

Abstract
Aurora kinases play a critical role in regulating mitosis and cell division and there over expression have been implicated in the survival and proliferation of human cancer. In this study we report the in vitro and in vivo activities of AZD1152, a compound that has selectivity for aurora B kinase, in acute myeloid leukemia (AML) cell lines, primary AML samples and cord blood cells. AZD1152 exerted anti-proliferative or cytotoxic effects in all cell lines studied, inhibited the phosphorylation of histone H3 (pHis H3) on ser10 in a dose dependent manner and resulted in cells with more than 4N DNA content. THP-1 cells treated with AZD1152 accumulated in a state of polyploidy and showed a senescent response to the drug in contrast to the apoptotic response seen in other cell lines. Accordingly, AZD1152 profoundly affected the growth of AML cell lines and primary AML in an in vivo xenotransplantation model. However, concentration-dependent effects on cell growth, apoptosis and cell cycle progression were also observed when human cord blood, primary lineage-negative stem and progenitor cells were analyzed in vitro and in vivo.
These data suggest that the inhibition of aurora B kinase may be a useful therapeutic strategy in the treatment of AML and that further exploration of dosing and treatment schedules is warranted in clinical trials.
Keywords: Leukemia, Xenotransplantation, Leukemic Stem Cells, Aurora, Aurora inhibitor
Introduction
The aurora kinases (Aur) have been implicated in the survival and proliferation of both hematological and solid malignancies (1, 2). These proteins play a critical role in regulating mitosis and cytokinesis, with the activity of both Aur-A and -B peaking during mitosis (1). The Aur genes map to chromosomal loci that are frequently altered in human cancers (2) and the expression of Aur-A and Aur-B is often increased in tumor cell lines and common primary tumors. Aur-A and B have distinct sub cellular localization during mitosis, reflecting their different roles in the process. Aur-A localizes to the centrosomes and mitotic spindle apparatus and regulates spindle formation (3), although it can also phosphorylate p53 facilitating Mdm2-mediated ubiquitination (1). In contrast Aur-B is a chromosomal passenger protein that regulates chromosome segregation and cytokinesis (4). Several aurora substrates have been described, including histone H3 phosphorylated at (ser10) (3). There is also a third aurora family member (Aur-C), another chromosome passenger protein, which has been implicated in the regulation of meiosis in testes (5).
The effects of inhibiting Aur-A and -B activity have been characterized in vitro. Suppression of Aur-A activity with the small molecule inhibitor MLN8054 leads to G2/M accumulation, spindle defects and anti-proliferative effects in tumor cells (6). Targeting Aur-B prevents chromosomal alignment, and compromises spindle checkpoint function, resulting in repeated rounds of DNA synthesis without cytokinesis, thereby generating polyploid cells and eventual loss of viability (7, 8). Interestingly, studies with mixed Aur (A and B) inhibitors results in a phenotype indicative of Aur-B rather than Aur-A inhibition (8, 9).
Aurora kinase expression in actively dividing cells makes them attractive therapeutic targets for the treatment of cancer. A number of small molecule inhibitors of aurora kinases have been developed and are currently in early clinical evaluation, including AZD1152 (10). AZD1152 is rapidly converted into the active moiety, AZD1152-hydroxyquinazoline-pyrazol-aniline (AZD1152-HQPA), following parenteral administration in vivo. AZD1152-HQPA, a reversible ATP-competitive inhibitor, is a highly potent and selective inhibitor of Aur B (Ki 0.36 nM) compared with Aur A (Ki 1369 nM) and has a high specificity in a panel of 50 additional serine-threonine and tyrosine kinases (11, 12). AZD1152 has demonstrated highly significant tumor growth inhibition in a diverse panel of solid human cancer tumor xenograft models, including lung and colorectal cancer (10).
Acute myeloid leukemia (AML) is characterized by a relentless accumulation of immature, abnormal hematopoietic cells in the bone marrow and peripheral blood. It has been postulated that AML is a disease maintained by leukemia stem cells (LSC). The immuno-deficient mouse xenotransplantation assay is currently the model of choice to assay LSC. This approach has been crucial to the understanding of human AML by providing reliable determination of the phenotypes of repopulating cells (13).
Despite the clear importance of the LSC in the genesis and perpetuation of leukemia, existing therapies largely target the bulk leukemic blasts. Since the survival of only a small number of LSC may facilitate disease relapse, any new treatment should be tested on the growth potential of these rare cells.
In this study, we have used AZD1152-HQPA and AZD1152 to evaluate the effects of inhibiting Aur B in vitro and in vivo respectively, in acute myeloid leukemia cell lines, primary AML cells and primary cord blood stem/progenitor cells.
