV (left column, 1) or +40 mV (right column, 2).
Table 1.
D1/D5-induced changes in the NMDAR-mediated synaptic responses
| Condition | Potentiation |
Depression |
Total |
|||
|---|---|---|---|---|---|---|
| n (EPSC) | % control | n (EPSC) | % control | n | % control | |
| SC | 14 | 164 ± 10 | 19 | 62 ± 5 | 33 | 106 ± 10 |
| TA | 12 | 153 ± 13 | 19 | 53 ± 6 | 31 | 91 ± 11 |
| SC + antagonista | 3 | 112 ± 11 | 4 | 95 ± 10 | 7 | 106 ± 8 |
| TA + antagonista | 3 | 110 ± 13 | 3 | 93 ± 12 | 6 | 104 ± 8 |
| SC + nimodipine | 3 | 164 ± 17 | 5 | 51 ± 6 | 8 | 93 ± 24 |
| TA + nimodipine | 3 | 139 ± 16 | 4 | 45 ± 7 | 7 | 85 ± 20 |
| SC + ifenprodil | 0 | 6 | 42 ± 9 | 6 | 42 ± 9 | |
| TA + ifenprodil | 0 | 5 | 38 ± 9 | 5 | 38 ± 9 | |
| SC-NR2A knock-outs | 7 | 186 ± 27 | 0 | 7 | 186 ± 27 | |
| TA-NR2A knock-outs | 7 | 166 ± 24 | 0 | 7 | 166 ± 24 | |
| SC-D1t3 | 4 | 145 ± 6 | 2 | 77 ± 3 | 6 | 122 ± 15 |
| TA-D1t3 | 3 | 150 ± 14 | 0 | 3 | 150 ± 14 | |
| SC-GDPβS | 2 | 107 ± 5 | 5 | 47 ± 17 | 7 | 67 ± 16 |
| TA-GDPβS | 1 | 127 | 5 | 47 ± 6 | 6 | 60 ± 14 |
| SC + amphetamine | 4 | 166 ± 95 | 5 | 37 ± 7 | 9 | 136 ± 50 |
| TA + amphetamine | 3 | 80 ± 54 | 6 | 40 ± 9 | 9 | 86 ± 26 |
aSKF-83566 or SCH-23390.
If the bath application of SKF-81297 was limited to 10 min, the minimum time required for SKF-81297 to have a measurable effect, then both the potentiating and depressing responses persisted for the duration of the recording (>30 min; n = 6 EPSCs for potentiation, n = 10 EPSCs for depression) (Fig. 2B). This suggests that the two polarities of modulation are forms of long-term potentiation (LTP) and long-term depression (LTD) of the NMDAR-EPSC.
Evidence for D1/D5 receptor-mediated effects was obtained with the addition of the selective D1/D5 antagonists R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH-23390; n = 5; 5–10 μm) or 8-bromo-2,3,4,5-tetrahydro-3-methyl-5-phenyl-1H-3-benza zepin-7-ol hydrobromide (SKF-83666; n = 8; 5–10 μm). The antagonists had no observable effect on their own, and both prevented SKF-81297-induced potentiation and inhibition of NMDAR-EPSCs. The degree of block produced by either agent ranged from 72 to 98%, and the degree of block was the same for both polarities of effect (Table 1). Furthermore, there was no difference between NMDAR-EPSCs evoked by TA or SC stimulation, and similar partial recoveries were obtained in all cases on washout and so we illustrated the antagonism using pooled data (Fig. 2A,D1–D4). Consistent with a block of opposite polarity D1/D5 effects, the coefficient of variance was markedly reduced in the presence of agonist plus antagonist compared with agonist alone when pooled data was used (data not illustrated).
Indirect modulation may have resulted from an effect of SKF-81297 on the hyperpolarizing current (IH) as reported in neocortical neurons (Wu and Hablitz, 2005; Rosenkranz and Johnston, 2006), by a receptor-independent, direct blockade of the NMDAR pore or by a modulation of voltage-gated inactivating calcium currents (Surmeier et al., 1995). None of these actions would be expected to occur at membrane potentials sufficiently depolarized to reverse the NMDAR synaptic current, so we repeated the experiments at a holding potential of +40 mV with a Cs-based internal solution to reduce potassium conductance-mediated shunt and improve membrane potential control. Under these conditions, both the potentiation and inhibition of the NMDAR were observed (Fig. 1A2,B2,C2,D2, Table 1).
Another potential indirect, postsynaptic effect might originate from a change in a voltage-insensitive potassium or chloride conductance. The substitution of the nonpermeant cation Cs+ for K+ in the recording electrode did not prevent the bidirectional modulation of NMDAR-EPSCs. Similar reversal potentials were observed for the NMDAR-EPSCs before (ENMDAR = 20 ± 2 mV; n = 8) and during (potentiation, ENMDAR = 21 ± 3 mV, n = 3; depression, ENMDAR = 20 ± 5 mV, n = 5) SKF81297 modulation, suggesting that neither a D1/D5 induced addition nor removal of a shunting current was a major contributing factor.
Finally, it has been shown previously that, in striatal neurons, D1/D5 agonists can produce facilitation of regenerative, high-voltage-activated calcium channels (Surmeier et al., 1995). L-type voltage-activated calcium channels are abundant on the distal dendrites in which the holding voltage cannot be well maintained, especially during fast transient current alterations originating on the distal dendrites. Thus, there may be an indirect effect on these channels by D1/D5 activation to potentiate NMDAR-EPSCs. To investigate this, we applied the L-type Ca channel blocker nimodipine (10 μm) to the bath and repeated the experiments. Under these conditions, we observed both significant potentiation and depression of NMDAR-EPSCs (Table 1). This does not rule out a partial contribution of L-type calcium channels. However, the majority of NMDAR-EPSC modulation cannot be accounted for by voltage-gated L-type calcium channel modulation.
In other CNS regions, there is both anatomical (Filloux et al., 1987; Hara and Pickel, 2005; Dumartin et al., 2007) and electrophysiological (Momiyama and Sim, 1996; Nicola et al., 1996; Gao et al., 2001; Ding et al., 2003; Paspalas and Goldman-Rakic, 2005; Hernández et al., 2007) evidence for D1-mediated presynaptic regulation of neurotransmission. However, examination of paired-pulse ratios for either SC (n = 4) or TA (n = 4) inputs over intervals of 50–300 ms showed no observable effect of SKF-81297, indicative of an absence of D1/D5-mediated presynaptic modulation of glutamate release onto CA1 pyramidal cells. Furthermore, as described below, the D1/D5 effects may be blocked by selective manipulation of the postsynaptic neuron. Together with the data from the D1/D5 antagonist blockade, the presence of effects at depolarized potentials, and the addition of nimodipine, it suggests that the bidirectional modulation of the NMDAR-EPSCs by the D1/D5 agonist SKF-81297 is direct.
