

Abstract
Sixteen of 22 low molecular weight integral membrane proteins from Mycobacterium tuberculosis with previously poor or undetectable levels of expression were expressed in Escherichia coli as fusions with both the maltose-binding protein (MBP) and a His8-tag. 68% of targeted proteins were expressed in high yield (≥30 mg/L) in soluble and/or inclusion body form. Thrombin cleavage of the MBP fusion protein was successful for 10 of 13 proteins expressed as soluble proteins and for three proteins expressed only as inclusion bodies. The use of autoinduction growth media increased yields over Luria-Bertani (LB) growth media in 75% of the expressed proteins. Expressing integral membrane proteins with yields suitable for structural studies from a set of previously low and non-expressing proteins proved highly successful upon attachment of the maltose-binding protein as a fusion tag.
Production of membrane proteins for structural and functional studies remains a difficult task due to their highly hydrophobic nature [1, 2]. During the last decade increasing efforts to express prokaryotic and eukaryotic membrane proteins have resulted in significant advancements for the expression of several bacterial transporter proteins [3–9], outer membrane proteins of bacteria [10], membrane protein complexes [11–13] and a few eukaryotic G-protein coupled receptors [14–21]. With others we have successfully expressed more than 70 membrane proteins from Mycobacterium tuberculosis in Escherichia coli using a His-tag [22]. However, a significant number of tested proteins (28%) were not expressed at detectable (Coomassie stain or Western) levels in E. coli. Expression of nonmembrane proteins by the Methanobacterium thermoautotrophicum structural genomics pilot project showed that almost 50% of cloned genes did not express when an N-terminal His-tag was used [23]. More importantly, 41% of our cloned membrane proteins with a His-tag that have a molecular weight less than 13.7 kDa did not express, and only 22% expressed well, such that a Coomassie stain was observed representing enough protein to proceed with structural studies. Here, we have set out to enhance the expression of small molecular weight proteins and to identify a robust protocol for doing this.
While many different vectors and tags have been used, the fusion construct with maltose binding protein (MBP) has been found to be an effective fusion partner for increased solubility and expression yields for a variety of recombinant water-soluble proteins [3, 24–28]. MBP has also been tried with a few eukaryotic and prokaryotic membrane proteins [3, 15, 29–31]. Grisshammer and colleagues have achieved membrane localization and functional expression in E. coli for neurotensin and neurokinin-2 G-protein coupled receptors upon fusion to the periplasmic MBP [1, 14]. Expression as non-periplasmic MBP fusions in E. coli followed by purification and structural characterization by NMR was reported for two small membrane proteins: phospholamban and sarcolipin [30].
Here we report a successful application of the MBP fusion expression system for expression of small molecular weight integral membrane proteins from M. tuberculosis in E. coli. Some of these proteins are expressed as soluble fusion proteins obviating the need for harsh solubilizing conditions in extracting membrane proteins from inclusion bodies, but many others were expressed in high yields in inclusion bodies from which we have demonstrated isolation and cleavage of the fusion.
Materials and Methods
Cloning
The DNA fragments encoding membrane proteins from M. tuberculosis were cloned into a modified pMALc2 plasmid [32] by PCR. Cloning was done according to standard techniques. Plasmid DNA encoding selected proteins were used as a template for PCR. All primers were purchased from IDT, Inc., USA. Pairs of gene specific primers were used to amplify DNA using standard PCR conditions and the enzyme Pfu Turbo DNA polymerase (Stratagene, USA), shown to have lower probability for introducing unwanted mutations. Two pairs of the restriction endonucleases were used to digest generated PCR fragments or vector DNA: BamHI and HindIII or BamHI and PstI. Sites for restriction endonucleases were introduced in the primer sequences. PCR products were gel purified after digestion with restriction enzymes and ligated into the prepared expression vector. Correct insertions into the vector were confirmed by PCR screening followed by DNA sequencing to check for in-frame insertion and lack of PCR-introduced point mutations.
Expression Constructs
The plasmid results in the expression of the MBP fusion protein with eight histidine residues and the thrombin protease cleavage site at the N-terminus of the protein of interest to improve cleavage of MBP (Figure 1). Importantly, two affinity tags, MBP and the His-tag, allow for a choice of the alternative purification approaches.
Figure 1.