Results
Effects of AZD1152-HQPA in AML cell lines in vitro and on the phosphorylation of histone H3 on ser10
The cytotoxic and anti-proliferative effects of AZD1152-HQPA were evaluated in exponentially growing HL-60, THP-1, U937 and MV411 cell lines treated with 0–1000 nM AZD1152-HQPA for up to 96hr. MV411 and THP-1 cells treated with 10nM show a marked inhibition in proliferation, as did HL-60 and U937 cells but to a lower extent (Fig 1A). At 100nM growth inhibition and cytotoxicity was observed in all cell lines except THP-1. The cytotoxic effect of AZD1152-HQPA was both concentration and time dependent (Fig 1B). In HL-60, MV411 and U937 cells, a 96hr exposure to 1 000 nM resulted in an approximate 80% loss of viability. Although AZD1152-HQPA had an anti-proliferative effect in THP-1 cells, the effect of the drug on cell viability was minimal.
Figure 1.
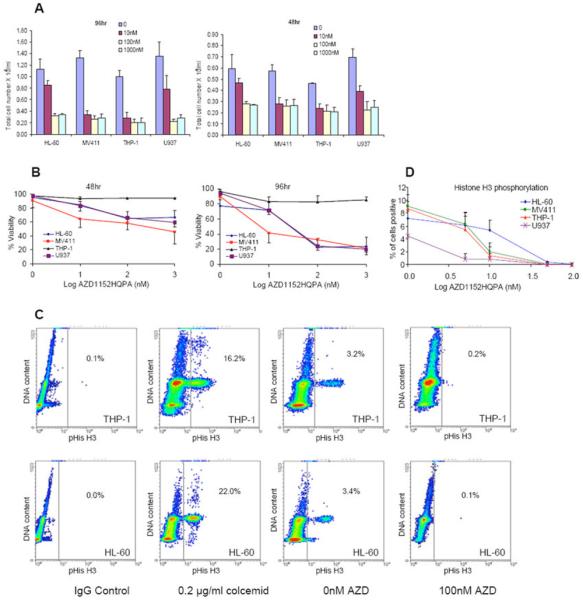
AZD1152-HQPA inhibited cell proliferation, induced cytotoxicity and inhibited phosphorylation of histone H3 (ser10) in AML cell lines. Effects of AZD1152-HQPA on cell number (Fig 1A) and cell viability (Fig 1B) at 48hr and 96 hrs; changes in histone H3 phosphorylation in HL-60 and THP-1 cells by flow cytometry at 18 hours (Fig 1C); IgG staining and colcemid treated cells were used as negative and positive controls respectively; the decrease in H3 phosphorylation was concentration-dependent in all cell lines and was fully inhibited at 100 nM AZD1152-HQPA (Fig 1D).
The inhibition of Aur-B activity was confirmed by a decrease in the phosphorylation of histone H3 ser10. This was observed in all cell lines studied, with complete inhibition of phosphorylation at 100 nM (Figs 1C and 1D).
Cell cycle effects of AZD1152-HQPA on AML cells
The effect of AZD1152-HQPA on cell cycle distribution was investigated in all cell lines, with data shown for HL-60 and THP-1 cells (Fig 2 A). The effects shown in HL-60 cells also reflect observations made in U937 and MV411 cells. AZD1152-HQPA induced polyploidy in all AML cell lines studied. By 48 hrs, cells had gone through a round of DNA replication without cytokinesis giving rise to a concentration dependent increase in polyploid (≥8N DNA content) cells (Fig. 2A). By 96 hours cells appeared to progress from polyploidy to apoptosis, with a concentration dependent increase in the proportion of cells with <2N DNA content (data not shown) and an increase in annexin-V staining (Fig 2B).
Figure 2.
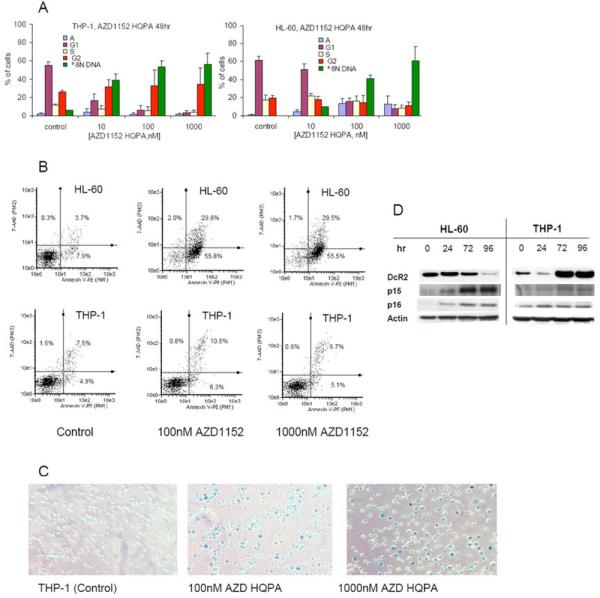
The induction of polyploidy by AZD1152-HQPA in HL-60 and THP-1 cells. Changes in cell-cycle distribution were investigated after exposure of HL-60 and THP-1 cells to the indicated concentrations of AZD1152-HQPA for 48hr (Fig 2A). HL-60 cells showed a marked increase in the apoptotic fraction after AZD1152-HQPA for 96 hr, whereas little change was seen in THP-1 cells (Fig 2B). AZD1152-HQPA treated THP1 showed the enlarged flattened morphology of senescent cells (Fig 2C), stained positive for senescence associated β-galactosidase activity (Fig 2C), and showed an increase in the anti-apoptotic TRAIL decoy receptor DcR2 with 100 nM AZD1152 (Fig 2D).