NMDAR subunit composition determines modulation polarity of D1/D5 effect
The D1/D5-mediated bidirectional modulation of NMDAR synaptic responses on the same cell type and, in some cases, even in the same cell (n = 4 of 22 cells) suggests that different mechanisms may be selectively activated and downstream from the D1/D5 receptor. NMDARs on CA1 pyramidal cells are composed of NR1/NR2A, NR1/NR2B, or NR1/NR2A/NR2B subunits (Monyer et al., 1994; Vicini et al., 1998). Opposite polarity modulation of NMDARs by D1/D5 activation has been demonstrated previously for specific NMDAR subunit compositions but in different neuronal preparations. In hippocampal cultured neurons, D1-induced depression of NMDAR-mediated currents can result from a direct protein–protein interaction between the NR2A subunit and the D1 receptor (Lee et al., 2002). In acute and culture preparations, D1 agonists induced an upregulation of surface expression of NR2B subunits in striatal neurons (Hallett et al., 2006; Gao and Wolf, 2008) and an increase in NMDAR currents involving NR2B subunits in dopamine neurons (Schilstrom et al., 2006).
Ifenprodil selectively antagonizes NMDARs composed of NR2B subunits (Kew et al., 1998; Mott et al., 1998) in an activity-dependent manner (Kew et al., 1996; Barria and Malinow, 2005). After either potentiation or depression of NMDAR-EPSCs by SKF-81297 (10 μm), ifenprodil (10 μm) was added to the bathing medium for a duration sufficient to attain steady-state effect. Ifenprodil had little additional effect on NMDAR-EPSCs that were already depressed by SKF-81297(n = 12) (Fig. 3A,C). In contrast, for the population of NMDAR-EPSCs that were potentiated by SKF-81297, ifenprodil elicited a significant reduction of the NMDAR-EPSC (n = 16) (Fig. 3B) of 84 ± 5% (p < 0.001, two-tailed test) (Fig. 3C). These results suggest that SKF-81297 potentiates NMDAR-EPSCs that are sensitive to ifenprodil and contain NR2B subunits. NMDAR-EPSCs that are depressed by D1/D5 activation are insensitive to ifenprodil and do not contain NR2B subunits.
Figure 3.
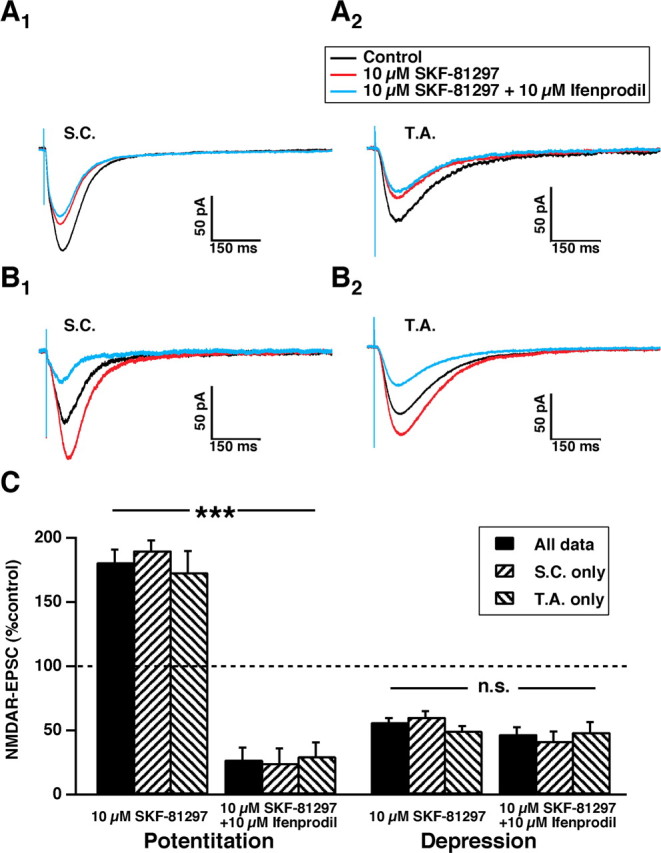
Ifenprodil antagonizes the NMDAR-EPSCs that are potentiated by D1/D5 activation but has little effect on those depressed by it. A, Averaged traces of NMDAR-EPSCs (n = 15 traces per average) from SC (A1) or TA (A2) stimulation that were depressed by SKF-81290 (10 μm for 30 min exposure) and then exposed to ifenprodil (10 μm for 15 min). B, As in A but for NMDAR-EPSCs that were potentiated by SKF-81290. C is a histogram of the NMDAR-EPSC responses to ifenprodil, grouped according to D1/D5 effect of either potentiation (on left; n = 16, ***p < 0.001) or depression (on right; n = 12; n.s., nonsignificant).
Hippocampal slices from genetically engineered animals lacking the gene encoding the NR2A subunit [a generous gift from Drs. Masayoshi (University of Tokyo, Tokyo, Japan) and Lovinger (National Institute on Alcohol Abuse and Alcoholism/National Institutes of Health, Rockville, MD)] were used to examine the response of the remaining NMDAR receptors in CA1 pyramidal cells, composed only of functional NR1/NR2B subunits. Previous observations that ifenprodil abolished NMDAR-EPSCs in NR2A knock-out mice were confirmed (Weitlauf et al., 2005). Bath application of SKF-81297 resulted in potentiation of the NMDAR-EPSC in every case (Fig. 4A, Table 1). Thus, the NR1/NR2B NMDARs are sufficient to support potentiation but not depression by D1/D5 activation.
Figure 4.
