

Summary
During cutaneous wound healing, epidermal keratinocytes play essential roles in the secretion of factors that promote angiogenesis. However, specific cues in the wound microenvironment that trigger the production of pro-angiogenic factors by keratinocytes, and the cellular receptors that mediate this response, remain unclear. In this study, we exploited a model of conditional integrin knockout to demonstrate impaired wound angiogenesis in mice that lack α3β1 integrin in epidermis. In addition, we used genetic and shRNA approaches to determine that α3β1-integrin deficiency in keratinocytes leads to reduced mRNA and protein expression of the pro-angiogenic factor mitogen-regulated protein 3 (MRP3; also known as PRL2C4), and to demonstrate that this regulation provides a mechanism of keratinocyte-to-endothelial-cell crosstalk that promotes endothelial-cell migration. Finally, we showed that the impaired wound angiogenesis in epidermis-specific α3-integrin-knockout mice is correlated with reduced expression of MRP3 in wounded epidermis. These findings identify a novel role for α3β1 integrin in promoting wound angiogenesis through a mechanism of crosstalk from epidermal to endothelial cells, and they implicate MRP3 in this integrin-dependent crosstalk. Such a mechanism represents a novel paradigm for integrin-mediated regulation of wound angiogenesis that extends beyond traditional roles for integrins in cell adhesion and migration.
Keywords: Integrin, MRP3 (Prl2c4), Proliferin, Keratinocyte, Wound healing
Introduction
Cutaneous wound healing is a complex process requiring collaborative interactions between distinct cell types that reside in different compartments of the skin, including epidermal keratinocytes, dermal fibroblasts, macrophages, neutrophils and vascular endothelial cells (Martin, 1997; Singer and Clark, 1999). Following wounding, the rapid regeneration of the epidermis, or re-epithelialization, is required to cover the denuded dermal surface of the wound and restore the barrier function and integrity of the skin. Keratinocyte migration and proliferation are crucial for this process (Santoro and Gaudino, 2005). In addition to their roles in re-epithelialization, wound keratinocytes secrete cytokines, growth factors and extracellular proteases that promote angiogenesis (Madlener et al., 1998; Salo et al., 1994; Santoro and Gaudino, 2005; Singer and Clark, 1999), thereby mediating crosstalk from keratinocytes to endothelial cells that promotes wound healing. Activation of wound-edge keratinocytes is probably triggered by newly encountered elements of the wound environment, including serum factors and exposed or newly synthesized cell-adhesion ligands in the provisional extracellular matrix (ECM) (Grinnell, 1992; Martin, 1997; Singer and Clark, 1999). However, specific extracellular cues in the wound environment that promote production of pro-angiogenic factors by keratinocytes remain unknown.
The mitogen-regulated proteins/proliferins (MRPs) are members of the prolactin growth hormone superfamily that comprise a group of highly homologous, growth-factor-inducible genes with roles in promoting endothelial cell proliferation, migration and angiogenesis during fetal and placental development (Corbacho et al., 2002; Fassett and Nilsen-Hamilton, 2001; Jackson et al., 1994; Soares et al., 2007; Toft et al., 2001). MRPs regulate endothelial cell migration and proliferation through interactions with cell-surface receptors, including the insulin-like growth factor 2 (IGF2)/mannose 6-phosphate receptor, MRP/proliferin receptor, or other unidentified receptors (Corbacho et al., 2002; Jackson et al., 1994; Nelson et al., 1995; Volpert et al., 1996). For example, endothelial cell migration can be induced by MRP binding to the IGF2 receptor and activation of a G-protein-coupled, mitogen-activated protein kinase (MAPK) pathway (Groskopf et al., 1997). Some studies have shown that increased MRP gene expression is associated with cellular immortalization and transformation (Malyankar et al., 1994; Parfett et al., 1985). Another study identified a novel role for MRP3, also known as PRL2C4 (prolactin family 2, subfamily c, member 4), as a likely regulator of angiogenesis during cutaneous wound healing (Fassett and Nilsen-Hamilton, 2001). Indeed, Mrp3 gene expression is induced in activated wound keratinocytes, and MRP3 protein continues to accumulate with peak expression occurring 4-5 days post-injury, corresponding well with the timing of wound angiogenesis and suggesting that MRP3 secretion by keratinocytes may promote angiogenesis during wound repair (Fassett and Nilsen-Hamilton, 2001). MRP3 expression can be induced in cultured keratinocytes by keratinocyte growth factor (KGF) (Fassett and Nilsen-Hamilton, 2001). However, extracellular cues that induce MRP3 secretion by keratinocytes during in vivo wound healing are not yet known.
A key to understanding the pathogenesis of chronic wounds, and age-related wound defects, is identifying cellular receptors that mediate responses of the epidermis to provisional wound matrix, and determining how changes in these receptors contribute to impaired wound healing. Integrins are the major cell-surface receptors for cell adhesion and migration (Hynes, 2002), and epidermal keratinocytes express several integrins that bind ECM ligands in provisional wound ECM (Litjens et al., 2006; Watt, 2002). Although epidermis-specific deletion of the β1 integrin subunit have caused severe wound healing defects (Grose et al., 2002), knockouts of individual integrins have had surprisingly mild effects on wound healing, and in vivo roles for some keratinocyte integrins remain unclear (Grenache et al., 2007; Litjens et al., 2006; Zweers et al., 2007). α3β1 integrin is expressed in both resting and wounded epidermis, where its main ECM ligand is laminin-332/laminin-5 (LN-332; previously known as laminin-5, kalinin, nicein or epiligrin). Newborn mice that lack α3β1 integrin owing to the Itga3 (which encodes α3 integrin)-null mutation display epidermal microblisters and organizational defects in the cutaneous basement membrane (DiPersio et al., 1997), which persist to some extent into adulthood of conditional Itga3-knockout mice lacking α3β1 integrin in epidermis (Margadant et al., 2009). In addition, studies in both mouse and human keratinocytes have supported an important role for α3β1 integrin and LN-332 in the regulation of polarized cell spreading and migration (Choma et al., 2004; Frank and Carter, 2004; Hamelers et al., 2005; Nguyen et al., 2000). Whereas keratinocyte migration is clearly facilitated by other integrins that also bind ligands present in the wound ECM, such as fibronectin and exposed dermal collagen (Clark et al., 1982; Nguyen et al., 2000; Pilcher et al., 1997), keratinocytes that are deficient in LN-332 show defects in migration even on collagen (Ryan et al., 1999), suggesting an important role for LN-332 deposition during epidermal migration over the provisional ECM of the wound bed. Consistently, de novo synthesis of precursor LN-332 is an early event in the activation of wound-edge keratinocytes (Litjens et al., 2006; Nguyen et al., 2000). However, two recent in vivo studies performed in distinct genetic models have indicated either pro-migratory or anti-migratory effects of α3β1 integrin during wound re-epithelialization (Margadant et al., 2009; Reynolds et al., 2008), suggesting that roles for this integrin in wound closure are more complex than previously anticipated (see Discussion).
