

Abstract
Rapid treatment (1 min) of rat striatal synaptosomes with low-dose amphetamine increases surface expression of the dopamine transporter (DAT). Using mouse neuroblastoma N2A cells, stably transfected with green fluorescent protein-DAT, we demonstrate the real-time substrate-induced rapid trafficking of DAT to the plasma membrane using total internal reflection fluorescence microscopy (TIRFM). Both the physiological substrate, dopamine, and amphetamine began to increase surface DAT within 10 s of drug addition and steadily increased surface DAT until removal 2 min later. The substrate-induced rise in surface DAT was dose-dependent, was blocked by cocaine, and abated after drug removal. Although individual vesicle fusion was not visually detectable, exocytosis of DAT was blocked using both tetanus neurotoxin and botulinum neurotoxin C to cleave soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins. Notably, the dopamine-induced increase in surface DAT was cocaine-sensitive but D2-receptor independent. TIRFM data were confirmed in human DAT-N2A cells using biotinylation, and similar effects were detected in rat striatal synaptosomes. A specific inhibitor of protein kinase C-β blocked the substrate-mediated increase in surface DAT in both DAT-N2A cells and rat striatal synaptosomes. These data demonstrate that the physiological substrate, dopamine, and amphetamine rapidly increase the trafficking of DAT to the surface by a mechanism dependent on SNARE proteins and protein kinase C-β but independent of dopamine D2 receptor activation. Importantly, this study suggests that the reuptake system is poised to rapidly increase its function during dopamine secretion to tightly regulate dopaminergic neurotransmission.
Introduction
Dopaminergic neurotransmission is crucial for normal physiological functions as well as neurological and psychiatric diseases including drug addiction (Giros and Caron, 1993) and is regulated by the level of synaptic dopamine. Extracellular dopamine is removed from the synapse by uptake into the nerve terminal through the Na+/Cl−-dependent dopamine transporter (DAT). The psychostimulant amphetamine is a DAT substrate which, when taken up into the nerve terminal, causes reversal of DAT, resulting in dopamine efflux, whereas cocaine blocks reuptake of dopamine.
Approximately 30–40% of total DAT is located on the plasma membrane of heterologous cells (Little et al., 2002; Loder and Melikian, 2003). Because surface DAT expression shapes dopaminergic neurotransmission, DAT trafficking can efficiently regulate neurotransmission. DAT undergoes both constitutive and substrate-mediated trafficking which alter its function (Kahlig and Galli, 2003; Melikian, 2004). Substrate-induced DAT trafficking has, until recently, focused on demonstrations of DAT internalization after prolonged, higher-dose substrate treatment. Both dopamine, the physiological substrate, and amphetamine induce DAT internalization in heterologous cells, Xenopus laevis oocytes, and striatal synaptosomes after 40–60 min of exposure (Saunders et al., 2000; Gulley et al., 2002; Chi and Reith, 2003). Similarly, a high-dose amphetamine injection reduced DAT function in rat striatum (Fleckenstein et al., 1999). Of interest, however, is how substrates alter DAT trafficking at times commensurate with their initial action at the transporter. To that end, our lab demonstrated rapid amphetamine-induced DAT trafficking to the surface at times corresponding to amphetamine-stimulated dopamine efflux. A 1 min treatment of rat striatal synaptosomes with 3 μm amphetamine induced a significant increase in DAT cell surface expression (Johnson et al., 2005b). By 30 min of treatment, DAT surface expression was reduced. Importantly, this rapid amphetamine-induced increase in DAT cell surface expression functionally enhanced amphetamine-induced dopamine efflux.
Although biotinylation and confocal microscopy are commonly used to monitor trafficking, they are limiting in temporal and spatial resolution, respectively. Investigation of plasma membrane processes is aided by live-cell imaging using total internal reflection fluorescence microscopy (TIRFM), which provides real-time resolution coupled with the ability to sensitively detect and analyze cytosol to plasmalemmal membrane movement of vesicles and granules. In the present study, we investigated, using the high temporal and spatial resolution available in TIRFM, the dynamics of DAT trafficking to the plasma membrane. We found that within seconds of addition of amphetamine or, notably, the physiological substrate dopamine, there were changes in surface expression of DAT with maximal increases within 1 min. The rapid increase in DAT was blocked by transfection of the light chains of botulinum neurotoxin C (BoNT C) and tetanus neurotoxin (TeNT), which cleave t- and v-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, respectively, and prevent vesicle fusion. The substrate-induced increase in DAT surface expression was modulated by a protein kinase C-mediated pathway and was independent of D2 receptor function. These data suggest a substrate-mediated trafficking mechanism which directly and rapidly regulates DAT surface expression to increase dopamine clearance.
Materials and Methods
Materials.
d-Amphetamine sulfate, dopamine, GBR12935, quinpirole, and sulpiride were purchased from Sigma. LY379196 was a generous gift from Eli Lilly and Company.
Generation of green fluorescent protein-DAT cDNA.
Rat dopamine transporter with a fluorescent tag [green fluorescent protein (GFP)-DAT] was created by fusing the coding region of enhanced GFP (EGFP) from pEGFP-C2 vector (Clontech) to the N terminus of the rat DAT cDNA between EcoRI and KpnI sites in the multicloning site of the vector.
Cell culture and stable cell lines.
GFP-DAT cDNA was transiently transfected into the N2A cell line using the Lipofectamine Plus reagent kit (Invitrogen). A stable cell line was generated through selection with Geneticin (Invitrogen) over several weeks. Stable human DAT N2A cells were a generous gift from Dr. Karley Little (University of Michigan, VA Hospital, Ann Arbor, MI). N2A cell lines were grown in Opti-MEM I supplemented with 10% bovine growth serum, 1% penicillin/streptomycin, and 400 μg/ml geneticin for stable maintenance. D2 receptor (D2R) short form DNA was kindly provided by Dr. Roger Sunahara (University of Michigan, Ann Arbor, MI) and was transiently transfected into GFP-DAT N2A cells using the Lipofectamine Plus reagent kit. In control cells, empty vector DNA was transiently transfected. Forty-eight hours after transfection, TIRFM experiments were performed. In experiments using neurotoxins, N2A cells were transiently cotransfected with GFP-DAT and the light chain form of TeNT or the light chain form of BoNT C. Control cells were cotransfected with GFP-DAT and empty vector.
TIRFM image acquisition and perfusion experiments.
GFP-DAT N2A cells were plated onto No. 1.5 glass coverslips coated with 0.5 mg/ml poly-d-lysine (Sigma). Coverslips were placed into a closed chamber (Harvard Apparatus) and Kreps–Ringer HEPES (KRH) buffer composed of the following (in mm): 25 HEPES, 125 NaCl, 4.8 KCl, 1.2 KH2PO4, 1.3 CaCl2, 1.2 MgSO4, and 5.6 glucose was added to cover the glass. Microperfusion of individual cells was performed at ∼27°C using a six-chamber perfusion apparatus equipped with a quartz pipette (inner diameter, 0.2 μm) that was positioned near the cell being imaged. Cells were continuously perfused throughout image collection with KRH buffer or various drugs dissolved in KRH using a computer-controlled perfusion apparatus (model DAD-6VM; ALA Scientific Instruments). Total internal reflection fluorescence was achieved using methods described previously with minor changes (Allersma et al., 2006). Prismless (through-the-objective) TIRFM was obtained using an Argon ion laser (488 line; Melles Griot) model 35-LAP-431-208 directed through a custom side port to a side-facing dichroic mirror Q495LPw/AR (Chroma Technology) and a HQ500 LP emission filter (Chroma Technology) on an Olympus IX70 (inverted) microscope (Olympus). The beam was focused on the periphery of the back focal plane of an oil-immersion objective (60× 1.49 numerical aperture; Olympus). The laser beam was incident on the coverslip at an angle that resulted in an evanescent field with a decay constant of ∼100 nm. Digital images were captured using a high-sensitivity charge-coupled device (CCD) camera (Andor iXon+, EM-CCD, BV, 512 × 512 pixels). Images were acquired at 0.5 Hz with exposure times of 50–100 ms. In general, cells were perfused with three solution changes: 120 s of KRH or test drug, 120 s of DAT agonist with or without test drug, and 60 s of KRH (150 frames total). For details, see individual experiments. For all TIRFM experiments, the first 100 s of baseline perfusion are omitted in the graphs.
TIRFM image quantification.