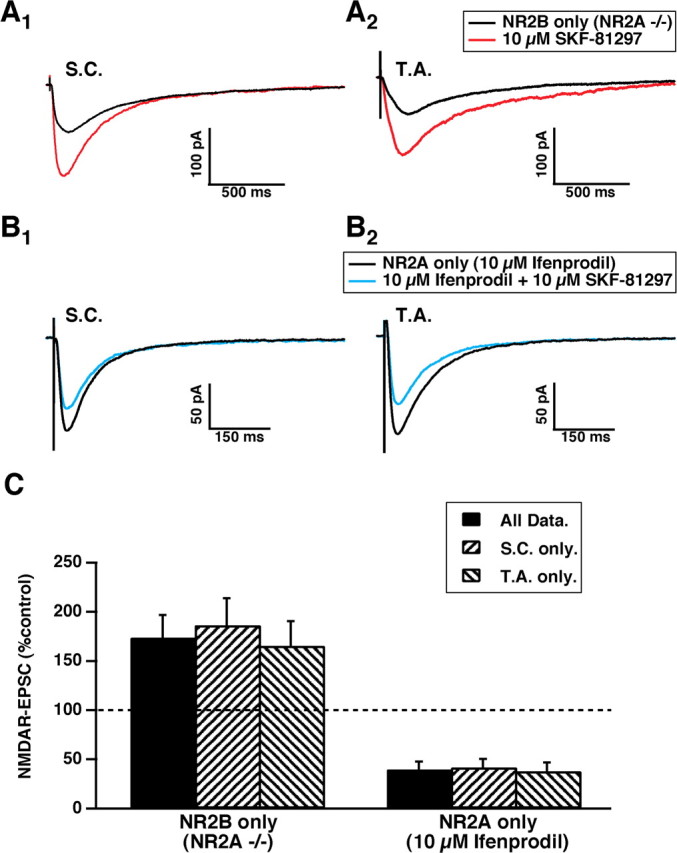
NMDAR subunit composition determines the polarity of response to D1/D5 modulation. A, Averaged traces of EPSCs (10 traces per average) evoked by SC (A1) or TA (A2) stimulation recorded from NR2A knock-out mutants showing SKF-81297-mediated enhancement after 30 min. B, As in A but preincubated in ifenprodil to antagonize NR2B-containing NMDAR-EPSCs. In this condition, only SKF-81297-mediated depression can be observed. C, A histogram showing pooled responses to SKF-81297 for NMDARs composed only of NR1/NR2B subunits (left; n = 14 EPSCs) or NMDARs with only functional NR1/NR2A subunits (right; n = 11 EPSCs).
The role of NR2A subunits in D1-mediated modulation of NMDAR-EPSCs was further examined by preincubation of the slice in ifenprodil (10 μm), followed by assessment of the effects of exposure to SKF-81297 (10 μm) (Fig. 4B). Under these conditions, the only functional NMDARs were composed of NR1/NR2A subunits. D1/D5 activation elicited only depression of the NMDAR-EPSC (Fig. 4C) (Table 1). This suggests that NR1/NR2A NMDARs are sufficient to support depression but not potentiation by D1/D5 activation.
D1/D5 activation decreases the ratio of NR2A/NR2B subunits
During development or after injury, the ratio of NR2A to NR2B subunits in synaptic NMDARs dramatically changes (Carmignoto and Vicini, 1992; Hestrin, 1992; Monyer et al., 1994; Kew et al., 1998; Roberts and Ramoa, 1999; Kawakami et al., 2003; Cull-Candy and Leszkiewicz, 2004). The well documented developmental increase of this ratio (NR1/NR2A heteromers predominate over heteromeric combinations containing NR2B subunits) has been associated with changes in dendritic morphology (Rajan et al., 1999; Ewald et al., 2008) and with altered metaplasticity that is necessary for experience-dependent plasticity in the visual system (Bear, 2003; Philpot et al., 2007; Yashiro and Philpot, 2008). Because SKF-81297 potentiates NMDAR-EPSCs with NR2B subunits and depressed NMDARs composed of NR2A subunits, the NR2A/NR2B ratio of NMDAR-EPSC subunit composition may be expected to be decreased. In control conditions, the addition of ifenprodil (10 μm; n = 20 EPSCs) reduced NMDAR-EPSCs to 64 ± 6% of control total charge transfer. As reported previously in rats (Arrigoni and Greene, 2004), ifenprodil reduced the TA NMDAR-EPSC to a lesser extent (74 ± 7% of control; n = 10 EPSCs) than its reduction of SC inputs (54 ± 7% of control; n = 10 EPSCs; p < 0.001, two-tailed t test), suggesting that the NR2A/NR2B ratio is greater for TA synapses compared with SC synapses.
In another series of experiments, neurons were first exposed to SKF-81297 (10 μm; n = 27) and then tested for ifenprodil sensitivity. For all of these NMDAR-EPSCs, the addition of ifenprodil reduced the synaptic total charge transfer to 42 ± 5% of control. Thus, for all inputs combined, preexposure to SKF-81297 resulted in a significant decrease in the NR2A/NR2B ratio (p < 0.01, two-tailed t test) compared with the control ratio (Fig. 5). The D1/D5-induced change of response to ifenprodil occurred primarily in the TA synapses (a change from 74 to 39 ± 8% of control amplitude; n = 12 EPSCs; p < 0.01, two-tailed t test) so that the TA NMDAR composition of subunits became similar to the SC NMDAR composition (Fig. 5). The SC subunit composition showed a trend toward an SKF-81297-induced decrease (a change from 54 to 45 ± 7%; n = 15 EPSCs) that was not significant.
Figure 5.

D1/D5-mediated decrease in NR2A/NR2B subunit ratio in synaptic NMDARs. Ifenprodil (10 μm) was used to assess the NR2A/NR2B ratio of subunit composition because, in its presence, only NR1/NR2A heteromers remain functional. The histogram shows that the ifenprodil antagonism of heteromers composed of NR2B subunits was significantly greater for all NMDAR-EPSCs recorded from CA1 pyramidal neurons (All; n = 27, **p < 0.01) and for NMDAR-EPSCs activated by stimulation of the TA input (n = 12, **p < 0.01) after D1/D5 activation. A nonsignificant trend for SC inputs (n = 15) was also observed. The greater ifenprodil effect indicates a reduced NR2A/NR2B subunit ratio in CA1 synapses.
Distinct D1/D5 intracellular signaling mechanisms
Because the findings described above show that specific NMDAR subunits determine the polarity NMDAR-EPSC modulation by D1/D5 activation, it seems plausible that distinct intracellular signaling mechanisms are also involved. A change in intracellular calcium is required for long-term, activity-dependent changes in AMPA receptor synaptic function (Malenka and Bear, 2004) and for long-term, activity-dependent, downregulation of NMDAR-EPSCs (Selig et al., 1995; Morishita et al., 2005). The role of intracellular Ca2+ in D1/D5 modulation was examined using intracellular application of the calcium chelator BAPTA (10 mm) before 10 μm SKF-81297 application. In the presence of BAPTA, D1/D5 modulation of NMDAR-EPSCs was not observed in either polarity, and there was no change in the coefficient of variance for NMDAR-EPSC pooled amplitudes from baseline values during 15 min of exposure to SKF-81297 (n = 5 EPSCs for SC, n = 5 EPSCs for TA; data not shown). Thus, changes in intracellular calcium concentration appear to be necessary for both inhibition and potentiation of NMDAR-EPSCs.