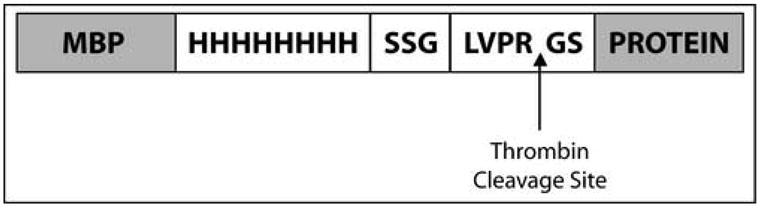
M. tuberculosis membrane proteins were expressed as fusions to the C-terminus of non-secreted MBP. The fusion construct has eight histidines and a thrombin recognition and cleavage site positioned between MBP and the fused polypeptide. The thrombin recognition site is composed of LVPRGS amino acids. Two amino acid residues (glycine and serine) remain with the N-terminus of the expressed protein after thrombin digest. Molecular weight of MBP with the C-terminal His-tag generated after thrombin digest is 42.5 kDa.
Protein Expression
E. coli BL-21(DE3)codonPlus-RP strain (Stratagene, USA) was used to express recombinant MBP-fusion proteins. This strain is designed to compensate for codon usage differences between M. tuberculosis and E. coli and was successfully used in our previous expression of membrane proteins.
The E. coli cultures were initially grown in standard liquid media with appropriate antibiotic [33]. Briefly, 20 ml of LB growth media supplemented with 50 mg/ml ampicillin was inoculated with a 1:100 dilution of an overnight bacterial culture and incubated with agitation at 37°C to an OD600 of ~ 0.5. Protein expression was induced with 0.4 mM IPTG. Cells were harvested 3 h after induction by centrifugation at 5,000 × g in an Avanti J-20 XP centrifuge (Beckman Coulter, Inc., USA). The cells were resuspended in 1 ml of water and lyzed by sonication using a Sonic Dismembrator, Model 100 (Fischer Scientific, Inc.). The lysate was clarified by centrifugation for 15 min at 10,000 × g using a Microfuge 18 Centrifuge (Beckman Coulter, Inc.). The pellet was resuspended in 1 ml of 1% SDS and 4% urea. Insoluble and soluble fractions were checked for expression by the appearance on the SDS-PAGE gel stained with Coomassie R-250. Samples were loaded on the gel on the basis of equal volume, and the expression outcomes were assessed by visual inspection. Furthermore, the amount of expressed fusion protein was estimated for each sample during the purification step when the fusion protein was eluted from the amylose or His-tag affinity columns. The concentration was determined via the absorbance at 280 nm.
Bacterial autoinduction growth system, which utilizes lactose for the induction of protein expression has also been used for all expressed fusion proteins in an effort to increase yields [34]. Briefly, TB growth media was supplemented with 50 mg/ml ampicillin, 100 mM Dibasic Sodium Phosphate, 50 mM Monobasic Potassium Phosphate, 25 mM Ammonium Sulfate, 0.5% Glycerol, 0.05% Glucose and 0.2% Lactose. 20 ml of prepared media were inoculated with a 1:100 dilution of an overnight bacterial culture and incubated with agitation at 37°C overnight (16 hours of growth). Cells were harvested by centrifugation followed by lysis and sample preparation as described above for the bacterial cells grown in LB media.
Protein purification
Membrane proteins from M. tuberculosis fused with the C-terminus of MBP were expressed in two forms: as soluble fusion proteins and as proteins directed into inclusion bodies. Dual affinity properties of the recombinant fusion protein, MBP and His-tag, allow the purification of both, a soluble recombinant protein using amylose affinity or metal affinity, as well as a protein solubilized from inclusion bodies and isolated via metal affinity chromatography.
200 ml of the LB or autoinduction media were used to grow E. coli cells expressing MBP fusion proteins under growth conditions identical to that described for the small expression screen. Cell debris was pelleted after lysis by centrifugation at 10,000 × g. A supernatant containing soluble MBP fusion protein in a loading buffer consisting of 20 mM Tris, pH 8.0 and 200 mM NaCl was applied to an amylose resin affinity column at 4°C. After extensive washing with loading buffer the fusion protein was eluted from the column with 10 mM maltose followed by thrombin digest without any additional buffer change.
Fusion proteins expressed in inclusion body form were reconstituted from the pellet obtained after lysis and 10,000 × g centrifugation in a buffer containing 20 mM Tris, pH 7.9, 5 mM imidazole, 500 mM NaCl and 6 M urea for at least two hours at 4°C. A sample was applied to the Chelating Sepharose Fast Flow column charged with Ni2+ and equilibrated with an identical buffer. After extensive washing with 20 mM imidazole, the fusion protein was eluted from the column with 300 mM imidazole. The isolated protein was dialyzed extensively to decrease the concentration of imidazole in the sample prior to thrombin digest maintaining the urea concentration at 1 M to prevent aggregation and precipitation of a fusion protein.