Using the ImageJ program (National Institutes of Health software), each cell was traced and analyzed for the mean fluorescence intensity over 150 frames. Background values were obtained by measuring a blank region from each frame and taking the average background of the frames. Data are plotted as mean arbitrary fluorescent units (AFU): mean cell intensity minus average background intensity. Cells are normalized to 100 AFU at t = −30 s before DAT agonist addition. Statistical significance was determined by two-way ANOVA using Prism 5 or by comparison of individual time points with buffer controls using two-tailed Student's t test.
Biotinylation of human DAT-N2A cells.
Cells were washed with KRH and treated with the substrates amphetamine or dopamine for 1 min at 37°C. In some cases, a pretreatment of 200 nm LY379196 was given before substrate treatment (see figure legends for detail). The reaction was quenched using excess 4°C PBS/Ca/Mg. All remaining steps were performed at 4°C to prevent further trafficking. Cells were washed with PBS/Ca/Mg, and surface expression of DAT was determined by reacting the surface proteins with 1.5 mg/ml sulfo-NHS-SS-biotin [sulfosuccinimidyl-2-(biotinamido)-ethyl-1,3-dithiopropionate; Pierce] at 4°C with constant rotation. Excess biotin was quenched by incubating cells with 100 mm glycine, followed by two washes of 100 mm glycine in PBS/Ca/Mg buffer. Cells were lysed in solubilization buffer (in mm: 25 Tris, 150 NaCl, 1 EDTA, 5 N-ethylmalemeide, phenylmethylsulfonyl fluoride, and 1% Triton X-100) containing a fresh protease inhibitor mixture tablet (Roche) and centrifuged at high speed to obtain soluble protein. Cells were analyzed for protein content, and ∼100–200 μg protein was reacted with streptavidin beads (Pierce) to isolate surface protein. Streptavidin beads were washed 3× with solubilization buffer and protein was eluted in sample buffer composed of 250 mm Tris, pH 6.8, 25 mm EDTA, 10% SDS, 25% sucrose, 0.5% bromophenol blue supplemented with 100 mm dithiothreitol. A portion of protein (∼10–20 μg) was saved for total DAT measurement (lysate). Surface and total DAT samples were resolved using SDS-PAGE (10–12% Tris-glycine gel). Western blot analysis was performed using a monoclonal anti-DAT antibody (MAB369; Millipore Bioscience Research Reagents). Protein was detected using West Pico (Pierce; biotin) or Amersham (lysate) electrochemiluminescence detection. Western blot bands were quantified using Scion Image software. Data are expressed as the optical density (OD) units of the biotinylated fraction/OD of the lysate.
Quinpirole-induced dopamine uptake.
Human DAT-N2A cells were treated with either KRH or 10 μm quinpirole for 15 min at 37°C. [3H]dopamine (30 nm, 34.4 μCi/μl; Perkin-Elmer) plus unlabeled dopamine was added to cells to initiate dopamine uptake in the presence or absence of the DAT blocker 10 μm GBR12935 to determine nonspecific binding. After 5 min of incubation, cells were filtered onto glass fiber C Whatman filters and washed three times with excess cold PBS. Filters were dried and counted on a Beckman scintillation counter.
Biotinylation of rat striatal synaptosomes.
Synaptosomes were prepared as described previously (Johnson et al., 2005b). Biotinylation of rat striatal synaptosomes was performed similarly in human DAT N2A cells but were conducted in KRB (in mm): 145 NaCl, 2.7 KCl, 1.2 KH2PO4, 1.2 CaCl2, 1 MgCl2, 10 glucose, 0.255 ascorbic acid, 24.9 NaHCO3. Synaptosomes were pretreated with KRB, 3 μm amphetamine, or 10 μm dopamine at 37°C (amphetamine) or room temperature (dopamine) for 1 min. To test the role of PKC-β, synaptosomes were incubated with 100 nm LY379196 added with the amphetamine. Reactions were stopped by adding 4°C PBS/Ca/Mg buffer and washed once with PBS/Ca/Mg to remove residual drugs. Synaptosomes were lysed in radioimmunoprecipitation assay/EDTA buffer (10 mm Tris, pH 7.4, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate and protease inhibitors). Surface DAT expression was determined as described above for human DAT-N2A cells. Immunoblot analysis was performed using MAB16, monoclonal anti-DAT antibody generously supplied by Dr. Roxanne Vaughan (University of North Dakota, Grand Forks, ND).
Amphetamine-stimulated dopamine efflux after amphetamine pretreatment.
Synaptosomes were pretreated with KRB, 3 μm amphetamine, 100 nm LY379196 or 100 nm LY379196 plus 3 μm amphetamine for 1 min at 37°C and then washed three times to remove all residual amphetamine. Amphetamine-stimulated dopamine efflux was performed in resuspended synaptosomes as described previously (Johnson et al., 2005b). Briefly, the resuspended synaptosomes were separated into two portions and incubated with either KRB or 3 μm amphetamine for 5 min at 37°C. The reactions were stopped by filtering the samples through a 0.2 mm syringe into an internal standard solution (ISS) containing 0.05 m HClO4, 4.55 mm dihydroxybenzylamine, 1 m metabisulfate, and 0.1 m EDTA. Dopamine was measured in the eluate by HPLC with electrochemical detection. Total dopamine was measured by lysing a portion of synaptosomes in ISS. Dopamine efflux was calculated as the percentage of total dopamine in the synaptosomes (dopamine in eluate/total dopamine in synaptosomes) after the pretreatment.
Statistical analysis was performed using Graph Pad Prism 5.
Results
Amphetamine increases surface DAT levels in DAT-N2A cells
Earlier biochemical studies demonstrated an increase in DAT surface expression within 1 min of amphetamine treatment in rat striatal synaptosomes using biotinylation (Johnson et al., 2005b). In this study, real-time DAT trafficking was visually monitored in live N2A neuroblastoma cells stably expressing GFP-DAT. We used TIRFM to analyze the spatial and temporal resolution of rapid DAT trafficking. TIRFM revealed that amphetamine had similar effects in GFP-DAT N2A cells as in synaptosomes. Individual cells were perfused with KRH for 120 s (baseline) followed by 120 s in the absence or presence of amphetamine and lastly with KRH for 60 s (washout). Figure 1 (top) shows a representative cell under KRH perfusion 20 s before amphetamine addition (−20 s) and 0, 60, and 120 s after amphetamine perfusion. Hot spots reflect high amounts of GFP-DAT and/or regions of the plasma membrane especially close to the glass interface. Fluorescence was stable for at least 90 s before perfusion with amphetamine. During perfusion with 5 μm amphetamine, GFP-DAT intensity increased, shown at 60 and 120 s after drug application in Figure 1. A supplemental movie of this cell is available in supplemental materials, available at www.jneurosci.org. Amphetamine addition increased GFP-DAT intensity in a dose-dependent manner. The curves were statistically different when analyzed by two-way ANOVA (see legend to Fig. 1). In post hoc Bonferroni testing, perfusion with 1 μm amphetamine significantly increased GFP-DAT intensity by 60 s of treatment, whereas a significant increase was attained after 42 s of perfusion with 5 μm amphetamine. GFP-DAT intensity remained significantly elevated with 5 μm amphetamine for another 60 s after washout by KRH, whereas 1 μm amphetamine treatment was not significantly different between KRH and 1 μm amphetamine just 18 s after washout.
Figure 1.