The D1/D5 agonist SKF-81297 has been shown to inhibit NMDAR-mediated currents by a direct physical interaction between the C terminus of the D1 receptor (the t3 domain) and NR2A NMDAR subunits in both HEK coexpression systems and in cultured hippocampal neurons (Lee et al., 2002). If inhibition of NMDA-EPSCs is mediated by a similar mechanism in acute slices, intracellular application of the C-terminal D1 receptor t3 fragment (D1t3; AA sequence, spalsvildydtdvslekiqpvthsgqhst) would be expected to interfere with the inhibitory interaction between the D1 receptor and NR2A subunits. When the D1t3 peptide was included in the intracellular solution of the recording pipette, D1/D5-induced depression of NMDA-EPSCSs was suppressed (Fig. 6A), leaving mainly potentiation (Table 1, Fig. 6C,D). Control experiments using a scrambled version of the peptide (n = 5 neurons) resulted in equal distribution of potentiation (n = 2 EPSCs) and depression (n = 3 EPSCs).
Figure 6.

Distinct intracellular signaling mechanisms mediate D1/D5 modulation of NMDAR-EPSCs. A, Averaged NMDAR-EPSC traces (n = 20 traces per average) recorded with D1t3 peptide fragment (30 μm) in the internal recording pipette solution. D1/D5 activation resulted in potentiation of the NMDAR-EPSC in the TA and SC pathways. B, Traces as in A but with the G-protein inhibitor GDPβS (200 μm) added to the recording solution. In this condition, D1/D5-mediated depression was observed. C, Amplitude distribution of NMDAR-EPSC modulation, after bath application of the D1/D5 agonist SKF-81297, with the three different internal solutions: control, D1t3, and GDPβS. D, Bar graph of the pooled, average modulation in NMDAR-EPSC elicited by D1/D5 activation with the three different internal solutions [control (ctr.), n = 18; D1t3, n = 9; GDPβS, n = 13; ***p < 0.001)].
D1 receptors are Gs-protein coupled, and it has been reported that D1 enhancement of NMDAR currents requires PKA activation (Surmeier et al., 1995; Gonzalez-Islas and Hablitz, 2003; Schilstrom et al., 2006). To test whether potentiation, but not depression, required the activation of G-proteins, the G-protein inhibitor GDPβS (200 μm) was added to the internal recording solution. Under these conditions, NMDAR-mediated synaptic currents remained stable for >30 min (n = 13). When SKF-81297 was added, a depression was induced in the majority of the synapses (Table 1, Fig. 6B–D). In another set of recordings, SKF-81297 was not added and the EPSCs were observed to remain stable (94 ± 8%; n = 6) for an additional 60 min. These findings suggest that D1/D5-mediated depression is G-protein independent but that potentiation requires intact G-protein signaling. Thus, the bidirectional effects of D1/D5 activation were doubly dissociated based on differential intracellular signaling mechanisms and different NMDAR subunit composition.
Endogenous activation of D1/D5 receptors
Amphetamine is known to release endogenous monoamines by blocking and reversing their reuptake transporters (Ritz and Kuhar, 1989; Sulzer et al., 1993). To examine the effects of activation of D1/D5 receptors by release of endogenous neurotransmitter, amphetamine (10 μm) was applied to the bath. We coapplied amphetamine and the D2 antagonist eticlopride (5 μm) during the recording of isolated NMDA-EPSCs evoked by stimulation of either the TA or SC pathways. Amphetamine, much like the D1/D5 agonist, produced bidirectional modulation of NMDA-EPSCs (n = 20 NMDAR-EPSCs) (Fig. 7A–C, Table 1). To determine whether the effects we observed were attributable to selective activation of D1/D5 receptors, we repeated the experiment in the presence of the D1/D5 antagonist SCH-23390 (n = 8 EPSCs). The effect observed under these conditions was a weak depression of NMDAR-EPSCs (73 ± 3% compared with 102 ± 5% for no drug at 30 min; p < 0.05, t test); this was a significantly different effect from that of amphetamine alone comparing the depressed and potentiated groups with and without SCH-23390 (p < 0.001, ANOVA single-factor analysis) (Fig 7C) or comparing only the depressed EPSC populations with and without antagonist (p < 0.001, t test). The coefficient of variance for the entire population of NMDAR-EPSCs exposed to amphetamine for 40 min was significantly different from the population exposed to amphetamine plus SCH-23390 (93 ± 3% compared with 23 ± 1%; p < 0.001, t test; data not shown). The latter was similar to the coefficient of variance for no drug (21 ± 2%). Together, these data indicate that amphetamine causes the activation of D1/D5 receptors in the same way as the D1/D5-selective agonist SKF-81297.
Figure 7.

Release of endogenous monoamines activates D1/D5 receptors to produce bidirectional modulation of NMDAR-EPSCs. A and B are averaged traces of NMDAR-EPSCs (n = 10 traces per average) evoked from stimulation of either the SC (A1, B1) or TA (A2, B2) inputs (control in black) that responded to amphetamine with either potentiation (A1, A2) or depression (B1, B2) of the NMDAR-EPSC. C is a graph of pooled data of NMDAR-EPSC amplitude (total charge transfer; n = 18 EPSCs) in response to amphetamine application for 30 min (potentiation, n = 7 EPSCs; depression n = 11 EPSCs). In the presence of D1/D5 antagonism, variability was reduced and only a small depression remained (in black; n = 9 EPSCs). D is a histogram of the differences between SC minus TA-evoked, NMDAR-EPSC amplitudes recorded from the same neuron (n = 9 neurons) before and during either amphetamine or amphetamine plus D1/D5 antagonist (n = 4 neurons, *p < 0.05).
Subunit-specific D1/D5 modulation of NMDARs correlates with input-specific NMDAR subunit distribution
It has been shown previously that an input-specific bias for the distribution of NMDA receptor subtypes exists in rat. This was assessed using NMDA-EPSCs recorded in CA1 stimulated from either the TA or SC pathways in conjunction with the selective NR2B antagonist ifenprodil. A larger ifenprodil-sensitive NR2B component was found in the SC-activated EPSC compared with the TA input (Arrigoni and Greene, 2004). To test for an input-specific distribution of NMDAR subunits in mouse, we applied ifenprodil (10 μm) to block NMDAR-EPSCs mediated by NMDARs containing either NR1/NR2B or NR1/NR2A/NR2B subunits. The inputs were stimulated at 0.05 Hz for a sufficient number of times to obtain an equilibrium response.
A similar bias in mice to that reported in rats was observed. For a particular CA1 pyramidal cell, the NMDAR-EPSC from the SC input was more strongly inhibited by ifenprodil (assessed by percentage reduction of the NMDAR-EPSC) than the NMDAR-EPSC from the TA input (Fig. 8A). The percentage ifenprodil-induced reduction of the TA input was then subtracted from the percentage ifenprodil-induced reduction of the SC input for each neuron, and the averaged differences were compared with average differences between SC and TA over the same period of time without the addition of ifenprodil (p < 0.001, Wilkinson's paired test, one tailed) (Fig. 8C). The differences in the presence of ifenprodil were much greater than control, suggesting that, in the mouse, TA inputs have a greater NR2A/NR2B ratio than the SC inputs. Because D1/5 modulation of NMDAR-EPSCs is determined by subunit composition of the NMDAR, an input-specific bias of NMDAR-EPSC modulation is also likely to exist.