In contrast to their widely studied roles in keratinocyte migration, proliferation and matrix assembly, potential roles for epidermal integrins in regulating the production of pro-angiogenic factors that promote wound angiogenesis have not been extensively explored. In the present study we have exploited a murine model of α3-integrin knockout to assess the effects of α3β1-integrin deficiency in epidermal keratinocytes on wound angiogenesis in vivo and endothelial cell function in vitro. We demonstrate that ablation of α3β1 integrin in the epidermis by conditional Itga3 knockout results in impaired wound angiogenesis in vivo. In addition, we use a combination of genetic and shRNA approaches to show that α3β1 integrin induces MRP3 mRNA expression and protein secretion in cultured keratinocytes, and that this regulation is required for keratinocyte-to-endothelial-cell crosstalk, which promotes endothelial-cell migration. Finally, we show that impaired wound angiogenesis in epidermis-specific α3-integrin-knockout mice is correlated with reduced expression of MRP3 in the wounded epidermis. These findings reveal a novel role for α3β1 integrin in promoting keratinocyte-to-endothelial-cell crosstalk by regulating expression in keratinocytes of a pro-angiogenic factor, MRP3, and they provide the initial evidence that this regulation promotes wound angiogenesis in vivo. Our findings may therefore represent a novel paradigm for the regulation of wound healing by certain integrins expressed in epidermis that extends beyond their traditional, well-documented roles in regulating cell adhesion and migration.
Results
Derivation of mice with epidermis-specific deletion of α3β1 integrin
To test the importance of α3β1 integrin expression in the epidermis for wound angiogenesis, we exploited conditional knockout mice with epidermis-specific deletion of the α3-integrin subunit (summarized in Fig. 1A). Mice with floxed Itga3 alleles (referred to here as Itga3flx/flx mice), in which exon 3 was flanked by loxP sites, were crossed with Itga3+/+ mice that express Cre recombinase under control of the keratin 14 promoter (K14-Cre), which restricts Cre expression to basal keratinocytes of the interfollicular and follicular epidermis, including the stem-cell compartment in the hair follicle (Castilho et al., 2007; Huelsken et al., 2001). The resulting mice of the K14-Cre:Itga3flx/+ genotype were then backcrossed to obtain K14-Cre:Itga3flx/flx mice. Where indicated, some experiments utilized mice of the K14-Cre:Itga3flx/– genotype, in which one of the Itga3 alleles corresponds to the conventional Itga3-null allele described previously (Kreidberg et al., 1996). Hereafter, K14-Cre:Itga3flx/flx mice or K14-Cre:Itga3flx/– mice are referred to as α3eKO mice; control mice were littermates that lacked K14-Cre and were of Itga3flx/flx or Itga3flx/– genotype, as appropriate.
Fig. 1.

α3-integrin expression is absent from epidermis of α3eKO mice. (A) Schematic representation of the strategy used to generate the targeted floxed Itga3 allele (see Materials and Methods for details). Frt and LoxP sites are indicated by red and blue triangles, respectively. NeoR, neomycin-resistance gene. (B,C) H&E histological sections from back skin of adult Itga3flx/– mice that either lack (B, control) or express (C, α3eKO) K14-Cre. Scale bar: 50 μm. (D-I) Frozen skin sections from adult control mice (Itga3flx/flx; D,F,H) or α3eKO mice (K14-Cre:Itga3flx/flx; E,G,I) were stained by immunofluorescence with anti-keratin 14 (K14), anti-α3-integrin (α3), or anti-LN-332 (LN). Arrowheads in D-G point to basal keratinocytes; arrows in H,I point to basement membrane; e, epidermis; d, dermis; f, hair follicle. Note that specific α3-integrin staining is absent from the basal cell layer of α3eKO epidermis [G; asterisks in F,G indicate non-specific staining in both control and α3eKO epidermis, which was also seen with the corresponding pre-immune serum (not shown)]. Scale bar: 25 μm.
Immunofluorescence of frozen skin sections from control mice that were of the Itga3flx/flx genotype, but lacked the K14-Cre transgene, showed that α3-integrin staining was easily detected in basal keratinocytes of the epidermis (Fig. 1F), as expected (DiPersio et al., 1997). By contrast, specific α3-integrin staining was absent from the epidermis of K14-Cre:Itga3flx/flx mice, confirming undetectable expression of α3β1 integrin in the epidermis of α3eKO mice (Fig. 1G; asterisks in Fig. 1F,G indicate non-specific staining in the cornified layer that was also seen with the corresponding pre-immune serum; not shown). Skin sections stained with Hematoxilin and Eosin (H&E) did not reveal gross differences in the morphology or thickness of the interfollicular epidermis between control and α3eKO mice (Fig. 1B,C), consistent with essentially normal development and stratification of interfollicular epidermis in previous studies of α3β1-integrin-deficient skin (Conti et al., 2003; DiPersio et al., 1997). However, we noticed a reduction in the size of hair follicles in α3eKO mice compared with control mice, as evident in the images of Fig. 1B,C. This observation is reminiscent of previously reported defects in hair follicle development in Itga3-null skin grafts (Conti et al., 2003), although less severe than the progressive loss of hair follicles that was reported in adult mice with epidermis-specific deletion of the β1 integrin subunit (Brakebusch et al., 2000). A recent study using a similar model of epidermis-specific deletion of α3 integrin reported inflammation around the eyes and local hair loss (alopecia) that appeared around 3 to 4 months after birth and progressed with age (Margadant et al., 2009). We observed similar phenotypes in α3eKO mice (not shown), although extensive alopecia was not usually obvious until several months later, possibly reflecting differences in genetic background.