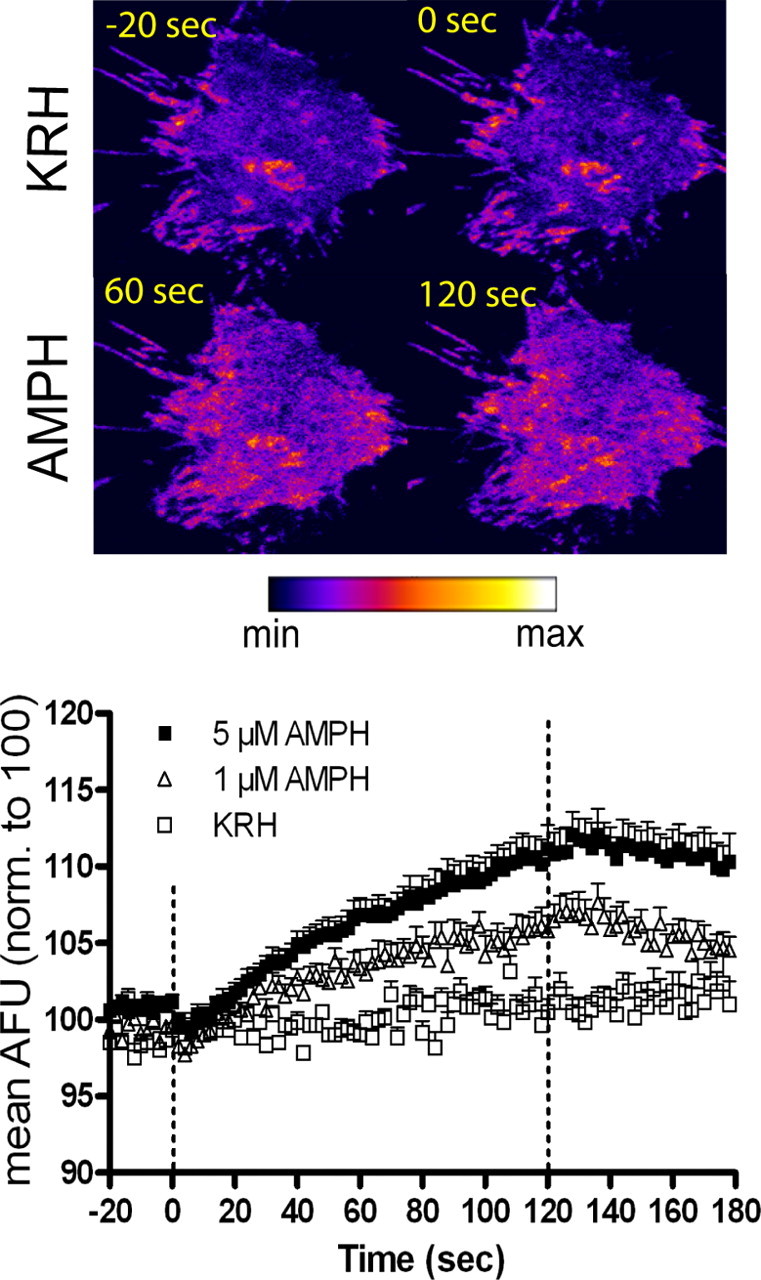
Rapid amphetamine (AMPH)-induced DAT trafficking to the plasma membrane: TIRFM analysis. Top, Pseudocolored images of a representative GFP-DAT N2A cell perfused with KRH for 120 s followed by 5 μm AMPH for 120 s. Images show cell under KRH perfusion 20 s before addition of AMPH (−20 s) and at 0, 60, and 120 s after addition of 5 μm AMPH. For movie, see supplemental Movie 1, available at www.jneurosci.org as supplemental material. Bottom, ImageJ quantitation of AMPH-induced increases in GFP-DAT pixel intensity. Cells were treated with KRH for 120 s (20 s before AMPH addition shown), followed by KRH (n = 5), 1 μm AMPH (n = 11), or 5 μm AMPH (n = 13) for 120 s. Data are expressed as mean AFU normalized to 100 AFU. Dashed lines represent addition of KRH or AMPH at 0 s, and perfusion is switched back to KRH at 120 s. Error bars represent SEM. p < 0.001 for dose and p < 0.0001 for time and interaction of dose and time: F(208,2808) = 6.61 by two-way ANOVA. In post hoc Bonferroni testing, statistical significance (p < 0.05) is achieved from 60 to 138 s with 1 μm AMPH and from 42 to 180 s with 5 μm AMPH compared with KRH values.
To confirm that the increase in fluorescence in TIRFM reflected increased DAT insertion into the plasma membrane, we measured surface expression by biotinylation. Biotinylation of DAT-N2A cells, as previously shown in rat synaptosomes, demonstrated an amphetamine-induced increase in DAT trafficking to the plasma membrane. Incubation with 3 μm amphetamine for 60 s at 37°C resulted in surface DAT that was 150% of control (KRH treatment; p < 0.01 by paired two-tailed t test; n = 4). These data temporally correlate with the TIRFM studies which demonstrate an increase in GFP-DAT intensity within 40–60 s of amphetamine perfusion.
To determine whether substrate transport was required for substrate-induced DAT trafficking, biotinylation experiments using a buffer absent of sodium were performed. When N-methyl-O-glucamine chloride was substituted for NaCl during the 1 min treatment of amphetamine or dopamine, no increase in DAT surface expression was detected (data not shown).
The physiological substrate dopamine increases surface DAT levels in DAT-N2A cells
The ability of the substrate, amphetamine, to rapidly increase DAT trafficking to the plasma membrane raised the possibility that the normally occurring substrate, dopamine, could have the same effect. Indeed, dopamine (10 μm) increased surface DAT with similar kinetics and efficacy as amphetamine. Figure 2 (top) shows a representative cell under KRH perfusion 20 s before dopamine addition (−20 s) and 0, 60, and 120 s after dopamine perfusion. Perfusion with 10 μm dopamine increased GFP-DAT intensity, shown at 60 and 120 s after dopamine (Fig. 2, top). In post hoc analysis of two-way ANOVA (see legend to Fig. 2), dopamine significantly increased GFP-DAT intensity within 80 s of treatment, although a noticeable increase can be seen ∼40 s after drug perfusion. After washout of dopamine, after 120 s of treatment, the intensity decreases to control (KRH) levels within 34 s of KRH addition. Comparison of the washout effects of dopamine with those of amphetamine suggests that amphetamine has the longer-lasting effect on DAT surface expression.
Figure 2.
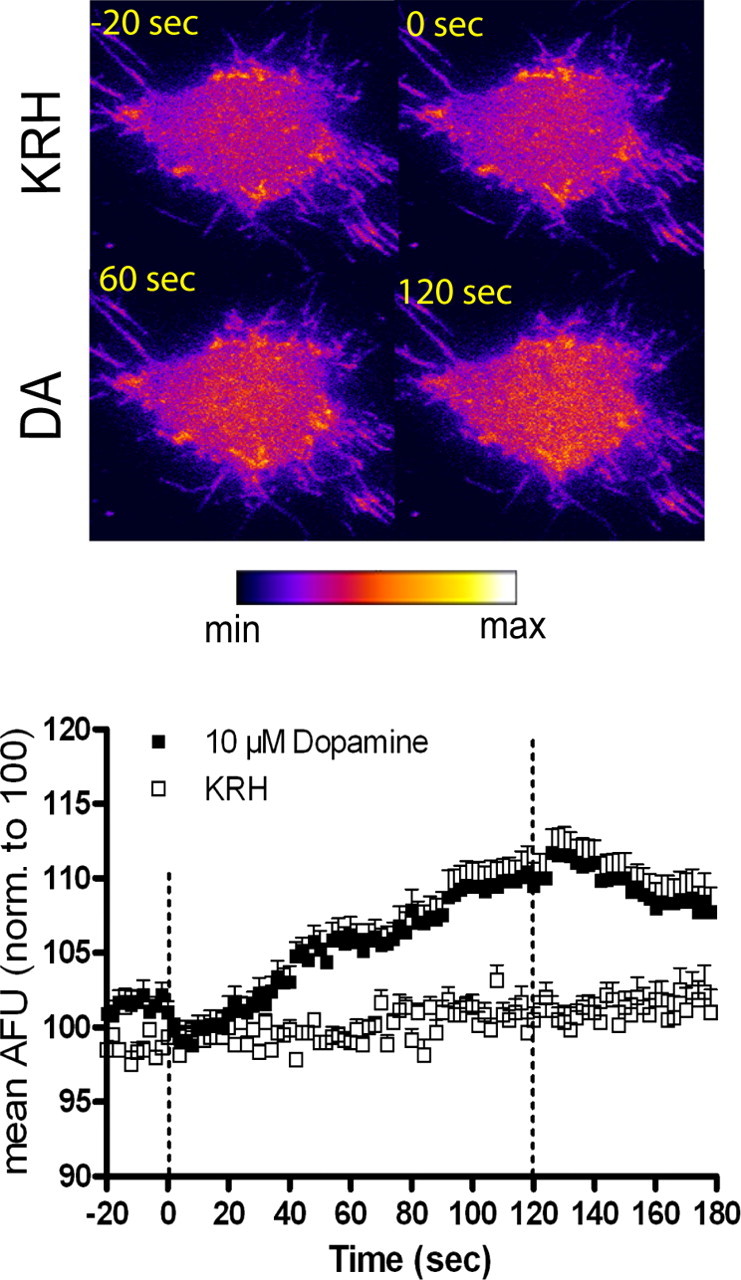
Rapid dopamine (DA)-induced DAT trafficking to the plasma membrane: TIRFM analysis. Top, Pseudocolored images of a representative GFP-DAT N2A cell perfused with KRH for 120 s followed by 10 μm DA for 120 s. Images show cell under KRH perfusion 20 s before addition of DA (−20 s) and at 0, 60, and 120 s after addition of 10 μm DA. Bottom, ImageJ quantitation of DA-induced increases in GFP-DAT pixel intensity. Cells were treated with KRH for 120 s (20 s before DA addition shown), followed by KRH (n = 5) or 10 μm DA (n = 14) for 120 s. Data are expressed as mean AFU normalized to 100 AFU. Dashed lines represent addition of KRH or DA at 0 s, and perfusion is switched back to KRH at 120 s. Error bars represent SEM. p < 0.01 for drug and p < 0.0001 for time and interaction of drug and time: F(104,1976) = 6.73 by two-way ANOVA. In post hoc Bonferroni testing, statistical significance (p < 0.05) is achieved from 80 to 154 s with 10 μm DA compared with KRH values.