Thrombin digest
Thrombin from human plasma (catalog # T6884) was purchased from Sigma, (USA). Thrombin digest of MBP-fusion proteins was selectively optimized for the amount of thrombin added per mg of fusion protein and the duration of the cleavage reaction. The reactions were supplemented with additives such as 0.01% SDS, 1 M urea, and 10% glycerol in order to facilitate the cleavage. The choice of additives was based on the thrombin digest recommendations developed by Novagen, USA. The completion of the thrombin digest was verified via the correct shifts in the positions of the protein bands on a polyacrylamide gel.
Results
Target Selection and Cloning
22 integral membrane proteins from M. tuberculosis were cloned into a modified pMALc2 plasmid for expression as fusions to the C-terminus of non-secreted MBP (Table 1). Previously, these proteins did not yield any detectable expression as fusions with short N-terminal His-tags [22] except for Rv0463, Rv0882, Rv0961, Rv1305 and Rv2076c which showed low level expression identified via western blots. Proteins selected for MBP fusions had a molecular weight range between 7.1 and 13.7 kDa and a number of putative transmembrane helices that varied between one and four. These proteins were selected for expression trials because they are attractive targets for structural characterization by NMR.
Table 1.
Relative expression levels shown as observed on Coomassie-stained denaturing polyacrylamide gels: “o” denotes no observed expression, and “+” though “++++” represents increasing expression level, where “+” corresponds to an expression yield between 10 mg and 30 mg of fusion protein per L of the bacterial culture, “++” corresponds to yields between 40 and 60 mg, “+++” corresponds to fusion protein yields between 70 and 90 mg, and “++++” corresponds to yields of fusion protein exceeding 100 mg. The amount of protein was determined by absorbance at 280 nm after eluting the sample from an amylose or metal affinity column. Abbreviations are as follows: #TM – the number of putative transmembrane helices; MW – molecular weight in kDa; n/a – not applicable; n/t – not tested.
| Membrane protein from M. tuberculosis | Fusion protein MW, kDa | Membrane protein MW, kDa | # TM | LB media growth | Autoinduction media growth | Thrombin Digest, % | |||
|---|---|---|---|---|---|---|---|---|---|
| sol | insol | sol | insol | sol | insol | ||||
| Rv3656c | 49.6 | 7.1 | 1 | o | o | o | o | n/a | n/a |
| Rv0476 | 51.7 | 9.2 | 2 | +++ | +++ | +++ | +++ | 10 | n/t |
| Rv0514 | 53.0 | 10.5 | 2 | o | + | + | +++ | 90 | n/a |
| Rv0879c | 52.1 | 9.6 | 2 | + | + | + | ++ | 100 | n/a |
| Rv1305 | 50.7 | 8.2 | 2 | + | ++ | o | +++ | 100 | n/a |
| Rv1440 | 50.7 | 8.2 | 2 | +++ | +++ | ++++ | ++++ | 0 | n/t |
| Rv1567c | 53.0 | 10.5 | 2 | + | ++ | + | ++++ | 100 | n/a |
| Rv2076c | 51.6 | 9.1 | 2 | o | o | o | o | n/a | n/a |
| Rv2128 | 50.0 | 7.5 | 2 | + | +++ | o | ++++ | 100 | n/a |
| Rv2144c | 54.5 | 12.0 | 2 | ++ | + | ++ | + | 0 | n/t |
| Rv2876 | 54.3 | 11.8 | 2 | + | ++ | ++ | ++ | 100 | n/a |
| Rv3346c | 51.5 | 9.0 | 2 | o | ++++ | o | ++++ | n/a | 30 |
| Rv0460 | 50.7 | 8.2 | 3 | o | + | + | ++ | 100 | n/a |
| Rv0463 | 52.7 | 10.2 | 3 | + | +++ | + | ++++ | 100 | n/a |
| Rv0882 | 52.3 | 9.8 | 3 | + | + | + | + | 100 | n/a |
| Rv0961 | 54.9 | 12.4 | 3 | o | o | o | o | n/a | n/a |
| Rv3155 | 53.4 | 10.9 | 3 | o | ++ | o | +++ | n/a | 50 |
| Rv3355c | 52.8 | 10.3 | 3 | o | + | o | ++ | n/a | 50 |
| Rv1824 | 55.2 | 12.7 | 4 | + | ++ | +++ | +++ | 100 | n/a |
| Rv2600 | 56.1 | 13.6 | 4 | o | o | o | o | n/a | n/a |
| Rv3065 | 53.6 | 11.1 | 4 | o | o | o | o | n/a | n/a |
| Rv3070 | 55.7 | 13.2 | 4 | o | o | o | o | n/a | n/a |
Expression
Sixteen of 22 (72%) M. tuberculosis membrane proteins were expressed as MBP fusions in E. coli. Results of the expression testing are shown in Table 1. We consider the expression of the MBP fusion protein in soluble form preferable because soluble proteins can be easily purified in one step using affinity chromatography and the thrombin digest can be done under nondenaturing conditions.