LN-332 staining was detected in the cutaneous basement membrane of both control and α3eKO mice (Fig. 1H,I), consistent with previous reports that α3β1 integrin is not required for LN-332 synthesis in vivo (DiPersio et al., 1997; DiPersio et al., 2000b). Margadant and co-workers reported occasional microblistering at the dermal-epidermal junction in α3β1-integrin-deficient epidermis of adult (1-year-old) mice (Margadant et al., 2009), a phenotype reminiscent of the microblistering that accompanies extensive basement membrane defects in Itga3-null neonatal mice (DiPersio et al., 1997). Although we did not observe such defects in the skin of young adult mice (<4 months) used in our current study, it seems likely that more extensive analysis over a greater age range may reveal occasional basement membrane defects and microblisters. The reduced incidence and severity of these phenotypes in mature α3eKO mice may indicate partial recovery in adult mice from basement membrane defects that are more prominent in Itga3-null neonatal mice (DiPersio et al., 1997). Alternatively, it is possible that the more extensive basement membrane defects seen in newborn Itga3-null mice reflect a requirement for α3β1 integrin in other skin cell types from which α3 integrin is also absent, or at a stage of prenatal development that precedes efficient K14-Cre-mediated deletion of the Itga3flx allele in α3eKO mice.
In a recent study of epidermis-specific deletion of the MET receptor, there was a substantial expansion in the wounded epidermis of a residual population of keratinocytes (∼5%) that had escaped Cre-mediated recombination and retained MET expression (Chmielowiec et al., 2007). This result illustrates the importance of assessing wound samples for compensation by residual cells that retain expression of a targeted gene that may have an essential role in wound healing. Therefore, we next determined if a similar expansion of residual α3β1-integrin-expressing cells occurs in wounded epidermis of α3eKO mice. To assess α3-integrin expression in wounded epidermis, frozen sections were prepared 5 days post-injury from 4-mm punch biopsy wounds of α3-integrin control and α3eKO mice and stained by immunofluorescence for α3 integrin. In control mice, α3 integrin was easily detected in keratinocytes of the migrating epidermal tongue (Fig. 2E). By contrast, α3-integrin expression remained undetectable in the wound-edge keratinocytes of α3eKO mice (Fig. 2F), and staining was comparable to that seen with the corresponding pre-immune serum (Fig. 2G,H), confirming that there was no expansion of residual cells that retained detectable levels of α3β1 integrin.
Fig. 2.

α3 integrin is absent from wound keratinocytes in α3eKO mice. (A,B) H&E-stained histological sections from 5-day excision wounds showing the epidermis migrating over the wound bed. Sections were from control mice (Itga3flx/–; A) or α3eKO mice (K14-Cre:Itga3flx/–; B); dotted lines outline the migrating epidermal tongue. Scale bar: 50 μm. (C-J) A series of adjacent cryosections from 5-day excision wounds of control mice (Itga3flx/flx; C,E,G,I) or α3eKO mice (K14-Cre:Itga3flx/flx; D,F,H,J) were stained by immunofluorescence with anti-keratin 14 (C,D), anti-α3-integrin (E,F), corresponding pre-immune serum (G,H), or anti-LN-332 (I,J). Arrowheads point to the leading edge of migrating epidermis. Scale bar: 50 μm. e, epidermis; d, dermis; s, eschar; wb, wound bed.
Previous studies in cultured Itga3-null keratinocytes (deHart et al., 2003; DiPersio et al., 2000a) and epidermis of Itga3-null mice (DiPersio et al., 2000b) showed that α3β1 integrin is not required for LN-332 expression by epidermal keratinocytes. However, it has been suggested that α3β1-integrin-mediated signaling in keratinocytes may regulate the deposition of LN-332 during wound repair in vivo (Hamelers et al., 2005). Therefore, we also examined LN-332 staining at the wound edge. LN-332 was present around the migrating epidermis of both control and α3eKO mice, and its distribution reflected that it was not yet organized into basement membrane (Fig. 2I,J), indicating that α3eKO keratinocytes are capable of depositing LN-332 into the wound and that α3β1 integrin is not essential for LN-332 expression. However, we cannot rule out the possibility that proteolytic processing of LN-332, or its incorporation into newly synthesized basement membrane, is altered in α3eKO wounds.
Mice that lack α3β1 integrin in the epidermis display reduced angiogenesis during wound healing
Epidermal keratinocytes are well known to secrete pro-angiogenic factors during wound healing (Santoro and Gaudino, 2005; Singer and Clark, 1999), although a role for integrins in this regulation has not been extensively explored. To test if α3β1 integrin in the epidermis is important for induction of wound angiogenesis in vivo, frozen sections were prepared 5 days post-injury from control and α3eKO mice and stained with anti-CD31/PECAM 1 to label blood vessels. As expected, control mice (Itga3flx/–) showed increased blood vessel density in dermis directly adjacent to the wound bed, compared with dermis distal to the wound (Fig. 3A, control), indicative of robust induction of angiogenesis during this phase of wound repair. Blood vessel density in these mice was similar to that observed in wounded skin of Itga3+/+ wild-type mice (data not shown), indicating that one functional Itga3 allele was sufficient for normal wound angiogenesis. By contrast, wounds from α3eKO mice (K14-Cre:Itga3flx/–) showed substantially reduced blood vessel density compared with wounds from control mice (Fig. 3A, α3eKO). Quantification of these data revealed that α3eKO mice displayed a statistically significant reduction in blood vessel density of about 60% compared with control mice (Fig. 3B). These results indicate that α3β1 integrin is required in epidermis for full induction of angiogenesis during wound healing.