AZD1152-HQPA also induced polyploidy in THP-1 cells (Fig 2A). However, more than 80% of the cells treated with 100nM AZD1152-HQPA remained polyploid at 96hr (data not shown), with less than 5% apoptotic cells (Fig 2B). As THP1 cells showed such a low apoptotic response to AZD1152 we investigated the effect of aurora kinase inhibition on the ability of these cells to form colonies. THP-1 cells treated with 100nM and 1000nM AZD1152 HQPA lost colony forming capability (data not shown). On further investigation 80% and 95% on THP1 cells treated at 100 nM and 1000 nM AZD1152 respectively for 72hr and then cultured in drug free media for 7 days showed clear senescence associated β-galactosidase activity (Fig 2C). No β-galactosidase activity was apparent in HL-60 cells. Senescence is commonly associated with an increase in the cell cycle inhibitory proteins p15 or p16 which was not seen for THP-1 cells treated with AZD1152 HQPA (Fig 2D). In contrast a marked increase in the senescence associated TRAIL decoy receptor Dcr2 was observed, whereas Dcr2 showed a marked decrease in HL-60 cells in which the AZD1152-HQPA induced apoptosis.
Effects of AZD1152-HQPA on AML primary cells in vitro
The effect of AZD1152-HQPA on viable cell number, cell cycle distribution and pHis H3 staining was studied in 12 human primary AML samples (see supplementary Table S1 for clinical parameters). After 96 hr exposure to 1000 nM AZD1152-HQPA viable cell number was reduced by a median of 34% (range 0-68%) relative to control (untreated cells). In most samples there was little effect on cell cycle distribution, or the appearance of an apoptotic population (results from 2 primary samples are shown in Fig S1). It was noticeable that pHis H3 staining in untreated primary AML cells was low (median 1.5%, range 0-4.4%) in comparison to AML cell lines, suggesting a low proliferative rate. Based on this limitation we went on to examine the effect of AZD1152 using an in vivo model of AML.
Effects of AZD1152 in vivo on mouse hematopoiesis
Preliminary experiments were performed to determine whether AZD1152 was tolerated in NOD/SCID mice. There was no significant negative effect on the weight of 12-week old NOD/SCID mice when AZD1152 was administered as an infusion on two consecutive weeks at 25mg/kg/day. The mean (± SD) weight of mice before drug delivery was 17.9 (±0.85) g (n=5) and after treatment was actually slightly higher at 19.5 (±1.9) g (n=5). This suggests that the overall health of NOD/SCID mice is not severely affected by AZD1152 treatment.
AZD1152 treatment had a minimal effect on the absolute numbers of CD45+ cells present in the marrows of NOD/SCID mice. In mice that received PBS pumps, there was a mean (± SD) of 2.5×107 ± 0.9 × 107 CD45+ cells. One week of AZD1152 treatment at 25mg/kg/day resulted in a reduction of absolute numbers of CD45+ cells to 1.7×107±0.3×107, (n=6 for each group, p=0.09, Figure S2).
A comparison of the percentage of various subsets of CD45+ cells found no significant difference in the percentage of Lineage−/Sca-1+/c-kit+ stem/progenitor cells, B220+ B-cells, CD2+ T-cells, Gr-1+ cells or CD11b+ cells between treated and untreated groups of mice (Supplementary Figure S2, n= 6 for all).
When combined, this initial toxicity assessment indicates that even in the presence of an apparent cytotoxic / cytostatic effect on murine hematopoietic cells, there does not seem to be any particular cell subset that is preferentially targeted and the mice tolerate the AZD1152 treatment.
AZD1152 rapidly and profoundly reduces an established xenograft of HL-60 cells
To explore the effect of AZD1152 on the growth of human AML cells in vivo, we first examined the growth of the AML cell line HL-60. The HL-60 cells grow aggressively in the NOD/SCID model.
As for all our in vivo experiments, we first established the xenograft before treatment with AZD1152 and subsequent analysis of the murine bone marrow for human cell content. Preliminary experiments identified 5 weeks as the maximum time of HL-60 marrow engraftment from 106 injected log-phase cells without serious adverse effects.
The results of one week of 25mg/kg/day AZD1152 treatment on HL-60 xenografts in vivo are summarized in Figure 3A. The mean (± SD) percentage of HL-60 engraftment in controls was 64.5% (±19.0%) (n=11). After one week of 25mg/kg/day AZD1152 treatment, the percentage of HL-60 cells was 0.29% (±0.74%) (n=9). An example of a FACS analysis for the human cell content is provided in Figure 3B. A small group of mice were kept for a further 2 weeks post AZD1152 treatment, to see if the xenograft would start to grow back. In all mice processed in this fashion, the xenograft did not re-grow (0.008±0.006, n=3); indicating that AZD1152 treatment effectively eradicated HL-60 engraftment (Figure 3A).
Figure 3.

