

Abstract
Evolution of high-throughput technologies has enabled us to quantify several thousands of gene expressions simultaneously. Several oncologic applications have emerged, two of which will be discussed: developing gene signatures and finding molecular targets. This article emphasizes the different nature of statisitical methods used and required for developing gene signatures and identifying molecular targets. Routine and careful consideration of validation methods are essential for developing gene signatures. Choice of model development methodology seems to be less critical. Because of the way gene signatures are developed, even after careful validation, it is unlikely that they will yield a direct link to molecular targets. Identifying molecular targets will require more careful and focused experimentation than large-sample microarray studies.
Gene expression profiling is one of the most significant technologic advances of this decade. In a typical research study, expression of several thousands of genes is measured simulatenously on a relatively few number of subjects. Currently, the primary reasons for limiting the number of subjects are availability of samples with usable clinical data (especially follow-up) and the cost of processing the microarrays. It is unlikely that we will see a significant increase in the sample sizes used in gene expression profiling studies before both of these issues are resolved.
Developing a gene signature and identifying molecular targets are common goals for gene expression studies. A gene signature is a rule to predict patient outcome, usually survival or progression, from the expression of a relatively small number of genes. A molecular target is a feature in a cancer cell microenvironment that contributes to malignant growth and that can be modified externally for treatment purposes.
Developing gene signatures involves sifting through thousands of genes and selecting a few that will predict the outcome. If the accuracy of these predictions is assessed on the same data that were used for developing the gene signature, the results will be biased. For this reason, each signature needs validation. It may appear that one needs to be as sophisticated as possible in gene signature development, even at the expense of the validation process. This article will present some counter-arguments that emphasize that model development is receiving more than its fair share of resources.
Because genes in a gene signature predict the outcome, it is natural to conclude that they will be appropriate molecular targets. It turns out that this is not necessarily the case: the way gene signatures are developed does not give priority to mechanistic or causal links between genes and outcome. Reasons for this will be discussed. The same reasoning also explains why there is little, if any, overlap between multiple gene signatures developed by different research groups.
GENE SIGNATURES
Recent high-profile publications reporting on the development of gene signatures for breast1 and lung2 cancers have attracted widespread interest from scientists and clinicians. The gene signature predicting response to FOLFIRI (5-fluorouracil, leucovorin, irinotecan) in patients with advanced colorectal cancer played a similar stimulating role for the community of gastrointestinal oncologists and researchers.3
In each of these three articles, a different technique was used to develop the signatures, a typical feature of the gene signature studies. The scientific community has been frenzied by the proliferation of methods to develop gene signatures. These methods are usually derivatives of statistical regression or machine learning techniques. Commonly used ones are proportional hazards regression, recursive partitioning, neural networks, and support vector machines. Despite the fanfare, there is little evidence that increased methodologic sophistication has resulted in substantial improvements in predictive accuracy. This can be explained by the possible abundance of “low-hanging fruit”: There are some, perhaps many, genes that are reasonably good predictors of outcome, and most sensible methods, including simpler ones, will capture a few of these genes. Sophisticated methods will include more genes in the mix, but they will make only small improvements in the overall predictive accuracy. This suggests that the return on investment on sophisticated methodology for making predictions is relatively low. For example, in the FOLFIRI gene signature study,3 there were several individual genes reaching 90% accuracy and the gene signature, which was developed using a support vector machine, improved this to approximately 95%. Presence of several individual genes with good accuracy suggests that a simple logistic regression model might have yielded the same results.
Various simulation studies comparing traditional regression approaches to these modern methods have concluded that there is little, if any, advantage to using a sophisticated method.4–7 These studies also found that the benefit of the sophisticated machinery is available only when the number of samples is in the thousands. This suggests that these methods may be more appropriate in the future when cost and data maturity problems are resolved.
Utilization of findings that claim good predictive accuracy requires a good deal of care about validation. Validation in this context means obtaining an estimate of predictive accuracy that is free of bias. Such unbiased estimates represent the performance of the signature when it is applied in practice to various patient populations. Model performance evaluated on the same set from which the data were derived usually overstates the true model performance. This is especially important in gene signatures: the number of genes is an order of magnitude greater than the number of samples and it is easy to overfit. To ensure that the incremental gains resulting from complex models are not sophistry, we should demand careful validation of gene signatures before we adopt them for routine use.
Approaches to Validation
There are three approaches to validation: independent-sample validation, split-sample validation, and internal validation. Independent- sample validation is considered the gold standard of model validation techniques. This requires an independently obtained data set that was not used in developing the signature. Independent validation studies are sometimes performed by investigators other than those who developed the signature first. A close cousin of independent-sample validation is splitsample validation. In this approach, the observations are randomly divided into two groups: one is used for model development and the other for validation. While this method gives an essentially honest picture of the predictive accuracy of a gene signature, it does not capture the betweenstudy variability that independent-sample validation can identify. For this reason, estimates obtained by split-sample methods remain slight overestimates of the true predictive performance.