Significant variations in the amount of expressed protein were observed in the different cellular compartments. In considering cultures in both LB and autoinduction media, 13 fusion constructs out of 16 (81%) were expressed as soluble proteins and all 16 were expressed in inclusion bodies. The highest levels of expression in soluble form were achieved for Rv0476, Rv1440, and Rv1824 with yields up to 120 mg of fusion protein per L of bacterial culture as estimated by measuring the absorbance at 280 nm after purifying the fusion proteins via an amylose affinity column. Most of the tested proteins showed moderate expression yields in the soluble form, between 10 and 30 mg of fusion protein per L of bacterial culture (e.g. Rv2876, Figure 2, panel B). For most of these fusion constructs the amount of expressed protein was substantially greater in the insoluble fraction than in the soluble fraction. Thus, the Rv1824 fusion expressed in LB media has three fold (based on A280 of purified protein) less protein expressed in the soluble fraction than in inclusion bodies (Figure 2, panel A). For three proteins (Rv3155, Rv3346c, and Rv3355c) expression was detected only in the insoluble fraction (Figure 2, panel C)
Figure 2.
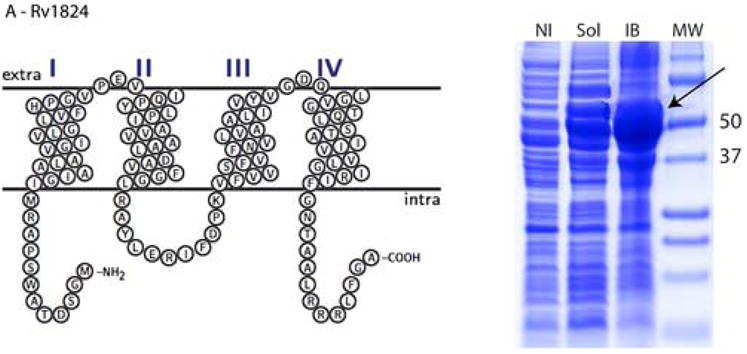

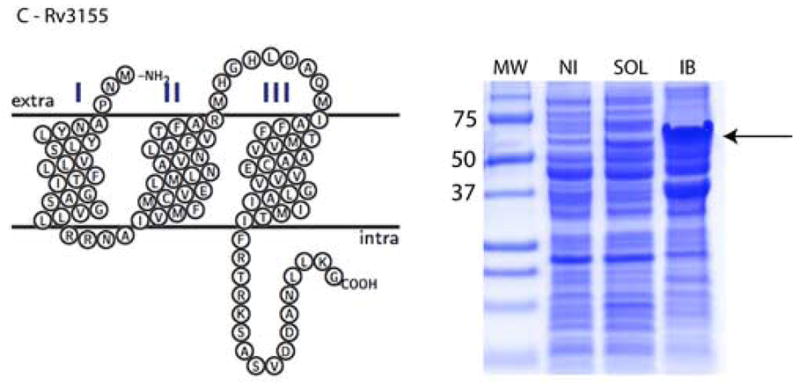
Topology plots and results of small-scale protein expression screening for (A) Rv1824, (B) Rv2876, and (C) Rv3155 MBP fusion proteins. Soluble and inclusion body fractions were separated on 12% Tricine SDS-PAGE (Schagger and von Jaggow, 1987) and stained with Coomassie Blue R-250. Equal volumes of each sample were loaded on the gel. Molecular Weight Markers (MW) are labeled with numbers (Precision Plus Protein Standards, BioRad, USA). Expressed proteins are shown with arrows. Lanes are as follows: NI – noninduced nonfractionated cells, Sol – soluble fraction, IB – inclusion body fraction.
The use of autoinduction media resulted in an increase in the expression yields for 12 of the tested proteins. While inclusion body expression increased for 11 of 12, the expression in soluble form increased for just five proteins: Rv0460, Rv0514, Rv1440, Rv1824, and Rv2876. Rv1824 fusion protein showed the most dramatic improvement with the use of autoinduction media as the amount of soluble protein was tripled (Figure 3). Importantly, Rv0460 and Rv0514 MBP fusions were only expressed in soluble form by the autoinduction media. While autoinduction improved the expression for a number of proteins, there were several proteins (Rv0476, Rv0882, Rv2144c, and Rv3346c) for which expression was not improved, and two(Rv1305 and Rv2128) for which expression in the soluble form was only achieved in LB media.
Figure 3.