Fig. 3.

Wound angiogenesis is reduced in mice with α3β1-integrin-deficient epidermis. (A) CD31/PECAM-1 immunostaining in the dermis in frozen skin sections away from (distal) or within (wound bed) 5-day wounds of control (Itga3flx/–) or α3eKO (K14-Cre:Itga3flx/–) mice. Arrows point to examples of CD31-positive blood vessels. Scale bar: 50 μm. (B) Quantification of blood-vessel density. Three 20× fields were collected from within each wound bed (see C), and total area of CD31-staining was determined per field. Average blood-vessel area was then determined for each wound; each wound was from a separate 6.5-week-old mouse. Graph shows CD31-positive area for control mice and α3eKO mice. Data are mean ±s.e.m.; five mice were analyzed for each genotype; Student's t-test, *P<0.01. (C) Schematic illustration of a wound, showing approximate positions of fields (boxes, not to scale) that were imaged for analysis of blood-vessel density in panel B. d, distal; e, epidermis; s, eschar; wb, wound bed.
α3β1 integrin in keratinocytes regulates the secretion of a pro-angiogenic factor(s) that promotes endothelial-cell migration
The impaired angiogenesis that we observed in wounds of α3eKO mice suggests a novel role for this integrin in promoting crosstalk between the epidermal and endothelial compartments of the skin. Therefore, we next tested if α3β1 integrin in epidermal keratinocytes is required for the crosstalk to endothelial cells that occurs through secretion of soluble factors that promote endothelial-cell migration. For these studies we utilized keratinocyte cell lines derived from a wild-type mouse (WT cells) or an Itga3-null mouse (α3– cells) (DiPersio et al., 2000a). As an additional control, α3β1-integrin expression was restored in the Itga3-null cell line by stable transfection with a cDNA encoding the human α3-integrin subunit (α3+ cells) (Iyer et al., 2005). Transwell assays were performed to measure relative migration of human umbilical vein endothelial cells (HUVECs) in response to factors secreted by keratinocytes. Serum-starved HUVECs were seeded into the upper chambers of gelatin-coated transwell filters, then conditioned media collected from 24 hour confluent cultures of WT, α3– or α3+ keratinocytes were added to the lower chambers and tested for effects on HUVEC migration over a 4-hour period. There was a significant stimulation of HUVEC migration in response to conditioned medium from WT keratinocytes, compared with basal migration seen in response to unconditioned medium. By contrast, HUVEC migration in conditioned medium from α3– keratinocytes was similar to basal levels (Fig. 4). Importantly, ability to stimulate HUVEC migration was restored in α3– cells that were rescued with human α3 integrin (Fig. 4). These data indicate that α3β1-integrin deficiency in keratinocytes leads to reduced secretion of factors that induce endothelial-cell migration.
Fig. 4.
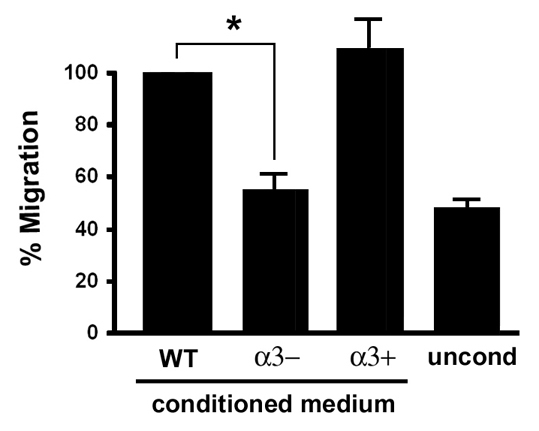
α3β1-integrin deficiency in keratinocytes leads to reduced secretion of soluble factors that induce endothelial-cell migration. Transwell migration assays were performed to compare relative cell migration of HUVECs in response to treatment with conditioned culture medium from wild-type keratinocytes (WT), Itga3-null keratinocytes (α3–), or the latter cells rescued with human α3 integrin (α3+). Bottom chambers of transwells contained keratinocyte-conditioned medium, or unconditioned medium (uncond) as a control to establish baseline migration. Graph shows average HUVEC migration as a percentage of that in cells treated with medium from wild-type cells; mean ±s.e.m.; n=3; *P<0.023, Student's t-test.
α3β1 integrin is required in keratinocytes for expression of the pro-angiogenic factor MRP3
We next wanted to identify the α3β1-integrin-dependent factor(s) involved in keratinocyte-mediated induction of endothelial-cell migration. Reduced wound angiogenesis observed in α3eKO mice (Fig. 3) prompted us to ask whether α3β1 integrin is required in keratinocytes for expression of known pro-angiogenic factors in wound healing. We focused our initial attention on VEGF, MMP9 and MRP3, all three of which are expressed in keratinocytes and have been previously implicated in wound angiogenesis (Fassett and Nilsen-Hamilton, 2001; Madlener et al., 1998; Salo et al., 1994; Santoro and Gaudino, 2005; Singer and Clark, 1999). First, total RNA was isolated from α3+ and α3– keratinocytes and analyzed for expression of these pro-angiogenic factors. Interestingly, Mrp3 mRNA was considerably more abundant in α3+ cells than in α3– cells (Fig. 5A). Mmp9 mRNA expression was also higher in α3+ cells than in α3– cells, which we have shown previously (Iyer et al., 2005), and served as a positive control for α3β1-integrin-dependent effects in this assay (Fig. 5A). We did not detect α3β1-integrin-dependent effects on expression of Vegf mRNA (Fig. 5A). In accordance with α3β1-integrin-dependent effects on Mrp3 mRNA, MRP3 protein was easily detected in cell lysates from both α3+ and WT keratinocytes, but it was barely detectable in lysates from α3– keratinocytes (Fig. 5B). An upper non-specific band, which was also detected by the corresponding pre-immune serum (not shown), served as an additional loading control (Fig. 5B, asterisk). Quantification of data from four independent experiments indicated that total MRP3 protein, after normalization to keratin 14, was decreased to approximately 5% in α3β1-integrin-deficient cells compared with either of the α3β1-integrin-expressing cell lines (Fig. 5C). Assessment of conditioned culture medium confirmed that α3β1-integrin-expressing keratinocytes secreted much higher levels of MRP3 than α3-integrin-deficient keratinocytes (Fig. 5B). These data indicate that α3β1 integrin promotes MRP3 expression and secretion in vitro, and they identify this pro-angiogenic factor as a candidate for mediating α3β1-integrin-dependent crosstalk from keratinocytes to endothelial cells that promotes wound angiogenesis.