In vivo effect of AZD 1152 treatment on HL-60 and primary AML. One million HL-60 cells were injected i.v. into sub-lethally irradiated mice. 4 weeks later, one group of mice were treated with AZD1152. One week after treatment the level of HL-60 xenograft were compared to untreated controls (Fig 3A). Example of FACS analysis that displays human, myeloid cell content in the murine marrows. Upper panel is from AZD1152-treated mice and lower panel is from untreated mice (Fig 3B). Three bar charts that summarize the frequency of human cells in murine marrows as a percentage of total nucleated cells. The number of mice in each group is represented by N. (Fig 3C-D). Summary of the effect of one-week (Fig 3C) and two-weeks (Fig 3D) of 25mg/kg/day AZD1152 treatment on the human cell content from various AML cases.
AZD1152 rapidly and extensively reduces primary AML xenografts in vivo
In an effort to mimic the presentation of human AML, we injected 107 primary AML MNCs and allowed them to establish as xenografts for 10 weeks before treating with AZD1152 for 1 week and analyzing the human cell content at the 11 weeks time point.
Five primary human AML samples (patients' details in Table S1) were analyzed in this fashion. After administration of 25mg/kg/day of AZD1152 for one week a rapid and extensive reduction in the xenografts were observed in all the AML samples analyzed. A summary of the human cell percentage human cell content immediately after 1 week of 25mg/kg/day of AZD1152 is shown in Figure 3C.
One cycle of AZD1152 treatment is sufficient to compromise AML xenografts in vivo
The experiment that featured the largest residual AML xenograft after AZD1152 treatment (AML-1) was selected for further analysis, the results of which are summarized in Figure 3D.
Four weeks after the first cycle of AZD1152 treatment, the xenograft had not grown back (2.9%±3.8% after one pump, n=12, analyzed at 11 weeks vs. 3.8%±3.7% 4 weeks after 1st pump, n=9 at 15 weeks, p=0.512). Further treatment with a second cycle of AZD1152 for one week at 14-weeks, did not further reduce the residual xenograft percentage (1.8%±2.3% p=0.628). The cohort of mice that were implanted with primary AML cells and then left untreated for 14 weeks before analysis featured a large percentage of human cells in their marrows (36.2%±29.7%, n=4). Treatment at this 14-week time point with one cycle of AZD1152 for a period of one week reduced this xenograft percentage to levels similar to those achieved with 2 rounds of AZD1152 treatment at weeks 11 and 14 (1.2%±1.7%, n=3).
Effects of AZD1152-HQPA on primary cord blood progenitor cells in vitro
To explore the effect of AZD1152 treatment on the growth of human normal hematopoietic stem / progenitor cells, we performed an in vitro assessment with AZD1152 HPQA. Cord blood (CB) lineage-negative cells were treated with 10nM, 100nM and 1000nM AZD1152 HPQA (results summarized in Figure 4). A dose dependent effect of AZD1152-HQPA on the growth and differentiation of progenitor cells was observed after a 2-week exposure. Control (PBS-supplemented) assays yielded 250± 53 colonies/ml from 1000 cells/ml (n=4), which was reduced to 126±35 with 10nM (n=4) and 34±18 with 100nM (n=4) AZD1152-HQPA (p<0.05, Fig 4A). At 1000nM AZD1152-HQPA, no colonies were detected after 2 weeks.
Figure 4.
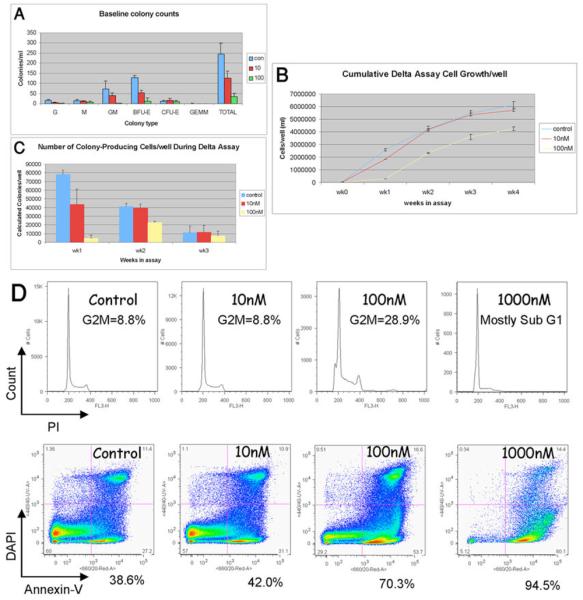
Summary of the number of progenitor colonies formed/ml of methylcellulose medium supplemented with differing concentrations of AZD1152 (Fig 4A). Summary of the accumulative cell count/well (ml) achieved for each AZD1152 condition. AZD1152 was added to the culture for the first week only (Fig 4B). Summary of the number of colonies produced each week displayed as the number of colonies produced/well per week (Fig 4C). Histogram analysis of PI fluorescence revealing the cell cycle status of CB Lin- cells after 1 week of culture in the presence of differing concentrations of AZD1152 (Fig 4D upper panel). Density-plot analysis of fluorescence due to Annexin-V and DAPI fluorescence. Displayed below each plot is the percentage of apoptotic cells (Annexin-V+/Dapi− cells) (Fig 4D lower panel).