Figure 8.

D1/D5 modulation of NMDAR-EPSCs is determined by the input-specific bias of the subunit composition of the NMDARs. A, Average current trace of an NMDAR-EPSC (n = 10 traces per average) for baseline (black) and during inhibition by 10 μm ifenprodil (light blue line) for the SC (A1) and TA (A2) inputs on the same neuron. B, Average NMDAR-EPSC (n = 10 traces per average) for baseline (black) and 10 μm SKF-81297 (red) for the SC (B1, B3) and TA (B2, B4) inputs obtained from the same neuron. C, A histogram showing the average difference between the SC and TA input for each neuron, during ifenprodil application (blue; n = 10 neurons) or in a separate set of experiments, during modulation by SKF-81297 (red; n = 22 neurons) or during sham perfusion (black; n = 7 neurons). **p < 0.01, ***p < 0.001.
For a particular neuron, if D1/D5-mediated potentiation was observed, it was greater for SC inputs than from TA inputs, and, if depression was observed, it was greater for NMDAR-EPSCs for TA inputs than from SC inputs (Fig. 8B). For each neuron, the percentage D1/D5-induced change of the NMDAR-EPSC evoked by TA stimulation was subtracted from the percentage D1/D5-induced change for NMDAR-EPSCs evoked by SC stimulation, and the average difference was significantly greater than the difference of the percentage changes of the NMDAR-EPSCs from the two inputs over the same period of time but without drug exposure (p < 0.05, Wilkinson's paired test, two tailed) (Fig. 8C). This suggests that DA acting on D1/D5 receptors can provide an input-specific bias toward enhanced NMDAR-mediated synaptic responsiveness for SC inputs compared with NMDAR synaptic responsiveness for TA inputs.
We next compared amphetamine modulation between the SC and TA pathways (Fig. 7D) and found that, as with exogenous D1/D5 activation, a significant bias existed for depression of TA inputs and potentiation of SC inputs for a given neuron (p < 0.05, Wilkinson's paired test, two tailed; n = 9 neurons). These results suggest that endogenous monoaminergic D1/D5 activation can modulate NMDA-EPSCs with an input-specific bias similar to exogenous D1/D5 activation.
Discussion
The NMDAR component of synaptic currents was either potentiated or depressed by activation of D1/D5 dopamine receptors in hippocampal CA1 pyramidal cells. The polarity of modulation was determined by the subunit composition of NMDARs. The NR1/NR2A subunit depression was mediated by a protein–protein interaction involving the C-terminal regions of the D1 receptor and NR2A NMDAR subunit. Potentiation of NMDARs required functional NR1/NR2B or NR1/NR2B/NR2A subunits, and, unlike the depression, potentiation also required G-protein activation.
Dopamine was shown previously to exert presynaptic inhibition of the TA but not SC inputs based on a dopamine-evoked increase in paired-pulse facilitation (Otmakhova and Lisman, 1999). Because we did not observe any evidence of D1/D5 mediated presynaptic effects on either the TA or SC inputs, this suggests that the effects observed by Otmakhova and Lisman (1999) are not mediated by D1/D5 receptors.
D1/D5 activation not only altered the amplitude of NMDAR-EPSCs but decreased the subunit NR2A/NR2B ratio as indicated by the D1/D5-elicited increased ifenprodil sensitivity (10 μm). Interestingly, only those NMDAR-EPSCs that were potentiated by SKF-81297 showed a significant change in ifenprodil sensitivity (Fig. 4), suggesting that the change in ratio was attributable primarily to an increase in NR2B-containing NMDAR synaptic current. Because D1 activation is also associated with increased surface expression of NR2B but not NR2A subunits (Gao and Wolf, 2008), it seems likely that the change in ifenprodil-sensitive current was mediated in largest part by an increase in NMDARs containing NR2B subunits, and, because the currents recorded in this study are synaptic, this increase includes synaptic NR2B-containing NMDARs.
The subunit composition of NMDARs imparts distinct properties to the receptor, including differences in kinetics, conductance, binding affinity, and intracellular signaling mechanisms (Cull-Candy and Leszkiewicz, 2004). NMDARs composed of NR1/NR2B subunits have slower kinetics than NR1/NR2A heteromer (Monyer et al., 1994; Vicini et al., 1998), and they flux a greater amount of calcium for an equal amount of NMDAR-mediated current (Sobczyk et al., 2005). Furthermore, NR1/NR2B receptors are differentially associated with intracellular signaling systems affecting plasticity, including CAMKII (Strack and Colbran, 1998; Barria and Malinow, 2005) and Ras-extracellular signal-regulated kinase (Kim et al., 2005). Together, these NR2B subunit properties are likely to impart an increased probability of activity and NMDAR-dependent, postsynaptic plasticity compared with NR1/NR2A receptors (Yashiro and Philpot, 2008).
The functional implications of NR2A versus NR2B subunit composition for NMDAR-dependent plasticity remain controversial. However, there is considerable converging evidence to suggest that the NR2A/NR2B ratio of NMDARs affects the relationship between neural activity and NMDAR-dependent LTP and LTD, termed metaplasticity (Bear, 2003). An increase in this ratio may be expected to broaden the low-frequency activity window for LTD and to raise the threshold frequency for switching from LTD-generating activity to activity that generates LTP (Bear, 2003; Barria and Malinow, 2005; Yashiro and Philpot, 2008). This increase occurs globally in the CNS during development (Carmignoto and Vicini, 1992; Hestrin, 1992; Monyer et al., 1994; Kew et al., 1998; Roberts and Ramoa, 1999; Cull-Candy and Leszkiewicz, 2004) and is necessary for the changes in experience-dependent plasticity or metaplasticity of the visual system in young animals preceding and during their critical period of visual development (Philpot et al., 2003, 2007). There are reports of D1 receptor-mediated slowly developing increases in AMPA receptor-mediated synaptic responses that are NMDAR-dependent yet are independent of high-frequency synaptic activation in neurons in the ventral tegmental nucleus (Argilli et al., 2008) and in the CA1 region of the hippocampus (Huang and Kandel, 1995). These findings are consistent with a significantly lowered threshold for LTP induction in association with a decreased NR2A/NR2B ratio.
A change in the NR2A/NR2B ratio in glutamatergic synapses was shown recently to be rapid, bidirectional, activity dependent and of sufficient degree to alter the integrative properties of the affected synapses, in neonatal but not mature neurons in an acute hippocampal preparation (Bellone and Nicoll, 2007). Neural activity evoking LTP of AMPA receptors could increase this ratio, and, once potentiated, the altered ratio could be reset by activity eliciting LTD-like, depotentiation.