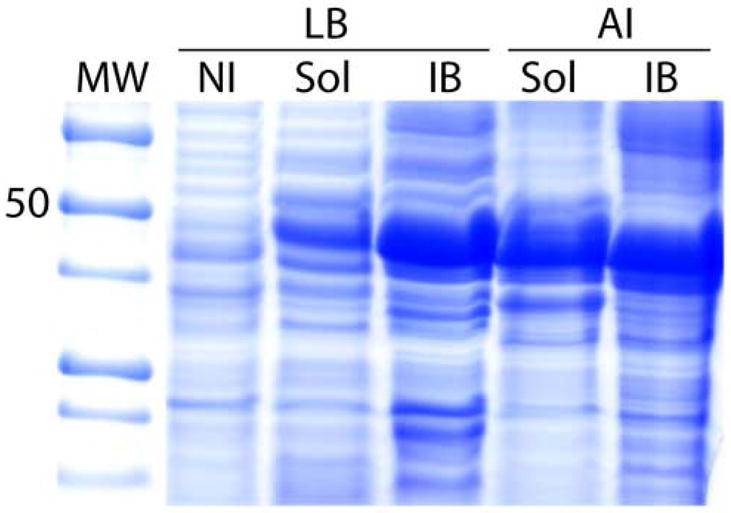
The results of small scale protein expression screening using 12% Tricine gels (as in Figure 2) for Rv1824 MBP fusion protein in LB and autoinduction (AI) media. The lanes are as follows: MW – molecular weight marker; NI – noninduced cells from LB media; Sol - soluble fraction; IB - inclusion bodies fraction.
Thrombin Digest of MBP Fusion Proteins
Our attempts to use a thrombin cleavage site were successful (≥ 30% cleavage) for 13 of the 16 (81%) expressed proteins (Table 1). However, fusion proteins showed significant variation in their ability to be cleaved with the protease (Table 1). The 13 soluble fusion proteins were purified using amylose affinity chromatography prior to thrombin digest. The concentration of fusion protein was assessed via protein absorbance at 280 nm and an appropriate amount of thrombin was added to the samples while still in the elution buffer. In most cases, 3–10 units of thrombin were used to cleave 1 mg of fusion protein at room temperature overnight. The additives such as 0.01 % SDS or 1 M urea as well as 10% glycerol were selectively added to the reaction mixtures to optimize the cleavage. An example of cleavage optimization efforts for Rv1824 fusion protein is presented in Figure 4. A low concentration of SDS was found to be most effective in facilitating the cleavage of fusion proteins. 9 of the 13 (69%) fusion proteins expressed in soluble form were completely cleaved.
Figure 4.

Polyacrylamide gel results of a thrombin digest of Rv1824 MBP fusion protein using gels as described in Figure 2. Thrombin digest results in two protein bands on the gel: 12.7 kDa for Rv1824 and 42.5 kDa for MBP with the addition of the His-tag. The lanes are as follows: NC – not cleaved; MW – molecular weight marker; SDS – 0.01% SDS added to reaction mixture; Urea – 0.8M urea added to reaction mixture; U – units of thrombin per mg fusion protein added to reaction mixture. In all cases, reaction mixture was incubated at room temperature for 14 hours.
If fusion proteins were expressed in soluble form, thrombin digest was performed only on the soluble fraction. Three fusion proteins, Rv3155, Rv3346c, and Rv3355c, were not expressed in soluble form and were isolated from inclusion bodies using a His-tag affinity column and digested with thrombin in the presence of 1 M urea. It required more protease to cleave the protein from inclusion bodies than protein expressed in soluble form; at least 15 units of thrombin were added for each mg of fusion protein. A further increase in the thrombin concentration did not improve the fraction cleaved. We estimate that only 30 to 50 % of the fusion protein was cleaved with the protease.
Discussion
A desirable expression system should produce a large amount of stable protein and permit efficient protein purification for subsequent structural or functional studies. Fusion protein approaches are popular and MBP fusions have been extensively utilized for water-soluble protein expression. However, relatively few transmembrane proteins and interfacial membrane proteins have been tested for expression as fusions with the large MBP [1, 14, 15, 29, 30]. Fusions of hydrophobic membrane proteins with the hydrophilic MBP domain have been shown to reduce toxicity and improve stability for bacterio-opsin and bacteriorodopsin [35, 36]. This study reports on the expression of multiple membrane proteins using MPB as a fusion partner. 16 membrane proteins from M. tuberculosis were expressed in E. coli here, only three of which had been expressed previously and these had poor yields. The achieved levels of expression with MBP fusions make functional and structural analyses feasible even though only a small fraction of the mg/L of culture is the desired protein product. Variation in expression yield is likely to be influenced by the size of the polypeptide, its amino acid content and three-dimensional conformation. It has been suggested that MBP has chaperon-like features and facilitates correct protein folding [24, 37, 38]. Thus, MBP may provide a protein scaffold that prevents the membrane protein portion of the fusion from aggregating, while MBP maintains its own structural integrity verified by its binding to an amylose column. Final separation of the membrane protein and its MBP partner after thrombin digest requires the addition of a detergent to create a membrane mimetic environment suitable for the membrane protein[39].