In a similar pattern to the baseline methylcellulose assays, a concentration-dependent effect of AZD1152-HQPA on the liquid culture growth of normal hematopoietic cells was observed. At the end of the first week, the mean total cell growth from 2 delta assays performed in triplicate was 2.6×106±5.4×105 cells/well for controls, 1.8×106±5.7×104 for 10nM, 2.9×105±7.1×104 for 100nM and below the threshold of accurate counting (<104 cells) for 1000nM AZD1152-HQPA treated wells.
Surviving cells on day 7 were re-seeded and cultured for an extra 4 weeks (not performed for 1000nM AZD1152-HQPA-treated cells, as cell number was too low). As summarized in Figure 4B, surviving cells continued to proliferate at a similar rate to non-treated cells.
To examine progenitor cell output, cells from the liquid culture were seeded into methylcellulose cultures at the end of each week. The number of colonies/well was reduced by AZD1152-HQPA in a concentration-dependent manner. The production of colonies continued after AZD1152-HQPA removal, indicating the presence of surviving progenitors at weeks 2 and 3 of the assay (Figure 4C). (Colony number reduces in all conditions with time as a result of the proliferation/differentiation cocktail used in the liquid culture).
At the end of the first week, cells were also harvested for analysis of cell cycle distribution and percentage apoptosis/necrosis. In a similar pattern to the in vitro effect of AZD1152 HPQA on AML, there was a concentration-dependent effect on the proportion of cells that were either undergoing apoptosis (Annexin-V+) or that were dead (Annexin-V+, DAPI+). This cell death occurred concurrently with a modest increase in the percentage of G2/M cells and polyploid cells. Representative results of this analysis are provided in Figure 4D.
Activities of AZD1152-HQPA in primary cord blood stem and progenitor cells in vivo
To explore a possible toxic affect of AZD1152 on the growth of normal hematopoietic cells in vivo, we examined its effects on xenografts derived from human CB Lineage negative cells. We established the xenograft before treatment with AZD1152 and subsequent analysis. The first cycle of one week's treatment with AZD1152 was carried out 8 weeks after implantation. One cohort of mice was analyzed 4 weeks after this AZD1152 treatment to determine whether the cord blood xenograft could recover from AZD1152 treatment. Another cohort was given an additional cycle of AZD1152 treatment 4 weeks after the first treatment. The results of this analysis are summarized in Figure 5.
Figure 5.

In vivo effect of AZD 1152 treatment on Umbilical Cord Blood Cells. One round of AZD1152 treatment at 25mg/kg/day severely reduced the level of human CB-derived xenografts. Although when left post treatment for 5 more weeks before analysis the level of CB-derived graft was slightly higher. Further treatment with another round of 1-week AZD1152 treatment did not completely eradicate the human grafts. Statistical significance (p<0.05) is indicated by *.
The level of engraftment after 9 weeks was significantly lower in the mice that were given AZD1152 than in the control mice (9.8%±6.6% (n=4) for AZD1152-treated mice vs. 62.5%±13.9% (n=5) for control mice (p<0.005)). Four weeks later, the percentage of human cells was slightly higher in mice that had received one cycle of AZD1152 at 9 weeks, but this did not reach statistical significance (16.7%±14.1% (n=3) at 13 weeks vs. 9.8%±6.6% (n=4) at 9 weeks, p=0.39). A second cycle of AZD1152 treatment reduced xenograft levels such that they were slightly lower levels than after the first round of treatment, but this was not statistically significant (7.9%±6.4% (n=3) indicating that residual cells might be more resistant to a second cycle of AZD1152.
Discussion
The aurora kinases are emerging as potential therapeutic targets in the treatment of cancer. Although several dual (Aur-A and Aur-B) inhibitors have been described, their effects may be more dependent on the inhibition of Aur- B than that of Aur-A (3). We have therefore investigated the activity of AZD1152, a selective inhibitor of Aur-B, on AML both in vitro and in vivo.
In vitro AZD1152-HQPA was shown to effectively inhibit Aur-B activity in AML cell lines and in some human primary leukemic cells, as evidenced by complete inhibition of His H3 phosphorylation at submicromolar concentrations. In all AML cell lines studied, AZD1152-HQPA also induced a marked anti-propliferative effect accompanied by the appearance of a polyploid population, which in most cases led to apoptosis. Similar observations have been reported recently with this compound by Yang et al (14). In THP-1 cells AZD1152-HQPA had little effect on viability or apoptosis, instead inducing a senescent phenotype. In THP-1 cells a senescent response was also induced by the topoisomerase II inhibitor doxorubicin, but not by other anti-leukemic agents, suggesting a drug-specific response (data not shown). This lack of apoptosis after exposure to AZD1152-HQPA in THP-1 cells may be associated with the marked increased expression of the senescence associated TRAIL decoy receptor Dcr2, which has previously been shown to reduce sensitivity to cytotoxic agents (15).
In primary AML cells the effect of AZD1152-HQPA was mainly cytostatic, possibly due to the low proliferative rate of primary cells ex vivo.