In cultured neurons, dendritic morphology is controlled by the NR2A/NR2B ratio in a manner consistent with an increase being associated with more fully developed, mature dendrites with more complex dendritic arbors (Ewald et al., 2008). Conversely, a reduced NR2A/NR2B ratio attributable to overexpression of NR2B subunits was shown by this same group to be associated with structurally labile dendritic branches.
Control of the NR2A/NR2B ratio has not been reported previously in mature neurons; however, our findings suggest that a decrease of this ratio can be induced by D1/D5 activation in mature neurons. Accordingly, the switch to a greater NR2A/NR2B ratio is not necessarily an irreversible developmental outcome but rather can be reversed by D1/D5 activation. We observed the greatest change in this ratio in synapses of the TA input that have the larger NR2A/NR2B ratio in adult hippocampus compared with the SC inputs. The apparently smaller effect in SC inputs may be attributable in part to a “floor effect” because the SC synapses have a reduced ratio under baseline conditions. The larger ratio observed in TA synapses is more comparable with other adult cortical regions.
D1/D5 activation decreased the NR2A/NR2B ratio toward that observed on less mature neurons that may undergo LTP with less stringent timing of inputs (Bellone and Nicoll, 2007) and have a lowered threshold for induction (Barria and Malinow, 2005; Philpot et al., 2007). This increase of effective period for the integration of synaptic inputs may increase plasticity at the expense of the precision of coincidence detection. A salient cue or amphetamine exposure may increase dopamine release and D1/D5 activation, reducing the NR2A/NR2B ratio. This change may result in more plastic synaptic responses and more plastic dendritic morphology compared with conditions in the absence of dopamine release. Accordingly, release of dopamine can modify the NMDAR subunit ratio toward that associated with more rapidly changing, developing neurons.
On a particular neuron, a bias in NMDAR subunit distribution was found favoring increased NR2A contribution to the TA input and increased NR2B contribution to the SC input. This NMDAR subunit bias was reflected as a pathway-specific D1/D5-mediated modulation of NMDAR-EPSCs. Accordingly, we observed a bias toward greater D1/D5-mediated inhibition of NMDAR-EPSCs in the TA input and for greater D1/D5-mediated potentiation of NMDAR-EPSCs in the SC input. This may seem at odds with our pooled data, showing an average D1/D5 effect on synaptic NMDAR total charge transfer of TA inputs of 91 ± 11% compared with SC inputs of 106 ± 10%, a very small difference. The small, pooled difference may be attributable to cell-to-cell variability of the magnitude of the effect; some cells showed large enhancements for both inputs, and some cells large reduction for both inputs, whereas others showed relatively small effects. Nevertheless, the relationship that on a particular cell the percentage change was greater for the SC than for the TA input was always observed. For example, if both the SC and TA inputs to a given cell responded with enhancement, the SC response was always enhanced to a greater degree. If both inputs responded with reduction, then the TA response was always reduced to a greater degree. This resulted in a significant bias for all the recorded cells.
It is also worth noting that the ifenprodil block of synaptic NMDAR activation reflects the antagonism of both NR2B/NR1 and NR2A/NR2B/NR1 subunit combinations. We do not know what the D1/D5 effect on the NR2A/NR2B/NR1 NMDAR is, and it is possible that the effect varies depending on the orientation of the heteromeric NMDAR to D1/D5 receptors. Our data only support the findings that D1/D5 effects on NR2A/NR1 composition are inhibitory and on NR2B/NR1 composition enhancing.
At a circuit level, D1/D5 modulation provides a mechanism for increasing the responsiveness of CA1 pyramidal cells to processed SC inputs from CA3 neurons of the trisynaptic pathway while decreasing responsiveness to direct TA inputs from the entorhinal cortex. Because CA3 ensemble firing patterns are determined by inputs from both dentate and CA3, D1/D5-mediated modulation can serve as a “gate” that alters CA1 ensemble firing patterns by redirecting NMDAR-dependent synaptic drive from TA to SC inputs. Thus, a signal that sufficiently activates D1/D5 receptors could bias CA1 ensemble firing toward synaptic influence exerted by the more highly processed CA3 SC inputs and away from the more direct TA influence.
Dopamine release from VTA neurons has been correlated with salience of environmental cues (Fiorillo et al., 2003), and it has also been shown that DA neurons from the A10 region of the VTA project to hippocampus (Swanson, 1982; Gasbarri et al., 1994). An effect of salience-induced release of DA in the hippocampus could be twofold in effect. First, a bias is exerted on CA1 toward influence from the highly processed entorhinal cortex–dentate–CA3–SC circuit and away from direct entorhinal cortical TA input. Second, the synapses, especially of the TA inputs, express a significantly decreased NR2A/NR2B ratio, associated with altered activity and NMDAR-dependent plasticity and dendritic stability, consistent with an increased plasticity of both direct and indirect pathways
Abnormally persistent D1/D5 activation may alter the ability of salience-induced DA release to switch gating of this circuit from less to more highly processed input and to facilitate increased plasticity for both inputs. Endogenous release of monoamines by amphetamine is sufficient to activate D1/D5 receptors and alter NMDAR-EPSCs to selectively alter the CA1 circuit function. Consistent with our findings, local intrahippocampal injections of amphetamine have been shown to selectively enhance hippocampal-dependent spatial memory (Packard et al., 1994). Accordingly, it is likely that dopamine signaling in the hippocampus can act via NMDARs to profoundly affect hippocampal-dependent learning and memory in both physiological and pathophysiological conditions.
Footnotes
This work was supported by National Institutes of Health Grants P50MH060450 and MH080297 (R.W.G.) and by the Department of Veterans Affairs.