Fig. 5.

α3β1 integrin regulates Mrp3 mRNA and protein expression in cultured keratinocytes. (A) α3β1-integrin-expressing (α3+) or α3-integrin-deficient (α3–) keratinocytes were cultured for 24 hours on LN-332 ECM in serum-free medium, and total RNA was isolated. Semi-quantitative RT-PCR was performed to assess mRNA levels for Mrp3, Mmp9, Vegf (bands correspond to 120 kD and 160 kD isoforms) and β-actin. Results shown are representative of three independent experiments. (B) Keratinocytes were cultured as in A, and total cell lysate, or equivalent proportions of conditioned culture media, were assessed for MRP3 protein levels by immunoblot. Cells assayed included wild type (WT), Itga3-null (α3–) or the latter cells stably transfected with human α3-integrin subunit (α3+). Note that MRP3 migrates as a diffuse band, probably due to heterogeneity in extent of glycosylation (Corbacho et al., 2002). The asterisk indicates a non-specific band, which was also detected with corresponding pre-immune serum (not shown). Immunoblot for anti-keratin 14 (K14) was performed to control for loading differences. (C) Quantification of MRP3 protein levels in cell lysates such as those from B. MRP3 protein levels are indicated as a proportion of levels in wild-type cells after normalization to keratin 14. Data are the mean ±s.e.m.; n=3; *P<0.001, Student's t-test.
α3β1-integrin-dependent expression of MRP3 in keratinocytes stimulates endothelial-cell migration
To test whether α3β1-integrin-dependent expression and secretion of MRP3 (Fig. 5) is involved in the keratinocyte-mediated induction of endothelial-cell migration (Fig. 4), α3β1-integrin-expressing keratinocytes were stably infected with lentiviruses that express shRNAs that target murine Mrp3, or non-targeting shRNA as a control. Mrp3 mRNA was decreased by 70% or more in cells expressing each of three independent shRNAs that target this transcript, compared with cells infected with control shRNA, whereas β-actin mRNA levels were unaffected by either control or targeting shRNAs (Fig. 6A). Immunoblot analysis of conditioned medium from these cells confirmed efficient suppression of secreted MRP3 protein levels by each Mrp3 shRNA (Fig. 6B). The HUVEC migratory response to conditioned medium from keratinocytes that express control shRNA was about 2.5-fold higher than the basal response to unconditioned medium. By contrast, the migratory response to conditioned medium from cells that express Mrp3-targeting shRNA was reduced to basal levels (Fig. 6C). Importantly, similar results with three different shRNAs that target distinct regions of the murine Mrp3 transcript indicate that this inhibition was not due to off-target RNAi effects. Poor transfection efficiency of the α3– keratinocytes precluded our ability to restore MRP3 expression to endogenous levels seen in WT cells. However, we found that conditioned medium from 293 cells that overexpress recombinant, His-tagged MRP3 at high levels induced HUVEC migration only 1.2-fold above levels seen in response to conditioned medium from control 293 cells (P<0.046, t-test; data not shown). Although we cannot rule out cell-specific modifications of MRP3 or inhibitory effects of the His-tag, this result may indicate that other keratinocyte factors in addition to MRP3 are required to induce a robust migratory response. Nevertheless, results in Fig. 6 show that α3β1-integrin-dependent MRP3 secretion in keratinocytes plays a crucial role in mediating crosstalk to endothelial cells that promotes their migration. These findings support a model wherein α3β1-integrin-mediated expression of MRP3 in the epidermis contributes to wound angiogenesis in vivo.
Fig. 6.

MRP3 expression is required in keratinocytes for the α3β1-integrin-dependent induction of endothelial-cell migration. α3β1-integrin-expressing keratinocytes were transduced with lentivirus expressing non-targeting shRNA (ctrl) or three different shRNAs that target murine Mrp3 (1-3), and stable populations were selected in puromycin. To confirm stable suppression of Mrp3 mRNA and MRP3 protein, transduced cells were cultured on collagen for 24 hours in serum-free medium, then (A) total RNA was isolated for RT-PCR analysis of Mrp3 mRNA, or β-actin mRNA as a control, and (B) conditioned culture medium was collected for immunoblot analysis of secreted MRP3 protein. (C) Transwell migration assays were performed as in Fig. 4 to compare HUVEC migratory responses to conditioned culture medium from keratinocytes that express the control shRNA (ctrl) or each of the Mrp3-targeting shRNAs (1-3); treatment with unconditioned medium was included to establish baseline migration. Graph shows relative HUVEC migration normalized to basal migration of cells treated with unconditioned medium (uncond). Data are mean±s.e.m.; n≥4 over two independent experiments; *P<0.05 compared to cells expressing control shRNA; one-way ANOVA followed by a Dunnett's post-hoc test.