Split-sample validation is simple to implement and communicate, but requires an initial sample that is of sufficient size, so when split into two parts, there will be enough observations in each part to effectively perform model development and validation. In addition, some investigators continue to find it irksome that their sample size is effectively reduced. Internal validation methods have been developed for these situtations. Internal validation can be performed via either cross-validation or bootstrap validation. Cross-validation is a widely used internal validation method that tries to mimic the split-sample approach by creating multiple splits on the same data set and repeating the process of model development and validation on the samples. Bootstrap validation is a variation on the same theme, relying on re-sampled data sets where subjects are sampled from the original data with replacement.
Internal validation is acceptable during the initial development of a signature.8 Before putting a gene signature to routine use, however, we should insist on independent- sample or split-sample validation. If this sounds like stating the obvious, it is sobering to read that substandard validation was one of the three common statistical errors in microarray profiling studies.9 It appears that many clinical journals have not yet adopted rules or policies that will ensure that gene signatures are valid tools for clinical use.
MOLECULAR TARGETS
While genes signatures are specifically developed for predicting outcome, resourceful scientists have used them for identifying molecular targets. This looks like a free lunch at first: if overexpression of a gene increases the likelihood of disease progression or death, a molecular intervention suppressing the expression of that gene might be a reasonable treatment strategy that will improve the outcome.
The fallacy in this thinking centers around the statistical concepts of correlation and causation. Suppose in a simple world there are two genes, Gene 1 and Gene 2, and the expression of Gene 1 is the only determinant of outcome. It also happens that the expression of Gene 1 also drives the expression of Gene 2 but the expression of Gene 2 has no mechanistic connection to the outcome. This is schematically depicted in Figure 1. In this setting, one can easily find Gene 2 to be correlated to the outcome because both of them follow from Gene 1. As long as this correlation is high enough, Gene 2 will be an accurate predictor of outcome and there is nothing wrong with using it as such, despite the fact that Gene 2 and the outcome are not mechanistically linked. Nevertheless, it would be incorrect to decide that Gene 2 is an appropriate target, because modifying Gene 2 will have no bearing on the outcome. On the other hand, Gene 1 is an appropriate target and a predictor. The problem is, using only clinical data in the absence of a mechanistic model, most methods will have difficulty distinguishing Gene 1 from Gene 2 as a preferable predictor. Thus, either gene is as likely as the other to be selected as a predictor in a gene signature. In contrast, Figure 2 shows Genes 1 and 2 with both directly linked to the outcome. In this case, both genes are potential predictors and targets. It has been observed previously that the scenario in Figure 2 is much more amenable to drug development.10
Figure 1:
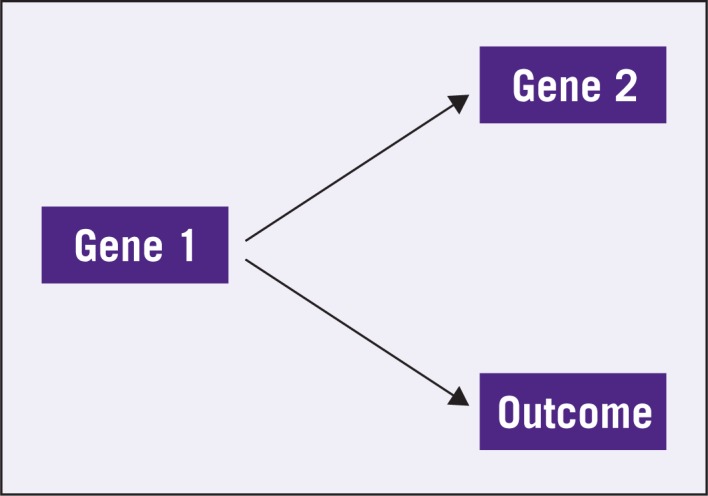
Only Gene 1 has a mechanistic connection with the outcome but Gene 2 might be identified as a predictor because of its connection with Gene 1.
Figure 2:
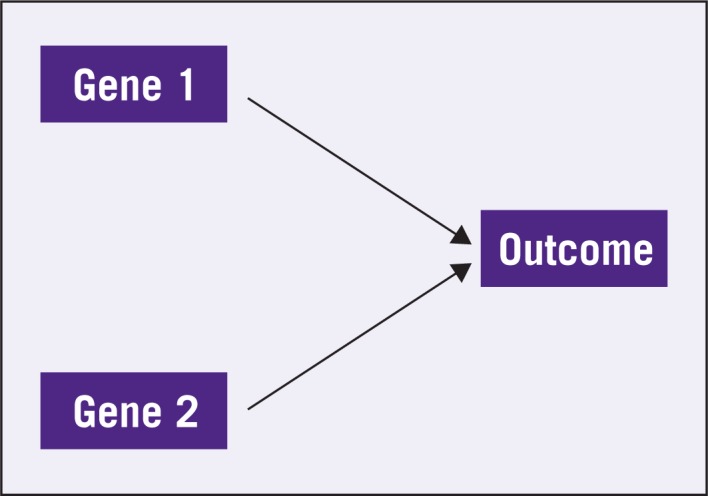
Both Gene 1 and Gene 2 are mechanistically connected to the outcome.