Because dopamine-induced DAT trafficking had not been previously demonstrated, we confirmed that DAT was physically being inserted into the plasma membrane using biotinylation. Biotinylation studies showed that 10 μm dopamine treatment resulted in surface DAT that was 250% of control (p < 0.05 by paired two-tailed t test; n = 5).
Amphetamine and dopamine increase GFP-DAT intensity through a DAT-dependent mechanism
To establish whether an interaction with DAT was required for the substrate-induced increase in GFP-DAT intensity, cells were pretreated with 30 μm cocaine for 2 min before addition of amphetamine or dopamine. For simplicity, data are presented at three time points: immediately before the addition of substrate (0 time) and 60 and 120 s after addition of substrate. At the 0 time, cells have been incubated for 2 min with either KRH or 30 μm cocaine. As shown in Figure 3, A and B, cocaine itself had no effect on surface DAT intensity. However, cocaine effectively blocked the increases in surface DAT fluorescence elicited by 5 μm amphetamine (Fig. 3A) or 10 μm dopamine (Fig. 3B) at 60 and 120 s.
Figure 3.
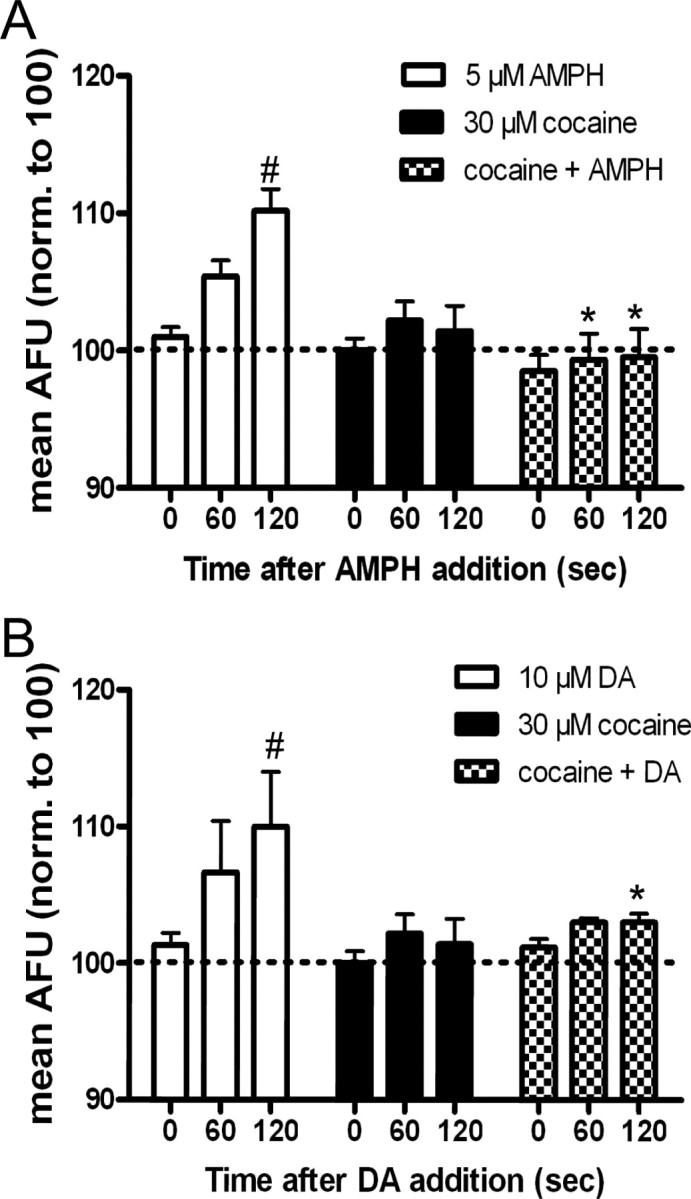
Cocaine blocks amphetamine (AMPH)- and dopamine (DA)-induced increases in GFP-DAT intensity in TIRFM. A, Cells were perfused for 2 min with KRH (white and black bars) or 30 μm cocaine (checkered bars) before addition of 5 μm AMPH (white bars; n = 5), 30 μm cocaine (black bars; n = 5), or 30 μm cocaine plus 5 μm AMPH (checkered bars; n = 5) at t = 0. Data are expressed as mean AFU and are normalized to 100 AFU (dashed line). Error bars represent SEM. Comparing across three drug groups, p < 0.0001 for drug and p < 0.05 for time by two-way ANOVA. *p < 0.05 by post hoc Bonferroni for 5 μm AMPH versus cocaine plus AMPH at t = 60 s and *p < 0.001 for 5 μm AMPH versus cocaine plus AMPH at t = 120 s. Comparing time points within each group, #p < 0.001 by two-way ANOVA: post hoc Bonferroni for 5 μm AMPH at t = 0 s versus 120 s. B, Cells were pretreated for 2 min with KRH (white and black bars) or 30 μm cocaine (checkered bars) before addition of 10 μm DA (white bars; n = 3), 30 μm cocaine (black bars; n = 5), or 30 μm cocaine plus DA (checkered bars; n = 5) at t = 0. Comparing across three drug groups, p < 0.01 for drug and p < 0.05 for time by two-way ANOVA. *p < 0.05 by post hoc Bonferroni for 10 μm DA versus cocaine plus DA at t = 120 s. Comparing time points within each group, #p < 0.05 by two-way ANOVA: post hoc Bonferroni for 10 μm DA at t = 0 versus 120 s.
SNARE proteins regulate GFP-DAT trafficking to the plasma membrane
Although monoamine transporter recycling to and from the membrane has been demonstrated, few studies have focused on the exocytosis of monoamine transporters. To determine whether DAT was undergoing exocytosis during substrate treatment, we used the specific neurotoxins, BoNT C and TeNT, which are known to proteolyze the SNARE syntaxin 1A and vesicle-associated membrane protein (VAMP)/synaptobrevin, respectively. Cells were transiently cotransfected with GFP-DAT and the light chain of either BoNT C or TeNT. All cells were perfused with KRH for 2 min followed by 10 μm dopamine for 2 min. As shown in Figure 4, in cells transfected with either BoNT C (gray bars) or TeNT (black bars), dopamine-induced DAT trafficking to the plasma membrane was significantly abrogated compared with cells transfected with vector alone. At no time did dopamine significantly increase the DAT intensity over the 0 time point in neurotoxin-treated cells. These data indicate that the substrate-induced increase in GFP-DAT intensity is an exocytotic event which is regulated by SNARE proteins.
Figure 4.

SNARE proteins regulate dopamine (DA)-induced DAT trafficking to the plasma membrane. N2A cells were cotransfected with GFP-DAT and empty vector (white bars; n = 13), the light chain of BoNT C (gray bars; n = 15), or the light chain of TeNT (black bars; n = 10). All cells were perfused for 2 min with KRH before addition of 10 μm dopamine for 2 min at t = 0. Data are expressed as mean AFU and are normalized to 100 AFU (dashed line). Error bars represent SEM. p < 0.05 for interaction and p < 0.0001 for time and treatment by two-way ANOVA. Post hoc Bonferroni analysis: *p < 0.01 for vector versus BoNT C at t = 120 s, p < 0.05 for vector versus TeNT at t = 60 s and p < 0.001 for vector versus TeNT at t = 120 s. Comparing time points within each group, #p < 0.05 for vector at t = 0 versus 60 s and p < 0.001 at t = 0 versus 120 s. There was no significant difference between any time points in BoNT C- or TeNT-treated cells.