Deletion of α3β1 integrin from epidermis leads to reduced MRP3 expression during wound healing in vivo
MRP3 is the major MRP gene expressed in the epidermis of wounds, and it has been identified as a likely pro-angiogenic factor during wound healing (Fassett and Nilsen-Hamilton, 2001). Mrp3 gene expression is induced soon after wounding, and the MRP3 protein continues to accumulate in the epidermis to peak levels 4-5 days post-injury, consistent with a role in promoting wound angiogenesis (Fassett and Nilsen-Hamilton, 2001). To test if α3β1 integrin regulates MRP3 expression in wounded epidermis, frozen skin sections from control or α3eKO mice were immunostained with anti-MRP3 antibody. MRP3 levels were low or absent in normal, unwounded skin of both control and α3eKO mice (data not shown). Immunostaining of epidermis adjacent to 5-day wounds indicated that MRP3 expression was detected in the majority of control wounds (7/8 wounds examined), within both wound-proximal hair follicles (Fig. 7A, upper panels) and the migrating epidermal tongue over the wound bed (Fig. 7B, upper panels), consistent with previous findings that MRP3 is induced in hair follicles near the wound and in wound-edge epidermis by 5 days post-injury (Fassett and Nilsen-Hamilton, 2001). Interestingly, specific MRP3 staining was markedly reduced or absent in wound-proximal hair follicles (Fig. 7A, lower panels) or wound-edge epidermis (Fig. 7B, lower panels) in the majority of α3eKO wounds (8/11 wounds examined). The lack of a complete correlation in α3eKO mice between absence of α3β1 integrin and reduced MRP3 is likely to reflect partial compensation by other factors present in the complex wound microenvironment that can also induce MRP3, such as KGF (Fassett and Nilsen-Hamilton, 2001). Nevertheless, our observations that α3β1-integrin deficiency leads to both reduced MRP3 expression in wounds (Fig. 7) and reduced wound angiogenesis (Fig. 3) support an in vivo role for α3β1-integrin-mediated induction of MRP3 during wound angiogenesis.
Fig. 7.

Reduced expression of MRP3 in wounded epidermis of α3eKO mice. (A) Adjacent cryosections from skin that is proximal to 5-day wounds (approximately 1000 μm), were obtained from 6.5-week-old control mice (Itga3flx/–) or α3eKO mice (K14-Cre:Itga3flx/–) and stained by immunofluorescence with anti-keratin 14 (K14) to label follicular and interfollicular epidermis, or with anti-MRP3 or corresponding pre-immune serum, as indicated. Arrows point to hair follicles. (B) Adjacent cryosections of the wound edge, showing the migrating epidermal tongue (e), were collected from wounds of the same mice assessed in A and stained with anti-keratin 14, anti-MRP3 or pre-immune serum. Arrowheads point to the leading edge of migrating epidermis. Scale bars: 50 μm.
Discussion
Although an extensive literature supports important roles for integrins in angiogenesis, most previous studies have focused on integrins expressed on endothelial cells that function within these cells to regulate essential processes such as differentiation, proliferation, migration and survival (for reviews, see Hynes, 2007; Silva et al., 2008). By contrast, results from the current study identify a distinct and novel role for α3β1 integrin in regulating crosstalk from the epidermis to the vasculature of the skin to promote wound angiogenesis (depicted in Fig. 8). Indeed, α3β1-integrin-mediated crosstalk between distinct tissue compartments of the skin may represent a novel paradigm of integrin control over angiogenesis that extends beyond their well-established roles in the regulation of cell adhesion and migration. We observed that wounds in mice with an epidermis-specific knockout of the α3-integrin subunit displayed markedly reduced blood-vessel density compared with wounds from control mice. Additionally, endothelial-cell migration in vitro was reduced in response to conditioned medium from α3β1-integrin-deficient keratinocytes, compared with α3β1-integrin-expressing keratinocytes, indicating that α3β1 integrin is required for keratinocyte-to-endothelial-cell crosstalk. Finally, our data demonstrate a novel function for α3β1 integrin in the induction of Mrp3 gene expression in keratinocytes that promotes endothelial-cell migration, and they support a role for this regulation during wound angiogenesis in vivo.
Fig. 8.

Model depicting α3β1-integrin-mediated functions of epidermis that contribute to wound healing. In vitro and in vivo studies indicate distinct and separable roles for α3β1 integrin in the epidermis during wound healing: (1) complex roles in epidermal migration and regeneration (see text for details and references), and (2) production of pro-angiogenic factors, such as MRP3, that promote wound angiogenesis (current study). α3β1-integrin-dependent MRP3 expression is indicated in keratinocytes within a wound-proximal hair follicle, as well as in epidermis that is migrating into the wound beneath the eschar and over the provisional ECM of the wound bed (shown in green). The basement membrane (shown in red) is indicated beneath the adjacent epidermis and around the hair follicle.
Several different integrins have been shown to be important for normal skin development or post-developmental skin functions such as wound healing (Litjens et al., 2006; Watt, 2002). Recent studies have shown that mice deficient for all β1 integrins in the epidermis have severe defects in wound re-epithelialization and hair-follicle morphogenesis (Brakebusch et al., 2000; Grose et al., 2002; Raghavan et al., 2000). One of the major α-subunit-binding partners for β1 integrins in the epidermis is the α3 subunit, and α3β1 integrin has important roles in maintaining basement membrane integrity in vivo and keratinocyte motility and survival in vitro (Choma et al., 2004; DiPersio et al., 1997; Frank and Carter, 2004; Manohar et al., 2004). However, two recent studies performed in distinct genetic models have reported either pro-migratory or anti-migratory effects of this integrin on wound re-epithelialization in vivo. Indeed, assessment of wound healing in full-thickness skin grafts from Itga3-null neonatal mice showed that absence of α3β1 integrin from skin results in reduced re-epithelialization, suggesting a pro-migratory role (Reynolds et al., 2008). By contrast, epidermis-specific deletion of α3 integrin caused slightly enhanced wound re-epithelialization, indicating that α3β1 integrin delays, rather than facilitates, wound closure in this model (Margadant et al., 2009). Although we did not perform an extensive analysis of wound re-epithelialization in the current study, our preliminary observations revealed no obvious reduction in wound re-epithelialization of α3eKO mice, consistent with the findings of Sonnenberg and co-workers (Margadant et al., 2009). It remains to be determined whether the disparate findings in the above-mentioned studies stem from inherent differences in the in vivo models that were used (i.e. total versus epidermis-specific α3-integrin knockout), or from different requirements for α3β1 integrin in neonatal skin (Reynolds et al., 2008) versus adult skin (Margadant et al., 2009). Nevertheless, the combined observations that α3eKO mice show reduced wound angiogenesis (current study), and that epidermis-specific deletion of α3β1 integrin does not impair (and appears to enhance) wound re-epithelialization (Margadant et al., 2009), suggest that α3β1-integrin-mediated wound angiogenesis is not crucial for the re-epithelialization step of wound healing in this model. This observation is consistent with a previous report that inhibiting angiogenesis did not affect the rate of re-epithelialization of full-thickness excisional wounds in mice (Michaels et al., 2005). It will be interesting in future studies to test if α3β1-integrin deficiency in epidermis alters other aspects of the wound-healing process that have been linked to angiogenesis, such as formation of granulation tissue or wound tensile strength.