CB Lin− cells responded to AZD1152-HQPA in vitro in a similar fashion to AML cell lines. There was a concentration-dependent toxic effect on cell cycle and cell growth that occurred alongside an increase in apoptosis and cell death. However, once AZD1152-HQPA was removed from the culture system, the surviving cord blood cells still had progenitor cell activity as illustrated by the in vitro proliferation and differentiation of lineage-restricted colony forming cells. Unfortunately, due to the poor performance of primary AML cells ex vivo, we could not perform the same comparison in AML cells. Therefore, although it is encouraging that non-malignant progenitors seem to survive AZD1152 treatment, we cannot conclude that there is a differential effect.
Once we had ascertained the effects of AZD1152-HQPA in vitro, we progressed to analyze the effects of the pro-drug AZD1152, on the growth of the AML cell line HL-60, primary AML cells and primary cord blood Lin− cells in the NOD/SCID xenotransplantation model. In all our in vivo assessments we first established xenografts before AZD1152 treatment and subsequent analysis. This approach should be a superior test to experiments where the xenograft and test drug are administered at the same time (16-18) as it better reflects the clinical situation where patients present with established leukemias.
AZD1152-HQPA had a profound, rapid and prolonged effect on the growth of HL-60 cell in vivo at the dose of 25mg/kg/day. In all five primary AML analyzed, one week of AZD1152 treatment lead to an extensive and rapid reduction of the xenograft content. Where analyzed, AML xenografts did not re-grow, suggesting that the SL-IC activity within the graft had been compromised.
There were however, small residual populations of primary AML xenografts that persisted after AZD1152 treatment and during a further round of AZD1152 treatment. Attempts at secondary engraftment from these cells were unsuccessful (data not shown), indicating that they may not be capable of repropagating the disease. However, due to the high frequency of false-negative results in secondary transplant experiments, we cannot rule out the existence of residual functional LSC populations.
Ongoing studies of the PK of AZD1152 administration in mice have revealed that at steady-state conditions, 25mg/kg/day of the pro-drug AZD1152, results in a free, unbound plasma concentration of approximately 16.9 to 18.7nM. Hence, the dose we used in vivo results in slightly higher plasma concentrations than the 10nM AZD1152 HPQA concentration we used in vitro (Astrazeneca unpublished data).
A 25mg/kg/day dose in vivo, and an in vitro 10 nM concentration was capable of inhibiting AML cell line and primary human AML cell growth in vivo and in vitro. Although, a larger proportion of cord blood-derived cells survived the same dose of AZD1152 treatment in vivo and in vitro, significant toxicity was observed; xenografts did not re-grow in 3 weeks after initial treatment and attempts at six-week secondary engraftment were unsuccessful (data not shown).
Attempts to obtain a clinically relevant therapeutic window with AZD1152 may therefore be limited by hemotoxicity. Phase 1 clinical data trial data supports this suggestion, as the dose limiting toxicity is reportedly neutropenia (20).
As a single agent, the effects of AZD1152 compare favourably with those of cytotoxic agents that have been tested on primary human AML cells grown as xenografts in NOD/SCID mice. For example, the effect of Ara-C on the NOD/SCID growth of the same number of primary AML cells as used in our own studies (107 cells) is reportedly to have a slight but not statistically significant inhibitory effect on the engraftment, after 4, 8 or 12 weeks of transplantation in NOD/SCID mice (19).
The present study describes the use of an established xenograft in the NOD/SCID model for the analysis of the efficacy of a novel anti-cancer treatment. This approach represents an advance for the pre-clinical development of novel anti-cancer treatments. A xenotransplantation model that utilizes human cells is more representative of the human disease than alternative animal models and can analyze a range of primary samples from patients with differing outcomes at different stages of the disease. In contrast, clinical trials early in the development of a novel anti-cancer compound are usually limited to heavily pre-treated patients with a poor prognosis.
In summary, we describe the anti-proliferative or cytotoxic effects of AZD1152, and the data presented from these studies supports the further clinical evaluation of AZD1152 in the treatment of AML and highlight the use of a valuable in vivo model for the preclinical investigation of novel anti-leukemic agents.
Materials and Methods
AZD1152
AZD1152 and the active moiety AZD1152-HQPA for in vitro studies were provided by AstraZeneca (Cheshire, UK).
AML cell lines and primary cell cultures
HL-60, MV411, THP-1 and U937 AML cell lines were obtained from the Cancer Research UK Cell Bank. These cell lines were cultured in RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 / 95% air. Primary AML cells were cultured in RPMI 1640 medium containing 17% heat inactivated fetal bovine serum, 2mM glutamine, penicillin and streptomycin, at 37°C in 5% CO2 / 95% air. Leukemic blast cell count in the primary sample prior to Histopaque separation was >80% for all samples.
Cord Blood and Primary AML cells
AML samples was collected from St Barts Hospital and Cord blood (CB) was collected from mothers attending the Royal London Hospital, London, UK, both after informed consent and via a protocol approved by the local Research Ethics Committees. Mononuclear cells (MNC) were obtained by Ficoll-Paque density centrifugation and ammonium chloride red cell lyzis. Density-separated cord blood MNCs were depleted for lineage marker positive cells via the StemSepTM system (Stem Cell Technologies, Canada) according to the manufacturer's instructions to generate Lin- cells.