References
- Amaral and Witter, 1989.Amaral DG, Witter MP. The three-dimensional organization of the hippocampal formation: a review of anatomical data. Neuroscience. 1989;31:571–591. doi: 10.1016/0306-4522(89)90424-7. [DOI] [PubMed] [Google Scholar]
- Ang et al., 2005.Ang CW, Carlson GC, Coulter DA. Hippocampal CA1 circuitry dynamically gates direct cortical inputs preferentially at theta frequencies. J Neurosci. 2005;25:9567–9580. doi: 10.1523/JNEUROSCI.2992-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilli et al., 2008.Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni and Greene, 2004.Arrigoni E, Greene RW. Schaffer collateral and perforant path inputs activate different subtypes of NMDA receptors on the same CA1 pyramidal cell. Br J Pharmacol. 2004;142:317–322. doi: 10.1038/sj.bjp.0705744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria and Malinow, 2005.Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Bear, 2003.Bear MF. Bidirectional synaptic plasticity: from theory to reality. Philos Trans R Soc Lond B Biol Sci. 2003;358:649–655. doi: 10.1098/rstb.2002.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone and Nicoll, 2007.Bellone C, Nicoll RA. Rapid bidirectional switching of synaptic NMDA receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Bikson et al., 2004.Bikson M, Inoue M, Akiyama H, Deans JK, Fox JE, Miyakawa H, Jefferys JG. Effects of uniform extracellular DC electric fields on excitability in rat hippocampal slices in vitro. J Physiol. 2004;557:175–190. doi: 10.1113/jphysiol.2003.055772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto and Vicini, 1992.Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Chen et al., 2004.Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–2600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy and Leszkiewicz, 2004.Cull-Candy SG, Leszkiewicz DN. Role of distinct NMDA receptor subtypes at central synapses. Sci STKE. 2004;2004:re16. doi: 10.1126/stke.2552004re16. [DOI] [PubMed] [Google Scholar]
- Ding et al., 2003.Ding L, Perkel DJ, Farries MA. Presynaptic depression of glutamatergic synaptic transmission by D1-like dopamine receptor activation in the avian basal ganglia. J Neurosci. 2003;23:6086–6095. doi: 10.1523/JNEUROSCI.23-14-06086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudman et al., 2007.Dudman JT, Tsay D, Siegelbaum SA. A role for synaptic inputs at distal dendrites: instructive signals for hippocampal long-term plasticity. Neuron. 2007;56:866–879. doi: 10.1016/j.neuron.2007.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumartin et al., 2007.Dumartin B, Doudnikoff E, Gonon F, Bloch B. Differences in ultrastructural localization of dopaminergic D1 receptors between dorsal striatum and nucleus accumbens in the rat. Neurosci Lett. 2007;419:273–277. doi: 10.1016/j.neulet.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Ewald et al., 2008.Ewald RC, Van Keuren-Jensen KR, Aizenman CD, Cline HT. Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J Neurosci. 2008;28:850–861. doi: 10.1523/JNEUROSCI.5078-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filloux et al., 1987.Filloux FM, Wamsley JK, Dawson TM. Presynaptic and postsynaptic D1 dopamine receptors in the nigrostriatal system of the rat brain: a quantitative autoradiographic study using the selective D1 antagonist [3H]SCH 23390. Brain Res. 1987;408:205–209. doi: 10.1016/0006-8993(87)90373-8. [DOI] [PubMed] [Google Scholar]
- Fiorillo et al., 2003.Fiorillo CD, Tobler PN, Schultz W. Discrete coding of reward probability and uncertainty by dopamine neurons. Science. 2003;299:1898–1902. doi: 10.1126/science.1077349. [DOI] [PubMed] [Google Scholar]
- Gao and Wolf, 2008.Gao C, Wolf ME. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J Neurochem. 2008;106:2489–2501. doi: 10.1111/j.1471-4159.2008.05597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao et al., 2001.Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasbarri et al., 1994.Gasbarri A, Verney C, Innocenzi R, Campana E, Pacitti C. Mesolimbic dopaminergic neurons innervating the hippocampal formation in the rat: a combined retrograde tracing and immunohistochemical study. Brain Res. 1994;668:71–79. doi: 10.1016/0006-8993(94)90512-6. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Islas and Hablitz, 2003.Gonzalez-Islas C, Hablitz JJ. Dopamine enhances EPSCs in layer II–III pyramidal neurons in rat prefrontal cortex. J Neurosci. 2003;23:867–875. doi: 10.1523/JNEUROSCI.23-03-00867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett et al., 2006.Hallett PJ, Spoelgen R, Hyman BT, Standaert DG, Dunah AW. Dopamine D1 activation potentiates striatal NMDA receptors by tyrosine phosphorylation-dependent subunit trafficking. J Neurosci. 2006;26:4690–4700. doi: 10.1523/JNEUROSCI.0792-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara and Pickel, 2005.Hara Y, Pickel VM. Overlapping intracellular and differential synaptic distributions of dopamine D1 and glutamate N-methyl-d-aspartate receptors in rat nucleus accumbens. J Comp Neurol. 2005;492:442–455. doi: 10.1002/cne.20740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser and Roth, 1997.Häusser M, Roth A. Estimating the time course of the excitatory synaptic conductance in neocortical pyramidal cells using a novel voltage jump method. J Neurosci. 1997;17:7606–7625. doi: 10.1523/JNEUROSCI.17-20-07606.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández et al., 2007.Hernández A, Sierra A, Valdiosera R, Florán B, Erlij D, Aceves J. Presynaptic D1 dopamine receptors facilitate glutamatergic neurotransmission in the rat globus pallidus. Neurosci Lett. 2007;425:188–191. doi: 10.1016/j.neulet.2007.08.026. [DOI] [PubMed] [Google Scholar]
- Hestrin, 1992.Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Huang and Kandel, 1995.Huang YY, Kandel ER. D1/D5 receptor agonists induce a protein synthesis-dependent late potentiation in the CA1 region of the hippocampus. Proc Natl Acad Sci U S A. 1995;92:2446–2450. doi: 10.1073/pnas.92.7.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge and Hasselmo, 2004.Judge SJ, Hasselmo ME. Theta rhythmic stimulation of stratum lacunosum-moleculare in rat hippocampus contributes to associative LTP at a phase offset in stratum radiatum. J Neurophysiol. 2004;92:1615–1624. doi: 10.1152/jn.00848.2003. [DOI] [PubMed] [Google Scholar]
- Kawakami et al., 2003.Kawakami R, Shinohara Y, Kato Y, Sugiyama H, Shigemoto R, Ito I. Asymmetrical allocation of NMDA receptor epsilon2 subunits in hippocampal circuitry. Science. 2003;300:990–994. doi: 10.1126/science.1082609. [DOI] [PubMed] [Google Scholar]
- Kew et al., 1996.Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol. 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew et al., 1998.Kew JN, Richards JG, Mutel V, Kemp JA. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim et al., 2005.Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Lee et al., 2002.Lee FJ, Xue S, Pei L, Vukusic B, Chéry N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F. Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell. 2002;111:219–230. doi: 10.1016/s0092-8674(02)00962-5. [DOI] [PubMed] [Google Scholar]