Comparing the expression outcomes in LB media and in the auto-induction media shows that there is no universal solution for protein expression of these low molecular weight membrane proteins. While it was rare that expression was observed in one media and not the other, the yields of both the soluble and insoluble forms of the fusion protein did vary substantially warranting an effort to “fine tune” the expression conditions.
Expression results suggest that proteins are more frequently expressed in soluble form as fusions with MBP when they have fewer transmembrane helices and hence fewer hydrophobic residues. In analyzing the combined results from LB and autoinduction media 9 out of 12 proteins (75%) with 1 or 2 transmembrane helices are expressed in soluble form while 4 out of 10 proteins (40%) with 3 or 4 transmembrane helices are expressed in soluble form. Even if the pool of proteins is restricted to just those that are expressed, the percentages still favor those with a small number of helices by 83% to 60%. There is no such trend for proteins expressed in inclusion bodies. Similarly, for proteins with only a His-tag there was no significant expression correlation with the number of transmembrane helices over a larger data set [22]. For soluble protein expression the data also suggests a correlation with molecular weight – 10 out of 13 proteins (70%) having a molecular weight less than 10.0 kDa are expressed in the soluble fraction in LB and/or autoinduction media while 6 out of 12 proteins (50%) having a molecular weight greater than 10.0 kDa are expressed in the soluble fraction. This correlation is likely to result from a correlation between molecular weight and the number of transmembrane helices which exists for such small membrane proteins and consequently the observed molecular weight correlation supports the transmembrane data analysis described above.
Separation of a target protein from the fusion partner may present a major challenge [37]. Incomplete enzymatic digestion with a protease is often due to inaccessibility of the protease site. Attempts to use thrombin cleavage site was successful in the case of MBP-sarcolipin fusion and failed in the case of MBP-phospholamban fusion proteins [30]. Poor results in using factor Xa to cleave the bacterio-opsin fusion with MBP led to trypsin digestion instead [35].
Typically, high yields (≥ 5 mg/L of the target protein) are desirable for structural studies, although there have been successful structural efforts even for membrane proteins at much lower expression levels. Out of 22 proteins that were not expressed at such levels with only a His-tag, five were expressed here at levels greater than 30 mg/L as an MBP fusion in solution.
Disappointingly, only two of these were successfully cleaved, Rv1824 and Rv2876. However, 14 of these 22 proteins were expressed at levels greater than 30 mg/L as an MBP fusion in inclusion bodies and all these high expression proteins for which thrombin cleavage was attempted (3) were substantially cleaved suggesting that an increased effort with the inclusion body fraction is warranted. Considerable success has been achieved with thrombin cleavage by using a modest detergent concentration and consequently the results suggest that this approach is a good choice for a first effort cleavage strategy, but others should be tried if thrombin fails. Conditions for detergent solubilization may need to be screened to optimize the percent cleavage. Here, only a low percentage of SDS has been tested.
We have shown in the present study that multiple small integral membrane proteins from M. tuberculosis are successfully expressed as fusions to the C-terminus of non-secreted MBP in E. coli. Most of the target proteins did not show any expression while fused with only an N-terminal His-tag in our previous study [22]. Expresssion of small molecular weight proteins was a clear weakness in the N-terminal His tag approach with only 22% of the cloned proteins expressing well enough for structural studies. Here the MBP fusion strategy represents an excellent second approach for this class of proteins, as the percent of well-expressed membrane proteins having a molecular weight ≤13.7 kDa is increased to 63%. As the hydrophobic portion of the fusion protein increases one might expect that proteins with a larger number of helices might not produce much protein in the soluble form and, in fact, there is some evidence for this. The results show that MBP fusions are an effective vector choice for low molecular weight membrane proteins and that the choice of media can significantly effect expression and expression yield. While LB media is a good choice for demonstrating expression, the yields of both soluble and insoluble forms were most frequently increased with autoinduction media.
Acknowledgments
This work was supported by the NIH Grant PO1 GM064676. JDM acknowledges support of a NIH predoctoral fellowship and an American Heart Association predoctoral fellowship. HN acknowledges support of an American Heart Association predoctoral fellowship.