Dopamine increases surface DAT through a D2R-independent mechanism
The experiments involving dopamine suggest that physiological levels of synaptic dopamine could interact with the DAT to rapidly regulate synaptic levels of dopamine. Rapid increases in surface DAT in response to dopamine D2R activation, however, have been demonstrated in human embryonic kidney cells coexpressing D2R-short and human DAT (Bolan et al., 2007). It is unlikely that the rapid increase in surface DAT in our experiments is mediated by D2Rs because the dopamine-stimulated increase was blocked by 30 μm cocaine. Nonetheless, we felt it important to examine whether any endogenous D2Rs in the GFP-DAT-N2A cells could be mediating the effect of dopamine. We could not unambiguously prove the presence or absence of D2Rs in the N2A cells using immunoblotting. A Western blot of N2A cell lysate with a D2 receptor antibody revealed a very faint band after overexposure (data not shown), suggesting there could be a small number of D2Rs in these cells. If there are D2Rs present, they appear not to be functionally coupled to dopamine transport. There was no increase in [3H]dopamine uptake in response to the D2R agonist quinpirole (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In addition, we pretreated GFP-DAT-N2A cells with 10 μm sulpiride, a D2R antagonist, for 2 min followed by 2 min of 10 μm dopamine and examined fluorescent intensity using TIRFM. Sulpiride alone did not affect GFP-DAT intensity and did not block the dopamine-induced increase when given simultaneously with dopamine (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Because previous experiments in DAT-N2A cells overexpressing D2Rs demonstrated a D2R-dependent increase in DAT activity (Bolan et al., 2007), we performed TIRFM experiments with cells overexpressing short form D2Rs, D2R-S. We confirmed surface presentation of D2Rs by performing binding assays with the D2R antagonist [3H]-spiperone (data not shown). TIRFM analysis demonstrated that there was no difference in the dopamine-induced increase in DAT surface expression between GFP-DAT cells transiently transfected with D2 receptors versus control cells transiently transfected with empty vector (supplemental Table 1, available at www.jneurosci.org as supplemental material). These data, combined with the lack of quinpirole-induced increase in [3H]dopamine uptake and the blockade of the dopamine effect by cocaine, demonstrate that the dopamine-stimulated rapid DAT trafficking is D2 receptor independent in the N2A cells within our experimental time frame.
Because the concept of a dopamine-induced, DAT-mediated increase in surface DAT is novel, we examined whether this could be demonstrated in a more physiologically relevant system, rat striatal synaptosomes. In a previous study, Johnson et al. (2005b) found no increase in surface DAT after treatment of rat synaptosomes with 30 μm dopamine at 37°C. At present, we chose to perform the experiments at room temperature, mimicking the conditions in which the TIRFM experiments were performed. Rat striatal synaptosomes were treated with KRB or 10 μm dopamine for 1 min, and surface DAT was measured by biotinylation. As shown in Figure 5, there was a significant 30% increase in surface DAT compared with vehicle after dopamine treatment that was blocked by simultaneous treatment with 1 μm of the DAT blocker, GBR12935. In some experiments, GBR12935 appeared to reduce surface DAT, but no significant difference between GBR12935- and KRB-treated cells was found.
Figure 5.

Dopamine (DA)-induced DAT trafficking in rat striatal synaptosomes. Top, Representative Western blot of biotinylated (surface) and lysate (total) of rat synaptosomes treated for 1 min with KRB, 10 μm DA, 1 μm GBR12935 (GBR), or GBR12935 plus DA (G+D). Bottom, Quantitation of biotinylation. Western blots were scanned and quantitated using Scion Image software. Data are expressed as fold KRB (OD biotin/OD lysate). Error bars represent SEM. p < 0.01 by one-way ANOVA, n = 4. *p < 0.05 by post hoc Bonferroni, DA versus GBR and DA versus GBR plus DA.
PKC-β inhibition blocks the substrate-induced increase in DAT surface expression
A previous report from our laboratory demonstrated that pretreatment with the specific PKC-β inhibitor, LY379196, attenuated amphetamine-stimulated dopamine efflux in rat striatal slices (Johnson et al., 2005a). We wished to examine whether PKC-β was affecting substrate-induced dopamine efflux by altering rapid DAT trafficking to the surface. To determine whether a PKC-β-dependent effect was also occurring in the N2A cell system, TIRFM and biotinylation experiments were performed. We found that these cells contain PKC-β1 (data not shown). Cells were pretreated for 5 min with KRH or 200 nm LY379196 and then treated with 5 μm amphetamine or 10 μm dopamine in the presence of KRH or LY379196. As shown in Figure 6A, LY379196 slightly but significantly decreased the amphetamine-induced increase in surface DAT after 2 min of treatment. Although the zero time point in the LY379196-plus-amphetamine group appears lower than that of the amphetamine-alone group, none of the three zero time groups are significantly different from one another. LY379196 had similar effects on dopamine-induced increases of surface DAT, although the blockade of the dopamine effect was more pronounced than with amphetamine (Fig. 6B). These data are further validated with biotinylation experiments which show complete blockade of amphetamine or dopamine-induced increases in surface DAT expression after pretreatment of LY379196 in DAT-N2A cells (Fig. 6C). The blocking effect of LY379196 appeared more complete in the biotinylation experiments than in the TIRFM experiments. This differential effect could indicate a delayed effect of the inhibitor when it is slowly perfused as opposed to being given as a bolus as in biotinylation.
Figure 6.
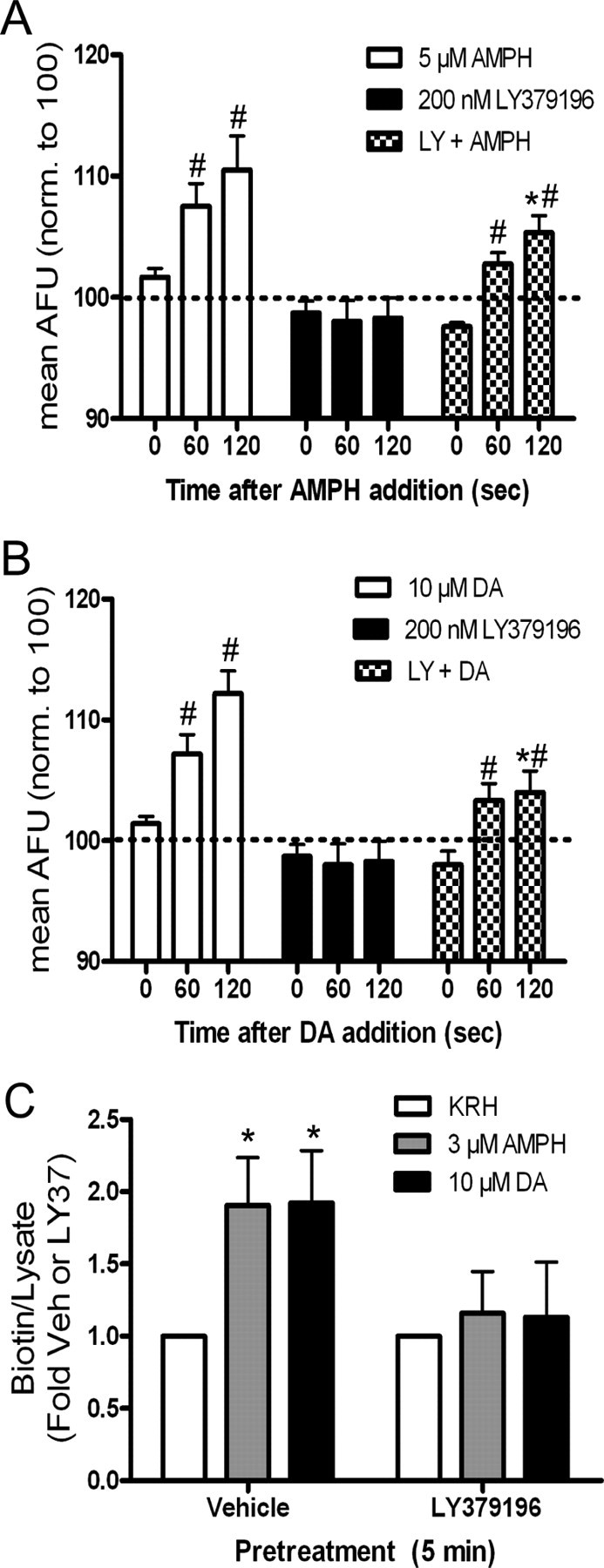
LY379196 (LY) inhibits substrate-induced DAT trafficking in DAT N2A cells. A, ImageJ quantitation of TIRFM. Cells were perfused for 2 min with KRH (white and black bars) or 200 nm LY379196 (checkered bars) before addition of 5 μm amphetamine (AMPH) (white bars; n = 6), 200 nm LY379196 (black bars; n = 7), or 200 nm LY379196 plus 5 μm AMPH (checkered bars; n = 12) at t = 0. Data are expressed as mean AFU and are normalized to 100 AFU (dashed line). Error bars represent SEM. Comparing across three drug groups, p < 0.0001 for drug and p < 0.05 for time by two-way ANOVA. *p < 0.05 by post hoc Bonferroni for 5 μm AMPH versus LY plus AMPH at t = 120 s. Comparing time points within each group, #p < 0.01 by two-way ANOVA: post hoc Bonferroni for 5 μm AMPH at t = 0 s versus 60 s and p < 0.001 at t = 0 s versus 120 s. Also, #p < 0.001 by two-way ANOVA: post hoc Bonferroni for LY plus AMPH at t = 0 s versus 60 s and p < 0.001 at t = 0 s versus 120 s. None of the zero time points were significantly different. B, ImageJ quantitation of TIRFM. Cells were perfused for 2 min with KRH (white and black bars) or 200 nm LY379196 (checkered bars) before addition of 10 μm dopamine (DA) (white bars; n = 5), 200 nm LY379196 (black bars; n = 7), or 200 nm LY379196 plus 10 μm DA (checkered bars; n = 6) at t = 0. Data are expressed as mean AFU and are normalized to 100 AFU (dashed line). Error bars represent SEM. Comparing across three drug groups, p < 0.0001 for drug and p < 0.001 for time by two-way ANOVA. *p < 0.01 by post hoc Bonferroni for 10 μm DA versus LY plus DA at t = 120 s. Comparing time points within each group, #p < 0.05 by two-way ANOVA: post hoc Bonferroni for 10 μm DA at t = 0 s versus 60 s and p < 0.001 at t = 0 s versus 120 s. Also, #p < 0.05 by two-way ANOVA: post hoc Bonferroni for LY plus DA at t = 0 s versus 60 s and p < 0.01 at t = 0 s versus 120 s. None of the zero time points were significantly different. C, Biotinylation. Cells were pretreated with Vehicle (Veh) or 200 nm LY379196 for 5 min followed by 1 min of KRH (n = 5), 3 μm AMPH (n = 5), or 10 μm DA (n = 6). Data are expressed as biotin/lysate and normalized to the respective pretreatment control (Veh or LY). Error bars represent SEM. p < 0.05 by one-way ANOVA for drug treatment. *p < 0.05 by post hoc Bonferroni for Veh versus AMPH and Veh. versus DA in vehicle pretreated cells. There was no significant difference between Veh (mean, 1.00 ± 0.27 OD) and LY37 (mean, 1.15 ± 0.32) raw values as measured by two-tailed paired t test (p = 0.455).