Cell proliferation, viability and apoptosis assay
Cell number and percentage viability were determined in 96-well plate format using the Guava ViaCount assay on a Guava PCA-96 system (Guava Technologies inc), following the manufacturer's instructions. Apoptotic cells were quantified by annexin V, 7-AAD dual labelling using the Guava Nexin assay.
Cell cycle distribution
Cells were collected by centrifugation; the pellet was washed with Hanks' balanced salt solution (HBSS), re-suspended in 80% ethanol and fixed overnight at −20°C. After fixation, cells were centrifuged to remove the ethanol, washed twice with HBSS, and re-suspended in staining solution containing 50μg/ml propidium iodide (PI) and 50μg/ml RNase A in HBSS. The cellular DNA content was then analyzed within 1hr on a FACSCalibur (Becton Dickinson, UK) flow cytometer.
Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase activity was determined using the senescence cell staining kit (Sigma, UK). Briefly, cells were seeded in 6-well plates followed by 72 hr drug treatment and then cultured in the absence of drug for 7 days. Cells were washed in HBSS, fixed for 6 minutes at room temperature. After fixing, cells were washed with HBSS and stained overnight at 37°C in the X-gal staining mixture. Cells were then washed and observed under the microscope.
Immunocytochemistry
Cells were collected by centrifugation; the pellet was washed with HBSS, re-suspended in 80% ethanol and fixed overnight at −20°C. After fixation the cells were centrifuged to remove the ethanol, permeabilized in a solution of 0.2% Tween-20, washed in a solution containing 2% BSA & 0.05% sodium azide and incubated in 5μg/ml anti-phospho Histone H3 (Ser10) antibody (Upstate, UK) for 2hr. Cells were then washed and incubated with FITC conjugated secondary antibody for 1hr in the dark. After incubation, cells were washed and applied onto a microscope slide, air dried and counter stained with DAPI stain. The percentage of cells positive for pHis H3 was assessed using fluorescence microscope and/or flow cytometry using PI, pHis H3 dual labelling.
Western Blot Analysis
Cell lyzates were prepared in Triton X-100 lysis buffer and 20 μg protein (determined using the Bradford reagent (Sigma-Aldrich Co., Poole, United Kingdom)) resolved by SDS-PAGE. Gels were then electro-blotted onto nitrocellulose membranes and probed for Dcr2, p15 (Upstate Biotechnology, NY) p16 and β-actin (Calbiochem, San Diego, CA). Protein bands were visualized using an enhanced chemo luminescence visualisation system (ECL Plus, Amersham Life Sciences, Little Chalfont, UK).
Cell line colony formation assay
Cells for colony forming assay were cultured in the presence of drug for 72hr. Surviving cells were then counted, washed and resuspended in drug-free media. 2 ×104 cells in 1.3% methylcellulose based media (R&D systems, UK) were then plated onto a 35mm culture dish and incubated for 9 days at 37°C in a humidified atmosphere of 5% CO2 / 95% air. At the end of the incubation period the numbers of colonies formed were counted under the microscope.
Xenotransplantation assays
All animal experiments were performed in compliance with Home Office and institutional guidelines. NOD/SCID mice were originally obtained from Dr Leonard Schultz (Jackson Laboratory, Bar Harbour, ME, USA) and bred at Charles Rivers Laboratories, London, UK. Mice aged 8-12 weeks were irradiated at 375 rads (137Caesium source) up to 24 hours before intravenous injection of human cells.
For analysis, the femurs, tibias and pelvis were dissected and flushed with PBS. Red blood cells were lyzed using ammonium chloride. Cells were stained with human specific FITC-conjugated anti-CD19, PE-conjugated anti-CD33 and PE-Cy5-conjugated anti-CD45 antibodies (all from Pharmingen, San Diego, CA). Dead cells and debris were excluded via DAPI (Sigma) staining. A BD LSR flow cytometer (BD Biosciences, Oxford, UK) was used for analysis. More than 100,000 DAPI negative events were collected.
AZD1152 was administered in vivo by the use of subcutaneous Alzet™ osmotic pumps according to the manufacturer's instructions (model 2001, http://www.alzet.com/). Pumps were weighed at the end of AZD1152 treatment to confirm delivery of the full dose. Treatment regimens are summarized in supplementary Figure S3.
Murine cell phenotyping
Antibody labelling was performed in PBS, 2%FCS, 10mM HEPES for 30 minutes with appropriate matched-isotype controls. The following antibodies were used (all from Pharmingen, San Diego, CA, USA): FITC conjugated anti-Thy-1, CD31 and CD34; PE conjugated anti-CD2, CD3, CD4, CD8, CD11b, CD135, B220, Ter-119, GR-1 and NK1.1; biotinylated anti-SCA-1; and APC conjugated anti-CD117. The lineage cocktail used throughout contained PE-conjugated anti-CD5, CD11b, B220, Gr-1 and Ter-119 antibodies.