- Lemon and Manahan-Vaughan, 2006.Lemon N, Manahan-Vaughan D. Dopamine D1/D5 receptors gate the acquisition of novel information through hippocampal long-term potentiation and long-term depression. J Neurosci. 2006;26:7723–7729. doi: 10.1523/JNEUROSCI.1454-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li et al., 2003.Li S, Cullen WK, Anwyl R, Rowan MJ. Dopamine-dependent facilitation of LTP induction in hippocampal CA1 by exposure to spatial novelty. Nature Neuroscience. 2003;6:526–531. doi: 10.1038/nn1049. [DOI] [PubMed] [Google Scholar]
- Malenka and Bear, 2004.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Momiyama and Sim, 1996.Momiyama T, Sim JA. Modulation of inhibitory transmission by dopamine in rat basal forebrain nuclei: activation of presynaptic D1-like dopaminergic receptors. J Neurosci. 1996;16:7505–7512. doi: 10.1523/JNEUROSCI.16-23-07505.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer et al., 1994.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Morishita et al., 2005.Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Mott et al., 1998.Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- Nicola et al., 1996.Nicola SM, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. J Neurosci. 1996;16:1591–1604. doi: 10.1523/JNEUROSCI.16-05-01591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Carroll et al., 2006.O'Carroll CM, Martin SJ, Sandin J, Frenguelli B, Morris RG. Dopaminergic modulation of the persistence of one-trial hippocampus-dependent memory. Learn Mem. 2006;13:760–769. doi: 10.1101/lm.321006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova and Lisman, 1999.Otmakhova NA, Lisman JE. Dopamine selectively inhibits the direct cortical pathway to the CA1 hippocampal region. J Neurosci. 1999;19:1437–1445. doi: 10.1523/JNEUROSCI.19-04-01437.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard et al., 1994.Packard MG, Cahill L, McGaugh JL. Amygdala modulation of hippocampal-dependent and caudate nucleus-dependent memory processes. Proc Natl Acad Sci U S A. 1994;91:8477–8481. doi: 10.1073/pnas.91.18.8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paspalas and Goldman-Rakic, 2005.Paspalas CD, Goldman-Rakic PS. Presynaptic D1 dopamine receptors in primate prefrontal cortex: target-specific expression in the glutamatergic synapse. J Neurosci. 2005;25:1260–1267. doi: 10.1523/JNEUROSCI.3436-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot et al., 2003.Philpot BD, Espinosa JS, Bear MF. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J Neurosci. 2003;23:5583–5588. doi: 10.1523/JNEUROSCI.23-13-05583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot et al., 2007.Philpot BD, Cho KK, Bear MF. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron. 2007;53:495–502. doi: 10.1016/j.neuron.2007.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan et al., 1999.Rajan I, Witte S, Cline HT. NMDA receptor activity stabilizes presynaptic retinotectal axons and postsynaptic optic tectal cell dendrites in vivo. J Neurobiol. 1999;38:357–368. doi: 10.1002/(sici)1097-4695(19990215)38:3<357::aid-neu5>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Ritz and Kuhar, 1989.Ritz MC, Kuhar MJ. Relationship between self-administration of amphetamine and monoamine receptors in brain: comparison with cocaine. J Pharmacol Exp Ther. 1989;248:1010–1017. [PubMed] [Google Scholar]
- Roberts and Ramoa, 1999.Roberts EB, Ramoa AS. Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol. 1999;81:2587–2591. doi: 10.1152/jn.1999.81.5.2587. [DOI] [PubMed] [Google Scholar]
- Rosenkranz and Johnston, 2006.Rosenkranz JA, Johnston D. Dopaminergic regulation of neuronal excitability through modulation of Ih in layer V entorhinal cortex. J Neurosci. 2006;26:3229–3244. doi: 10.1523/JNEUROSCI.4333-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilström et al., 2006.Schilström B, Yaka R, Argilli E, Suvarna N, Schumann J, Chen BT, Carman M, Singh V, Mailliard WS, Ron D, Bonci A. Cocaine enhances NMDA receptor-mediated currents in ventral tegmental area cells via dopamine D5 receptor-dependent redistribution of NMDA receptors. J Neurosci. 2006;26:8549–8558. doi: 10.1523/JNEUROSCI.5179-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans et al., 2001.Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selig et al., 1995.Selig DK, Hjelmstad GO, Herron C, Nicoll RA, Malenka RC. Independent mechanisms for long-term depression of AMPA and NMDA responses. Neuron. 1995;15:417–426. doi: 10.1016/0896-6273(95)90045-4. [DOI] [PubMed] [Google Scholar]
- Sobczyk et al., 2005.Sobczyk A, Scheuss V, Svoboda K. NMDA receptor subunit-dependent [Ca2+] signaling in individual hippocampal dendritic spines. J Neurosci. 2005;25:6037–6046. doi: 10.1523/JNEUROSCI.1221-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed and Dobrunz, 2009.Speed HE, Dobrunz LE. Developmental changes in short-term facilitation are opposite at temporoammonic synapses compared to Schaffer collateral synapses onto CA1 pyramidal cells. Hippocampus. 2009;19:187–204. doi: 10.1002/hipo.20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward and Scoville, 1976.Steward O, Scoville SA. Cells of origin of entorhinal cortical afferents to the hippocampus and fascia dentata of the rat. J Comp Neurol. 1976;169:347–370. doi: 10.1002/cne.901690306. [DOI] [PubMed] [Google Scholar]
- Strack and Colbran, 1998.Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Sulzer et al., 1993.Sulzer D, Maidment NT, Rayport S. Amphetamine and other weak bases act to promote reverse transport of dopamine in ventral midbrain neurons. J Neurochem. 1993;60:527–535. doi: 10.1111/j.1471-4159.1993.tb03181.x. [DOI] [PubMed] [Google Scholar]
- Surmeier et al., 1995.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Swanson, 1982.Swanson LW. The projections of the ventral tegmental area and adjacent regions: a combined fluorescent retrograde tracer and immunofluorescence study in the rat. Brain Res Bull. 1982;9:321–353. doi: 10.1016/0361-9230(82)90145-9. [DOI] [PubMed] [Google Scholar]
- Vervaeke et al., 2006.Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini et al., 1998.Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Weitlauf et al., 2005.Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25:8386–8390. doi: 10.1523/JNEUROSCI.2388-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter et al., 1988.Witter MP, Griffioen AW, Jorritsma-Byham B, Krijnen JL. Entorhinal projections to the hippocampal CA1 region in the rat: an underestimated pathway. Neurosci Lett. 1988;85:193–198. doi: 10.1016/0304-3940(88)90350-3. [DOI] [PubMed] [Google Scholar]
- Wu and Hablitz, 2005.Wu J, Hablitz JJ. Cooperative activation of D1 and D2 dopamine receptors enhances a hyperpolarization-activated inward current in layer I interneurons. J Neurosci. 2005;25:6322–6328. doi: 10.1523/JNEUROSCI.1405-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, 2000.Yang SN. Sustained enhancement of AMPA receptor- and NMDA receptor-mediated currents induced by dopamine D1/D5 receptor activation in the hippocampus: an essential role of postsynaptic Ca2+ Hippocampus. 2000;10:57–63. doi: 10.1002/(SICI)1098-1063(2000)10:1<57::AID-HIPO6>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Yashiro and Philpot, 2008.Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]