We next examined whether PKC-β similarly affects substrate-induced DAT trafficking in rat synaptosomes. Synaptosomes were treated with vehicle, 3 μm amphetamine, 100 nm LY379196, or amphetamine plus LY379196 for 1 min, and surface DAT expression was then measured using biotinylation (Fig. 7A). In the absence of LY379196, amphetamine increased DAT surface expression 1.5-fold over vehicle, but in the presence of drug, this effect was attenuated. LY379196 alone did not alter basal DAT surface expression, indicating that there is a blockade of amphetamine-induced DAT trafficking as opposed to a change in basal DAT trafficking. To determine whether the inhibition of amphetamine-induced DAT trafficking by PKC-β was sufficient to block amphetamine-stimulated dopamine efflux, synaptosomes were pretreated with KRB, 3 μm amphetamine, 100 nm LY379196, or amphetamine plus LY379196 for 1 min as described in the biotinylation experiment. Synaptosomes were subsequently washed at 4°C to prevent trafficking, and amphetamine-induced dopamine efflux was measured (see Materials and Methods). As we demonstrated previously (Johnson et al., 2005b), a 1 min pretreatment with amphetamine alone (followed by washout) significantly enhanced amphetamine-stimulated dopamine efflux (Fig. 7B). However, this enhanced efflux was blocked when amphetamine, and LY379196 were given simultaneously during the 1 min pretreatment. This demonstrates that the enhanced amphetamine-stimulated dopamine efflux is likely attributable to increased DAT surface expression that is PKC-β dependent. A 1 min pretreatment of LY379196 alone, followed by washout, did not block subsequent amphetamine-stimulated dopamine efflux (Fig. 7B).
Figure 7.
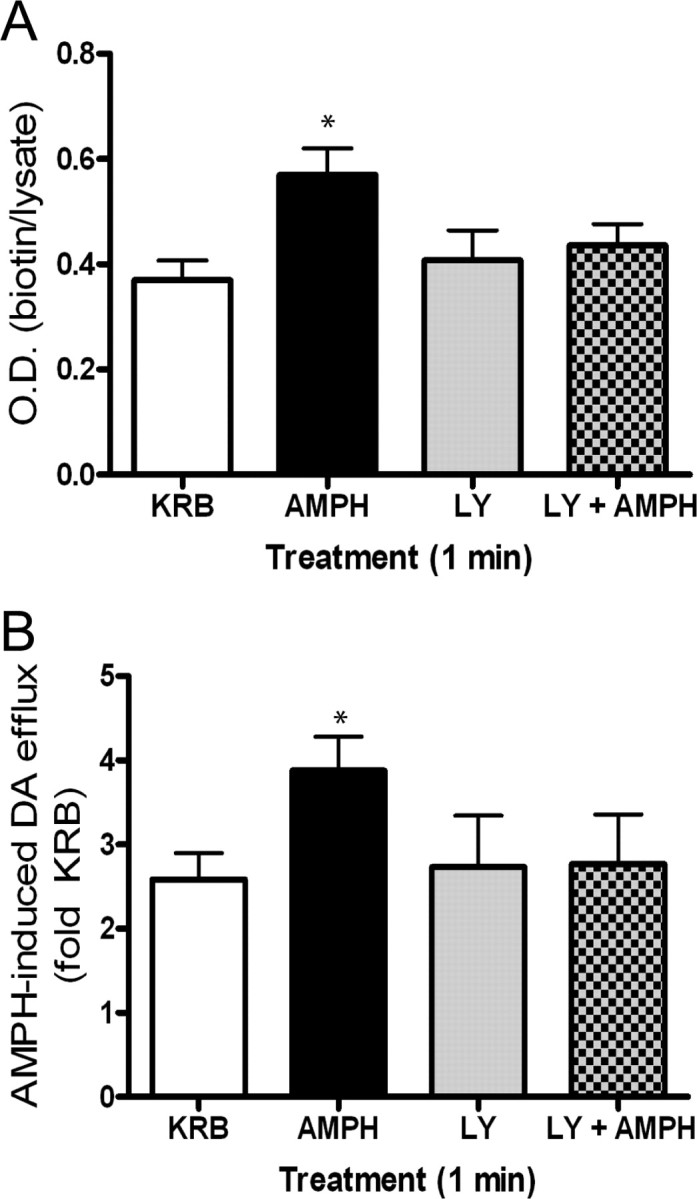
Role of PKC-β in amphetamine-induced DAT trafficking in rat synaptosomes. A, Biotinylation of DAT in rat synaptosomes treated for 1 min with vehicle (Veh; KRB), 3 μm AMPH, 100 nm LY379196 (LY), or AMPH plus LY379196. Error bars represent SEM. *p < 0.05 by one-way ANOVA, n = 3. Post hoc Bonferroni analysis shows a significant difference between amphetamine (AMPH) and Veh (p < 0.05) B, Synaptosomes were pretreated using same protocol as in A. Synaptosomes were washed three times at 4°C then further separated and treated with vehicle or AMPH. Data are expressed as AMPH-induced dopamine (DA) efflux (fold KRB): DA efflux with AMPH/DA efflux with KRB. Error bars represent SEM. *p < 0.01 by one-way ANOVA, n = 4. Post hoc Bonferroni analysis shows a significant difference of AMPH versus Veh (p < 0.01), AMPH versus LY (p < 0.05), as well as AMPH versus LY plus AMPH (p < 0.05).
Discussion
Using total internal reflection microscopy, we have documented real-time trafficking of DAT to the cell surface in response to the substrates, amphetamine, and dopamine. This is the first report to describe rapid trafficking of DAT in response to the physiological substrate dopamine independent of dopamine D2R stimulation. To our knowledge, this is the first report to demonstrate that rapid substrate-induced exocytosis of DAT is dependent on the SNARE proteins, syntaxin 1A, and VAMP/synaptobrevin. Moreover, the results obtained in the DAT-N2A cells are verified in rat synaptosomes using biotinylation. We also demonstrated the involvement of PKC-β in the rapid trafficking of DAT in both systems.