Abbreviations
- LB
Luria-Bertani growth media
- MBP
maltose-binding protein
- IPTG
isopropyl-β-D-thiogalactoside
- MW
molecular weight
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Grisshammer R, Tate CG. Overexpression of integral membrane proteins for structural studies. Q Rev Biophys. 1995;28:315–422. doi: 10.1017/s0033583500003504. [DOI] [PubMed] [Google Scholar]
- 2.Essen L. Structural genomics of “non-standard” proteins: a chance for membrane proteins? Gene Function & Disease. 2002;3:39–48. [Google Scholar]
- 3.Wang DN, Safferling M, Lemieux MJ, Griffith H, Chen Y, Li XD. Practical aspects of overexpressing bacterial secondary membrane transporters for structural studies. Biochim Biophys Acta. 2003;1610:23–36. doi: 10.1016/s0005-2736(02)00709-5. [DOI] [PubMed] [Google Scholar]
- 4.Liang WJ, Wilson KJ, Xie H, Knol J, Suzuki S, Rutherford NG, Henderson PJ, Jefferson RA. The gusBC genes of Escherichia coli encode a glucuronide transport system. J Bacteriol. 2005;187:2377–2385. doi: 10.1128/JB.187.7.2377-2385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saidijam M, Bettaney KE, Szakonyi G, Psakis G, Shibayama K, Suzuki S, Clough JL, Blessie V, Abu-Bakr A, Baumberg S, et al. Active membrane transport and receptor proteins from bacteria. Biochem Soc Trans. 2005;33:867–872. doi: 10.1042/BST0330867. [DOI] [PubMed] [Google Scholar]
- 6.Bonander N, Hedfalk K, Larsson C, Mostad P, Chang C, Gustafsson L, Bill RM. Design of improved membrane protein production experiments: quantitation of the host response. Protein Sci. 2005;14:1729–1740. doi: 10.1110/ps.051435705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khademi S, O’Connell J, 3rd, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 A. Science. 2004;305:1587–1594. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 8.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem Rev. 2004;104:3587–3606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang DN, Lemieux MJ, Boulter JM. Purification and characterization of transporter proteins from human erythrocyte membrane. Methods Mol Biol. 2003;228:239–255. doi: 10.1385/1-59259-400-X:239. [DOI] [PubMed] [Google Scholar]
- 10.Bannwarth M, Schulz GE. The expression of outer membrane proteins for crystallization. Biochim Biophys Acta. 2003;1610:37–45. doi: 10.1016/s0005-2736(02)00711-3. [DOI] [PubMed] [Google Scholar]
- 11.Alexandrov A, Vignali M, LaCount DJ, Quartley E, de Vries C, De Rosa D, Babulski J, Mitchell SF, Schoenfeld LW, Fields S, et al. A facile method for high-throughput co-expression of protein pairs. Mol Cell Proteomics. 2004;3:934–938. doi: 10.1074/mcp.T400008-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Mileni M, Haas AH, Mantele W, Simon J, Lancaster CR. Probing heme propionate involvement in transmembrane proton transfer coupled to electron transfer in dihemic quinol:fumarate reductase by 13C-labeling and FTIR difference spectroscopy. Biochemistry. 2005;44:16718–16728. doi: 10.1021/bi051034s. [DOI] [PubMed] [Google Scholar]
- 13.Mileni M, Macmillan F, Tziatzios C, Zwicker K, Haas AH, Mantele W, Simon J, Lancaster CR. Heterologous production in Wolinella succinogenes and characterisation of the quinol:fumarate reductase enzymes from Helicobacter pylori and Campylobacter jejuni. Biochem J. 2005 doi: 10.1042/BJ20051675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grisshammer R, Little J, Aharony D. Expression of rat NK-2 (neurokinin A) receptor in E. coli. Receptors Channels. 1994;2:295–302. [PubMed] [Google Scholar]
- 15.Stanasila L, Massotte D, Kieffer BL, Pattus F. Expression of delta, kappa and mu human opioid receptors in Escherichia coli and reconstitution of the high-affinity state for agonist with heterotrimeric G proteins. Eur J Biochem. 1999;260:430–438. doi: 10.1046/j.1432-1327.1999.00187.x. [DOI] [PubMed] [Google Scholar]
- 16.Grisshammer R, White JF, Trinh LB, Shiloach J. Large-scale expression and purification of a G-protein-coupled receptor for structure determination -- an overview. J Struct Funct Genomics. 2005;6:159–163. doi: 10.1007/s10969-005-1917-6. [DOI] [PubMed] [Google Scholar]
- 17.White JF, Trinh LB, Shiloach J, Grisshammer R. Automated large-scale purification of a G protein-coupled receptor for neurotensin. FEBS Lett. 2004;564:289–293. doi: 10.1016/S0014-5793(04)00195-4. [DOI] [PubMed] [Google Scholar]