Now consider a pathway with several genes in a map, which is essentially interconnected versions of Figures 1 and 2. It quickly becomes obvious that there are several possible mechanistic configurations and it is impossible for any data analysis mechanism to sift through them with moderate sample sizes. It is important to keep in mind that a gene signature has no reason to uncover all (or even one) of the mechanistic links in a pathway. Yet, without some understanding of the links, it is impossible to identify targets.
This reasoning explains another phenomenon that has baffled some oncologists. It is entirely possible to have two (in fact several) gene signatures that have no common genes.10,11 Either due to slight variations in methodology or sampling error, different genes may be used to represent the information contained in one set of genes. In Figure 1 or 2, for example, a method might pick Gene 1 and another method might pick Gene 2 for inclusion in a signature, and it is unlikely that both will be picked. The resulting signatures will contain either of the two genes and not both, resulting in a non-overlapping signature. Hence, gene signatures are not unique and existence of other non-overlapping gene signatures does not necessarily invalidate one of the signatures.
DISCUSSION
This article emphasized the different nature of statistical methods used and required for developing gene signatures and identifying molecular targets.
Many sophisticated methods for developing gene signatures are available. Each has various advantages over simple methods, but large sample sizes are needed to fully realize the benefit of this machinery. While using these methods is certainly welcome, it is not essential. Instead, resources can be directed to careful validation of the gene signature.
The relative ignorance of validation in this field underscores a psychological aspect of model development as well. Finding a model that predicts well is a creative exercise and requires skills that scientists are trained to have. Because the audience is also of a similar mindset, model development gets the lion’s share of attention and credit. By comparison, validation requires discipline and organization more than creativity and may not strike some as an attractive line of research. It is the collective responsibility of all researchers as reviewers, readers, and users of gene signature works to make sure that validation is properly performed.
Even the most carefully validated gene signatures will not necessarily reveal novel molecular targets. This is due to the nature of prediction: a good predictor merely needs to be correlated with outcome. A causal link between the two is helpful but not necessary. Thus, a gene signature does not a target make. In addition, gene signatures are hardly unique. It is possible to have two well-developed and well-validated signatures with no overlap. This is also due to the fact that many genes are correlated with the outcome — not because of a mechanistic connection but through other genes. Two non-overlapping signatures simply capture different such genes. In terms of target identification, one should be cognizant of the fact that, while genes in a well-validated signature may form starting points for further exploration, identifying molecular targets will require more careful and focused experimentation than large-sample microarray studies.
Rapid technologic developments combined with scientific advances have made it possible to turn genetic information into prognostic and treatment knowledge. Gene signatures and molecular targets are two promising ways of achieving this goal. Understanding the shortcomings and fallacies of current practices will help us build and choose better signatures and identify more appropriate molecular targets.
Footnotes
Disclosures of Potential Conflicts of Interest
Dr. Gönen has indicated no potential conflicts of interest.
REFERENCES
- 1.van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999– 2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 2.Liu R, Wang X, Chen GY, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007;356:217– 226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 3.Del Rio M, Molina F, Bascoul-Mollevi C, et al. Gene expression signature in advanced colorectal cancer patients select drugs and response for the use of leucovorin, fluorouracil, and irinotecan. J Clin Oncol. 2007;25:773–780. doi: 10.1200/JCO.2006.07.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sargent DJ. Comparison of artificial neural networks with other statistical approaches: results from medical data sets. Cancer. 2001;91(suppl 8):1636–1642. doi: 10.1002/1097-0142(20010415)91:8+<1636::aid-cncr1176>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 5.Kattan MW. Comparison of Cox regression with other methods for determining prediction models and nomograms. J Urol. 170:S6–S9. doi: 10.1097/01.ju.0000094764.56269.2d. discussion S10, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Linder R, König IR, Weimar C, et al. Two models for outcome prediction—a comparison of logistic regression and neural networks. Methods Inf Med. 2006;45:536–540. [PubMed] [Google Scholar]
- 7.Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for classification of tumors using gene expression data. J Am Stat Assoc. 2002;97:77–87. [Google Scholar]
- 8.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J Clin Oncol. 2005;23:7332–7341. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 9.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99:147–157. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 10.Massague J. Sorting out breast-cancer gene signatures. N Engl J Med. 2007;356:294–297. doi: 10.1056/NEJMe068292. [DOI] [PubMed] [Google Scholar]
- 11.Parmigiani G, Garrett-Mayer ES, Anbazhagan R, et al. A cross-study comparison of gene expression studies for the molecular classification of lung cancer. Clin Cancer Res. 2004;10:2922–2927. doi: 10.1158/1078-0432.ccr-03-0490. [DOI] [PubMed] [Google Scholar]