Substrate-induced DAT trafficking to the plasmalemmal membrane occurred at times commensurate with activation of DAT by substrate. Previously, using biotinylation of rat striatal synaptosomes, we demonstrated that incubation with amphetamine increased DAT surface expression within 30–60 s (Johnson et al., 2005a). Functional significance of this rapid transit was established by an enhancement in amphetamine-stimulated dopamine efflux, as shown again in this study. We also find that pretreatment of synaptosomes with amphetamine, followed by washing out of the drug, elicits an increase in [3H] dopamine uptake (R. Chen and M. Gnegy, unpublished observations). Together, these results suggest a mechanism by which a DAT substrate rapidly recruits the transporter to the surface, increasing both substrate uptake and, in response to amphetamine, efflux. During continued exposure to substrate, the transporter undergoes internalization which reduces uptake of substrate.
We sought to determine the mechanism of DAT trafficking toward the surface by visualizing the movement of GFP-DAT containing vesicles near the plasmalemmal membrane. TIRFM provides real-time resolution coupled with the ability to sensitively detect and analyze cytosol to plasmalemmal membrane movement of vesicles and granules. TIRFM selectively illuminates the aqueous phase immediately adjacent to a glass interface with an exponentially decaying excitation (Axelrod, 1981, 2003). TIRFM has been used to discern tethering of GLUT4 containing vesicles to the rat adipocyte membrane before fusion (Lizunov et al., 2005), as well as the multiple tethering states and significant motion of chromaffin granules immediately preceding exocytosis (Holz and Axelrod, 2008).
We were unable to detect fusion of individual puncta on the plasma membrane in these cells, which may be attributable to the diffuse pattern of DAT on the membrane, a low incorporation of DAT molecules into individual vesicles, or the small size of the individual vesicles. The GABA transporter, GAT1, for example, is found in clear synaptic vesicles with a diameter of 50 nm (Deken et al., 2003). Therefore, to determine whether the substrate-stimulated increase in DAT surface intensity was attributable to a fusion event, we used neurotoxins known to proteolyze SNARE proteins and inhibit exocytosis. A role for vesicle fusion in the substrate-induced increase in GFP-DAT intensity is strongly suggested by the fact that neurotoxins affecting either a t-SNARE (syntaxin 1A) or a v-SNARE (VAMP/synaptobrevin) abolished the substrate-induced increase in fluorescence. VAMP2 was detected as a component of small GAT-1-containing vesicles in rat cortices (Deken et al., 2003), and the present study suggests it is also a component of DAT-containing vesicles. In addition to playing an essential role in the docking and fusion of neurotransmitter-containing vesicles to the presynaptic membrane of neurons, syntaxin 1A regulates function and localization of transporters (Quick, 2006). Furthermore, we demonstrated that amphetamine increases the binding of syntaxin 1A to DAT and that syntaxin 1A promotes amphetamine-stimulated dopamine efflux in hDAT-expressing cells (Binda et al., 2008).
Despite the sensitivity of TIRFM, we found distinct differences between TIRFM and biotinylation in quantitation of the degree of change in surface DAT after substrate treatment. Biotinylation showed amphetamine and dopamine treatment to result in DAT surface expression that was 150 and 250% of control, respectively, whereas measurement by TIRFM showed ∼112% of control for both substrates. One major factor that likely contributes to this difference is that biotin has access only to transporter that is physically within the membrane and not to compartments that are continuous with the membrane (invaginations). TIRFM, however, may detect DAT-containing compartments that reside immediately underneath the membrane and could contribute to background fluorescence. The evanescent field of our TIRFM setup is ∼100 nm, and the intensity of fluorescence falls off exponentially with distance from the surface. If the DAT-containing vesicles are similar in size to the ∼50 nm GAT1-containing vesicles, background fluorescence could represent some DAT that is not integrally located in the plasmalemmal membrane. Fluorescence of GFP-DAT >200 nm from the plasma membrane would be attenuated at least 90%, so the source of the rapidly trafficked DAT is likely within 100 nm of the membrane. The increase in intensity after substrate, however, represents GFP-DAT that is moving closer to the membrane and would involve fusion events as suggested by the experiments with the neurotoxins. The “background” fluorescence detected in TIRFM could potentially represent the source of the DAT that moves to the surface after substrate stimulation. More experiments are underway to determine where this rapidly translocated DAT is coming from within the cell. Carneiro and Blakely (2006) found that serotonin stimulation elicited a trafficking of the serotonin transporter, SERT, from cytoskeletal fractions to membrane fractions in platelets within 30 min (Carneiro and Blakely, 2006). A similar, but more rapid, change in association could occur with DAT vesicles residing directly below the plasma membrane.
Other factors reported to increase surface DAT relatively rapidly are cocaine treatment and dopamine D2R activation. Cocaine increased surface DAT in DAT overexpressed cells (Daws et al., 2002; Little et al., 2002). An increase in dopamine uptake was seen after 5 min of 10 μm cocaine treatment (Daws et al., 2002). Cocaine alone had no effect on DAT trafficking within our experimental time frame but blocked substrate-mediated DAT trafficking in both synaptosomes and heterologous cells demonstrating that, in our system, substrate binding to DAT was required to elicit trafficking. Activation of D2Rs also regulate surface DAT and dopamine uptake (Meiergerd et al., 1993; Mayfield and Zahniser, 2001). Bolan et al. (2007) demonstrated a D2R-dependent increase in DAT surface expression in human DAT-N2A cells overexpressing the D2R after 1 min of activation by the D2R agonist quinpirole. Our human DAT-N2A cells were not overexpressing D2Rs but may contain low but active levels of endogenous D2 receptors. Our experiments clearly demonstrate, however, that the dopamine-induced increase in surface DAT in human DAT-N2A cells was not attributable to D2R activation because D2R overexpression in N2A cells did not change the dopamine-induced increase in DAT surface expression. Moreover, our experiments in rat striatal synaptosomes demonstrate that interaction of the substrate, dopamine, with DAT is required for the very rapid transport of DAT to the plasmalemmal membrane in a more physiological system. However, more experiments in rat synaptosomes using specific D2R antagonists are needed to determine whether D2Rs are directly or indirectly involved.
PKC appears to play an integral role in DAT trafficking. Analogous to substrate treatment, a more prolonged exposure of synaptosomes or DAT-overexpressed heterologous cells to PKC-activating phorbol esters induces a downregulation of DAT (Melikian, 2004; Sorkina et al., 2005). However, evidence from our laboratory suggested that PKC could also enhance some DAT activities. PKC inhibitors blocked amphetamine-stimulated dopamine efflux from rat striatum, whereas phorbol esters themselves increased efflux (Kantor and Gnegy, 1998; Cowell et al., 2000). Johnson et al. (2005a) identified PKC-β as a PKC isozyme contributing to amphetamine-stimulated dopamine efflux (Johnson et al., 2005a). We have used the selective PKC-β inhibitor, LY379196, to demonstrate that PKC-β plays a role in the rapid substrate-induced trafficking in both DAT-N2A cells using TIRFM as well as biotinylation and in synaptosomes using biotinylation. Furthermore, using synaptosomes, we demonstrated functional consequences of the PKC-β effect in that the enhancement in amphetamine-stimulated dopamine efflux after amphetamine pretreatment was blocked by LY379196.
Together, our results suggest that there are dual, biphasic regulations of DAT both by substrate and PKC activation. Within seconds of interacting with DAT, substrates induce exocytosis of DAT-containing vesicles into the plasmalemmal membrane. Substrate transport is likely required because experiments where sodium was removed failed to show increased DAT surface expression during substrate exposure. The movement of DAT to the surface is dependent on PKC-β activation. The source of DAT may be “readily releasable pools” in a subplasmalemmal membrane compartment that are primed for translocation into the plasmalemmal membrane during substrate stimulation. Work by Schmidt et al. (1997) revealed a synaptic-like microvesicle compartment that is continuous with the plasmalemmal membrane in neuroendocrine cells (Schmidt et al., 1997).
The rapid translocation of DAT-containing vesicles to the surface has functional consequences (Johnson et al., 2005b). Longer treatment with substrate, or PKC activators, leads to internalization of the transporter, although amphetamine-stimulated internalization may not require PKC (Boudanova et al., 2008). It is possible that different isozymes of PKC could be responsible for the rapid transport and internalization of DAT, although it is likely that a “classical” Ca2+-dependent PKC is also important for PKC-dependent internalization (Doolen and Zahniser, 2002). These studies are important for understanding the responsiveness of the dopamine transporter to both the physiological substrate and to drugs of abuse.