As keratinocytes migrate across the wound bed, they secrete multiple cytokines and growth factors that facilitate the healing process, including pro-angiogenic factors that can act upon endothelial cells to induce angiogenesis (Santoro and Gaudino, 2005; Singer and Clark, 1999). Our data identify MRP3 as a potentially important α3β1-integrin-dependent factor secreted by keratinocytes to promote angiogenesis. Consistently, MRPs are known to regulate angiogenesis during development (Corbacho et al., 2002; Jackson et al., 1994), and MRP3 expression in wound keratinocytes and hair follicles adjacent to the wound is thought to promote wound angiogenesis (Fassett and Nilsen-Hamilton, 2001). Our combined findings that MRP3 expression was α3β1-integrin dependent in keratinocytes, and that shRNA-mediated suppression of MRP3 in α3β1-integrin-expressing keratinocytes eliminated the ability to stimulate endothelial-cell migration, identify MRP3 as a mediator of α3β1-integrin-dependent keratinocyte-to-endothelial-cell crosstalk. Interestingly, shRNA-mediated suppression of MMP9, another α3β1-integrin-dependent pro-angiogenic factor (Iyer et al., 2005), did not consistently alter endothelial-cell migration in response to keratinocyte-conditioned medium (data not shown), suggesting that MMP9 is not required for this response, at least in vitro. Importantly, our in vivo model revealed a correlation between reduced MRP3 expression and absence of α3β1 integrin in wound-proximal hair follicles and wound-edge keratinocytes of α3eKO mice. These findings indicate that α3β1 integrin regulates MRP3 expression in vivo and support a role for MRP3 in the α3β1-integrin-dependent wound angiogenesis that we observed in these mice. It is well known that hair follicles include a reservoir of stem cells that may contribute to wound re-epithelialization, as cells from the hair follicle bulge migrate outward to repair the damaged epidermis (Ito et al., 2005; Levy et al., 2005). It is possible that activated keratinocytes in wound-proximal hair follicles secrete MRP3, which then diffuses into the adjacent wound and acts upon endothelial cells at the site of injury to promote their migration and drive angiogenesis.
To the best of our knowledge, the current study is the first to show that MRP3 can be regulated by integrins, and it remains to be determined whether the ability to induce MRP3 expression is specific to α3β1 integrin or also extends to other integrins. Importantly, expression of other integrins is not altered in the α3β1-integrin-deficient keratinocytes used in this study (DiPersio et al., 2000a). In addition, several studies have indicated that in vivo deletion of α3 integrin does not alter other integrins in the epidermis (DiPersio et al., 2000b; Hodivala-Dilke et al., 1998; Margadant et al., 2009). Based on these previous studies, together with our current findings that in the absence of α3β1 integrin other endogenous keratinocyte integrins do not support robust wound angiogenesis in vivo (Fig. 3), stimulation of endothelial-cell migration in vitro (Fig. 4) or MRP3 expression (Fig. 5), we think compensation by other integrins is an unlikely explanation for the MRP3 expression that we observed in a small proportion of α3β1-integrin-deficient wounds. Instead, we suggest that residual MRP3 expression may result from soluble factors present in the wound microenvironment that can also induce MRP3 in keratinocytes, such as KGF (Fassett and Nilsen-Hamilton, 2001). Thus, it seems likely that α3β1 integrin and certain growth factors are both required to achieve consistent and efficient induction of MRP3 during wound healing.
In addition to functioning as an adhesion receptor for laminins in the ECM, there is evidence that α3β1 integrin can function as a signaling component from within cell-cell junctional complexes that include E-cadherin (also known as cadherin 1) and the tetraspanin CD151 (Chattopadhyay et al., 2003). Importantly, α3β1-integrin expression in quiescent epidermis (where it is localized largely to cell-cell junctions) does not induce MRP3 expression or an angiogenic response, whereas our results show that α3β1 integrin promotes these responses in wounded epidermis. These observations suggest that wound-associated changes in α3β1 integrin, such as ligation of newly synthesized LN-332 (Nguyen et al., 2000), may be required for induction of MRP3. Direct testing of a requirement for LN-332 is complicated by the fact that cultured keratinocytes rapidly deposit abundant LN-332 onto their ECM substrate (Nguyen et al., 2000). However, future studies using laminin-binding mutants of α3β1 integrin should help determine whether ligation to LN-332 is required for MRP3 induction. Interestingly, LN-332 expression is also elevated in invasive squamous cell carcinomas (Marinkovich, 2007), and recent studies in our lab indicate that α3β1 integrin promotes subcutaneous tumor growth of oncogenically transformed keratinocytes (Lamar et al., 2008). Given the role for α3β1 integrin in wound angiogenesis that we have identified in the current study, it is possible that this integrin plays a similar role in mediating crosstalk between carcinoma cells and endothelial cells to promote tumor angiogenesis. Indeed, previous studies have implicated MRPs in promoting tumor angiogenesis (Bengtson and Linzer, 2000; Corbacho et al., 2002; Toft et al., 2001).