Primary cell progenitor assays
To assay human primary hematopoietic progenitors, Lin− cells were resuspended in 300μl of IMDM and added to 2700μl of MethoCult H4434 (Stem Cell Technologies, Canada). Cultures were plated in duplicate. Plates were incubated at 37°C, 5% C02 for 14 days and colonies were scored according to standard criteria.
Delta assay
5×104 Lin− cells were seeded in 1ml of IMDM 10% FCS supplemented with SCF, IL-3, IL-6, EPO and G-CSF. Cells were incubated at 37°C in 5% CO2 for one week. At the end of the first week, cells were enumerated, tested for methylcellulose progenitor assays (104/ml), PI cell cycle analysis and annexin-V analysis. Each week, 5×104 cells were seeded back into the assay for re-growth in cytokine-supplemented IMDM 10% FCS as before. At weeks 2 and 3 of the assay, 3×104 cells/ml and 1×105 cells/ml respectively were plated in methylcellulose.
Annexin-V labelling
Annexin V labelling was used to quantify the effects of AZD1152 on apoptosis. 100μl of 10× Annexin V binding buffer (BD Pharmingen, San Jose, CA, USA) was added to 900μl of the resuspended cells and mixed. Then 5μl of directly conjugated Annexin V Alexa Fluor® 647 (Molecular Probes, Leiden, The Netherlands) was added to the cells before incubation at 37°C for 15 minutes. DAPI was added to the cells, as above, prior to analysis on a BD LSR-2 flow cytometer.
Statistics
The student's paired t-test was used to compare the effects of different treatments.
Supplementary Material
Acknowledgements
This work would not have been possible without the assistance of staff of both the Biological Research Unit and the Flow cytometry Unit at Cancer Research UK. We thank Nicholas Keen, Kate Byth, Jim Growcott, Kirsten Mundt and Jane Robertson (AstraZeneca) for helpful discussions.
References
- 1.Katayama H, Sasai K, Kawai H, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet. 2004;36:55–62. doi: 10.1038/ng1279. [DOI] [PubMed] [Google Scholar]
- 2.Meraldi P, Honda R, Nigg EA. Aurora kinases link chromosome segregation and cell division to cancer susceptibility. Curr Opin Genet Dev. 2004;14:29–36. doi: 10.1016/j.gde.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Crosio C, Fimia GM, Loury R, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–85. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams RR, Carmena M, Earnshaw WC. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol. 2001;11:49–54. doi: 10.1016/s0962-8924(00)01880-8. [DOI] [PubMed] [Google Scholar]
- 5.Tang CJ, Lin CY, Tang TK. Dynamic localization and functional implications of Aurora-C kinase during male mouse meiosis. Dev Biol. 2006;290:398–410. doi: 10.1016/j.ydbio.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 6.Manfredi MG, Ecsedy JA, Meetze KA, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ditchfield C, Johnson VL, Tighe A, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–80. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girdler F, Gascoigne KE, Eyers PA, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–75. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 9.Harrington EA, Bebbington D, Moore J, et al. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10:262–7. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 10.Wilkinson RW, Odedra R, Heaton SP, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res. 2007;13:3682–8. doi: 10.1158/1078-0432.CCR-06-2979. [DOI] [PubMed] [Google Scholar]
- 11.Jung FH, Pasquet G, Lambert-van der Brempt C, et al. Discovery of novel and potent thiazoloquinazolines as selective Aurora A and B kinase inhibitors. J Med Chem. 2006;49:955–70. doi: 10.1021/jm050786h. [DOI] [PubMed] [Google Scholar]
- 12.Mortlock AA, Foote KM, Heron NM, et al. Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem. 2007;50:2213–24. doi: 10.1021/jm061335f. [DOI] [PubMed] [Google Scholar]
- 13.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 14.Yang J, Ikezoe T, Nishioka C, et al. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007;110:2034–40. doi: 10.1182/blood-2007-02-073700. [DOI] [PubMed] [Google Scholar]
- 15.Liu X, Yue P, Khuri FR, Sun SY. Decoy receptor 2 (DcR2) is a p53 target gene and regulates chemosensitivity. Cancer Res. 2005;65:9169–75. doi: 10.1158/0008-5472.CAN-05-0939. [DOI] [PubMed] [Google Scholar]
- 16.Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–9. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guzman ML, Swiderski CF, Howard DS, et al. Preferential induction of apoptosis for primary human leukemic stem cells. Proc Natl Acad Sci U S A. 2002;99:16220–5. doi: 10.1073/pnas.252462599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hogge DE, Feuring-Buske M, Gerhard B, Frankel AE. The efficacy of diphtheria-growth factor fusion proteins is enhanced by co-administration of cytosine arabinoside in an immunodeficient mouse model of human acute myeloid leukemia. Leuk Res. 2004;28:1221–6. doi: 10.1016/j.leukres.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Schellens JH, Boss D, Witteveen PO, Zandvliet A, Beijnen JH, Voogel-Fuchs M, Morris C, Wilson D, Voest EE. Phase I and pharmacological study of the novel aurora kinase inhibitor AZD1152. Journal of Clinical Oncology. 2006;24-18S:3008. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.