Footnotes
This work was supported by National Institutes of Health Grants DA011697 (M.G.), T32 GM07767 (C.A.F.), and RO1 NS-038129 (R.W.H.). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of General Medical Sciences. This work used the DNA sequencing core(s) of the Michigan Diabetes Research and Training Center funded by NIH5P60 DK20572 from the National Institute of Diabetes and Digestive and Kidney Diseases. We thank Dr. Miriam Allersma and Charles Lo for helpful discussions and technical assistance.
References
- Allersma et al., 2006.Allersma MW, Bittner MA, Axelrod D, Holz RW. Motion matters: secretory granule motion adjacent to the plasma membrane and exocytosis. Mol Biol Cell. 2006;17:2424–2438. doi: 10.1091/mbc.E05-10-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod, 1981.Axelrod D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J Cell Biol. 1981;89:141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod, 2003.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. doi: 10.1016/s0076-6879(03)61003-7. [DOI] [PubMed] [Google Scholar]
- Binda et al., 2008.Binda F, Dipace C, Bowton E, Robertson SD, Lute BJ, Fog JU, Zhang M, Sen N, Colbran RJ, Gnegy ME, Gether U, Javitch JA, Erreger K, Galli A. Syntaxin 1A interaction with the dopamine transporter promotes amphetamine-induced dopamine efflux. Mol Pharmacol. 2008;74:1101–1108. doi: 10.1124/mol.108.048447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan et al., 2007.Bolan EA, Kivell B, Jaligam V, Oz M, Jayanthi LD, Han Y, Sen N, Urizar E, Gomes I, Devi LA, Ramamoorthy S, Javitch JA, Zapata A, Shippenberg TS. D2 receptors regulate dopamine transporter function via an extracellular signal-regulated kinases 1 and 2-dependent and phosphoinositide 3 kinase-independent mechanism. Mol Pharmacol. 2007;71:1222–1232. doi: 10.1124/mol.106.027763. [DOI] [PubMed] [Google Scholar]
- Boudanova et al., 2008.Boudanova E, Navaroli DM, Melikian HE. Amphetamine-induced decreases in dopamine transporter surface expression are protein kinase C-independent. Neuropharmacology. 2008;54:605–612. doi: 10.1016/j.neuropharm.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro and Blakely, 2006.Carneiro AM, Blakely RD. Serotonin-, protein kinase C-, and Hic-5-associated redistribution of the platelet serotonin transporter. J Biol Chem. 2006;281:24769–24780. doi: 10.1074/jbc.M603877200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi and Reith, 2003.Chi L, Reith ME. Substrate-induced trafficking of the dopamine transporter in heterologously expressing cells and in rat striatal synaptosomal preparations. J Pharmacol Exp Ther. 2003;307:729–736. doi: 10.1124/jpet.103.055095. [DOI] [PubMed] [Google Scholar]
- Cowell et al., 2000.Cowell RM, Kantor L, Hewlett GH, Frey KA, Gnegy ME. Dopamine transporter antagonists block phorbol ester-induced dopamine release and dopamine transporter phosphorylation in striatal synaptosomes. Eur J Pharmacol. 2000;389:59–65. doi: 10.1016/s0014-2999(99)00828-6. [DOI] [PubMed] [Google Scholar]
- Daws et al., 2002.Daws LC, Callaghan PD, Morón JA, Kahlig KM, Shippenberg TS, Javitch JA, Galli A. Cocaine increases dopamine uptake and cell surface expression of dopamine transporters. Biochem Biophys Res Commun. 2002;290:1545–1550. doi: 10.1006/bbrc.2002.6384. [DOI] [PubMed] [Google Scholar]
- Deken et al., 2003.Deken SL, Wang D, Quick MW. Plasma membrane GABA transporters reside on distinct vesicles and undergo rapid regulated recycling. J Neurosci. 2003;23:1563–1568. doi: 10.1523/JNEUROSCI.23-05-01563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolen and Zahniser, 2002.Doolen S, Zahniser NR. Conventional protein kinase C isoforms regulate human dopamine transporter activity in Xenopus oocytes. FEBS Lett. 2002;516:187–190. doi: 10.1016/s0014-5793(02)02554-1. [DOI] [PubMed] [Google Scholar]
- Fleckenstein et al., 1999.Fleckenstein AE, Haughey HM, Metzger RR, Kokoshka JM, Riddle EL, Hanson JE, Gibb JW, Hanson GR. Differential effects of psychostimulants and related agents on dopaminergic and serotonergic transporter function. Eur J Pharmacol. 1999;382:45–49. doi: 10.1016/s0014-2999(99)00588-9. [DOI] [PubMed] [Google Scholar]
- Giros and Caron, 1993.Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Gulley et al., 2002.Gulley JM, Doolen S, Zahniser NR. Brief, repeated exposure to substrates down-regulates dopamine transporter function in Xenopus oocytes in vitro and rat dorsal striatum in vivo. J Neurochem. 2002;83:400–411. doi: 10.1046/j.1471-4159.2002.01133.x. [DOI] [PubMed] [Google Scholar]
- Holz and Axelrod, 2008.Holz RW, Axelrod D. Secretory granule behaviour adjacent to the plasma membrane before and during exocytosis: total internal reflection fluorescence microscopy studies. Acta Physiol (Oxf) 2008;192:303–307. doi: 10.1111/j.1748-1716.2007.01818.x. [DOI] [PubMed] [Google Scholar]
- Johnson et al., 2005a.Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME. Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. J Biol Chem. 2005a;280:10914–10919. doi: 10.1074/jbc.M413887200. [DOI] [PubMed] [Google Scholar]
- Johnson et al., 2005b.Johnson LA, Furman CA, Zhang M, Guptaroy B, Gnegy ME. Rapid delivery of the dopamine transporter to the plasmalemmal membrane upon amphetamine stimulation. Neuropharmacology. 2005b;49:750–758. doi: 10.1016/j.neuropharm.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Kahlig and Galli, 2003.Kahlig KM, Galli A. Regulation of dopamine transporter function and plasma membrane expression by dopamine, amphetamine, and cocaine. Eur J Pharmacol. 2003;479:153–158. doi: 10.1016/j.ejphar.2003.08.065. [DOI] [PubMed] [Google Scholar]
- Kantor and Gnegy, 1998.Kantor L, Gnegy ME. Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. J Pharmacol Exp Ther. 1998;284:592–598. [PubMed] [Google Scholar]
- Little et al., 2002.Little KY, Elmer LW, Zhong H, Scheys JO, Zhang L. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol. 2002;61:436–445. doi: 10.1124/mol.61.2.436. [DOI] [PubMed] [Google Scholar]
- Lizunov et al., 2005.Lizunov VA, Matsumoto H, Zimmerberg J, Cushman SW, Frolov VA. Insulin stimulates the halting, tethering, and fusion of mobile GLUT4 vesicles in rat adipose cells. J Cell Biol. 2005;169:481–489. doi: 10.1083/jcb.200412069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loder and Melikian, 2003.Loder MK, Melikian HE. The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J Biol Chem. 2003;278:22168–22174. doi: 10.1074/jbc.M301845200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield and Zahniser, 2001.Mayfield RD, Zahniser NR. Dopamine D2 receptor regulation of the dopamine transporter expressed in Xenopus laevis oocytes is voltage-independent. Mol Pharmacol. 2001;59:113–121. doi: 10.1124/mol.59.1.113. [DOI] [PubMed] [Google Scholar]
- Meiergerd et al., 1993.Meiergerd SM, Patterson TA, Schenk JO. D2 receptors may modulate the function of the striatal transporter for dopamine: kinetic evidence from studies in vitro and in vivo. J Neurochem. 1993;61:764–767. doi: 10.1111/j.1471-4159.1993.tb02185.x. [DOI] [PubMed] [Google Scholar]
- Melikian, 2004.Melikian HE. Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol Ther. 2004;104:17–27. doi: 10.1016/j.pharmthera.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Quick, 2006.Quick MW. The role of SNARE proteins in trafficking and function of neurotransmitter transporters. Handb Exp Pharmacol. 2006:181–196. doi: 10.1007/3-540-29784-7_9. [DOI] [PubMed] [Google Scholar]
- Saunders et al., 2000.Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci U S A. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt et al., 1997.Schmidt A, Hannah MJ, Huttner WB. Synaptic-like microvesicles of neuroendocrine cells originate from a novel compartment that is continuous with the plasma membrane and devoid of transferrin receptor. J Cell Biol. 1997;137:445–458. doi: 10.1083/jcb.137.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorkina et al., 2005.Sorkina T, Hoover BR, Zahniser NR, Sorkin A. Constitutive and protein kinase C-induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic. 2005;6:157–170. doi: 10.1111/j.1600-0854.2005.00259.x. [DOI] [PubMed] [Google Scholar]