- 18.Lundstrom K. Structural genomics of GPCRs. Trends Biotechnol. 2005;23:103–108. doi: 10.1016/j.tibtech.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Tian C, Breyer RM, Kim HJ, Karra MD, Friedman DB, Karpay A, Sanders CR. Solution NMR spectroscopy of the human vasopressin V2 receptor, a G protein-coupled receptor. J Am Chem Soc. 2005;127:8010–8011. doi: 10.1021/ja051161b. [DOI] [PubMed] [Google Scholar]
- 20.Kiefer H, Vogel R, Maier K. Bacterial expression of G-protein-coupled receptors: prediction of expression levels from sequence. Receptors Channels. 2000;7:109–119. [PubMed] [Google Scholar]
- 21.Luca S, White JF, Sohal AK, Filippov DV, van Boom JH, Grisshammer R, Baldus M. The conformation of neurotensin bound to its G protein-coupled receptor. Proc Natl Acad Sci U S A. 2003;100:10706–10711. doi: 10.1073/pnas.1834523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korepanova A, Gao FP, Hua Y, Qin H, Nakamoto RK, Cross TA. Cloning and expression of multiple integral membrane proteins from Mycobacterium tuberculosis in Escherichia coli. Protein Sci. 2005;14:148–158. doi: 10.1110/ps.041022305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christendat D, Yee A, Dharamsi A, Kluger Y, Savchenko A, Cort JR, Booth V, Mackereth CD, Saridakis V, Ekiel I, et al. Structural proteomics of an archaeon. Nat Struct Biol. 2000;7:903–909. doi: 10.1038/82823. [DOI] [PubMed] [Google Scholar]
- 24.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J. Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci U S A. 2002;99:2654–2659. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammarstrom M, Hellgren N, van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shih YP, Kung WM, Chen JC, Yeh CH, Wang AH, Wang TF. High-throughput screening of soluble recombinant proteins. Protein Sci. 2002;11:1714–1719. doi: 10.1110/ps.0205202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eshaghi S, Hedren M, Nasser MI, Hammarberg T, Thornell A, Nordlund P. An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins. Protein Sci. 2005;14:676–683. doi: 10.1110/ps.041127005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berthold DA, Stenmark P, Nordlund P. Screening for functional expression and overexpression of a family of diiron-containing interfacial membrane proteins using the univector recombination system. Protein Sci. 2003;12:124–134. doi: 10.1110/ps.0223703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buck B, Zamoon J, Kirby TL, DeSilva TM, Karim C, Thomas D, Veglia G. Overexpression, purification, and characterization of recombinant Ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Protein Expr Purif. 2003;30:253–261. doi: 10.1016/s1046-5928(03)00127-x. [DOI] [PubMed] [Google Scholar]
- 31.Laage R, Langosch D. Strategies for prokaryotic expression of eukaryotic membrane proteins. Traffic. 2001;2:99–104. doi: 10.1034/j.1600-0854.2001.020204.x. [DOI] [PubMed] [Google Scholar]
- 32.Korepanova A, Douglas C, Leyngold I, Logan TM. N-terminal extension changes the folding mechanism of the FK506-binding protein. Protein Sci. 2001;10:1905–1910. doi: 10.1110/ps.14801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a laboratory manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 34.Grabski A, Mehler M, Drott D. Unattended high-density cell growth and induction of protein expression with overnight express autoinduction system. InNovations. 2003;17:3–8. [Google Scholar]
- 35.Chen GQ, Gouaux JE. Overexpression of bacterio-opsin in Escherichia coli as a water-soluble fusion to maltose binding protein: efficient regeneration of the fusion protein and selective cleavage with trypsin. Protein Sci. 1996;5:456–467. doi: 10.1002/pro.5560050307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pompejus M, Friedrich K, Teufel M, Fritz HJ. High-yield production of bacteriorhodopsin via expression of a synthetic gene in Escherichia coli. Eur J Biochem. 1993;211:27–35. doi: 10.1111/j.1432-1033.1993.tb19866.x. [DOI] [PubMed] [Google Scholar]
- 37.Baneyx F. Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol. 1999;10:411–421. doi: 10.1016/s0958-1669(99)00003-8. [DOI] [PubMed] [Google Scholar]
- 38.Sachdev D, Chirgwin JM. Solubility of proteins isolated from inclusion bodies is enhanced by fusion to maltose-binding protein or thioredoxin. Protein Expr Purif. 1998;12:122–132. doi: 10.1006/prep.1997.0826. [DOI] [PubMed] [Google Scholar]
- 39.Keifer PA, Peterkofsky A, Wang G. Effects of detergent alkyl chain length and chemical structure on the properties of a micelle-bound bacterial membrane targeting peptide. Anal Biochem. 2004;331:33–39. doi: 10.1016/j.ab.2004.03.074. [DOI] [PubMed] [Google Scholar]