In summary, this study identifies a novel role for α3β1 integrin in the epidermis in promoting wound angiogenesis, and it identifies α3β1-integrin-mediated induction of MRP3 expression as a novel mechanism of keratinocyte-to-endothelial-cell crosstalk. As these findings extend the roles of epidermal integrins beyond their traditional roles in regulating cell adhesion and migration, they further our understanding of how integrins mediate epidermal responses to provisional wound ECM that contribute to normal wound healing. Integrins are particularly attractive targets for therapeutic agents aimed at enhancing wound healing, as they are accessible on the cell surface and can be manipulated with relative ease. The novel pro-angiogenic function of α3β1 integrin described here, together with previously studied functions of α3β1 integrin in keratinocyte migration, basement membrane assembly and wound re-epithelialization (Choma et al., 2004; deHart et al., 2003; Frank and Carter, 2004; Hamelers et al., 2005; Margadant et al., 2009; Reynolds et al., 2008), suggest multiple and complex roles for this integrin that may be simultaneously exploitable in therapeutic strategies to facilitate wound repair.
Materials and Methods
Mice
Generation of mice that carry the conventional Itga3-null mutation was described previously (Kreidberg et al., 1996). The targeting vector to generate the floxed Itga3 allele (Itga3flx) was constructed in the pDELBOY vector, in which exon 3 of the Itga3 gene was flanked by loxP sites, and a Frt-NeoR-Frt cassette was placed downstream of exon 2 (see Fig. 1A). Following homologous recombination in embryonic stem cells and generation of mice that carry the Itga3flx allele in the germ line, the Frt-NeoR-Frt cassette was excised by mating with mice that express FlpE (a gift from Susan Dymecki, Harvard Medical School). Further details regarding generation of the Itga3flx allele can be found in Liu et al. (Liu et al., 2009). To generate mice with an epidermis-specific deletion of α3 integrin, Itga3flx/flx mice or Itga3flx/– mice were crossed with mice that carry a Cre recombinase transgene under the control of the keratin 14 promoter (K14-Cre; a gift from Andrew McMahon, Harvard University), and offspring were backcrossed to obtain mice of the K14-Cre:Itga3flx/flx or K14-Cre:Itga3flx/– genotypes. PCR genotyping for the Itga3-null allele was described previously (DiPersio et al., 1997). PCR genotyping for the Itga3flx allele was performed using the primers 5′-TGATGACTATACCAACCGGAC-3′ (forward) and 5′-ACTCCAAGCCACATATCCTC-3′ (reverse) and the following reaction conditions: denaturation at 94°C for 30 seconds; extension at 58°C for 60 seconds; annealing at 72°C for 120 seconds; 30 amplification cycles. PCR generates a 542 bp product for the wild-type Itga3 allele, and a 623 bp product for the Itga3flx allele. PCR genotyping for the K14-Cre transgene was performed using the primers 5′-ATGTCCAATTTACTGACCGT-3′ (forward) and 5′-CGCCGCATAACCAGTGAAAC-3′ (reverse) and the following reaction conditions: denaturation at 93°C for 60 seconds; extension at 49°C for 60 seconds; annealing at 72°C for 90 seconds; 32 amplification cycles. PCR generates a 370 bp product for the Cre transgene. Cre-mediated excision of exon 3 from the Itga3flx allele results in translational termination within exon 4. Itga3flx/flx or Itga3flx/– littermates that lacked K14-Cre served as controls in experiments. Itga3flx/flx or Itga3flx/– mice, with or without K14-Cre expression, were born in normal Mendelian ratios and were healthy and viable with no overt phenotypes as young adults. However, progressive hair loss and inflammation around the eyes was observed frequently in older mice of the K14-Cre:Itga3flx/flx or K14-Cre:Itga3flx/– genotypes.
In vivo wounding and tissue preparation for histology and immunofluorescence
Age-matched adult mice were anaesthetized and shaved, and two or four full-thickness wounds were made on the back with a sterile 4-mm biopsy punch. Mice were euthanized by CO2 narcosis 5 days following injury, and wounds were surgically excised and bisected along the anteroposterior axis. For each wound, one half was frozen in OCT compound, and the other half was embedded in paraffin, sectioned (3 μM), and stained with H&E (Mass Histology Service, Worcester, MA). For immunofluorescence, 10-μm frozen sections were cut and rehydrated in PBS with 0.2% Tween-20 for 10 minutes, then stained with the following rabbit polyclonal antisera: anti-α3-integrin or corresponding pre-immune serum (1:100 dilution) (DiPersio et al., 1995); anti-keratin 14 (1:250 dilution) (Covance, Richmond, CA); anti-LN-332 (β3VI/V), (1:100 dilution) (generous gift from Dr Rupert Timple, Max-Planck Institute). Following treatment with primary antibody, sections were treated with fluorescein-conjugated goat anti-rabbit IgG (Pierce, Rockford IL). For MRP3 staining, sections were permeabilized for 20 minutes in 0.2% Triton X-100, then stained with anti-MRP3 or the corresponding pre-immune sera (1:100 dilution) using the Zenon Alexa Flour 488 Rabbit IgG Labeling Kit Tag, according to the manufacturer's instructions (Invitrogen Corporation, Carlsbad, CA). Images were collected on a Nikon Eclipse 80i using a Spot camera (Diagnostic Instruments, Sterling Heights, MI).
For analysis of wound angiogenesis, cryosections were prepared as described above and stained with anti-CD31/PECAM-1 (BD Pharmingen, Franklin Lakes, NJ), followed by Alexa Flour 488 goat anti-rat IgG (Molecular Probes, Eugene, OR). To quantify blood-vessel density, three 20× fields were collected from within each wound bed, and the total area of CD31 staining was determined per field using Slidebook (Intelligent Imaging Innovation). Average blood-vessel area (pixels/field) was then determined for each wound, as described (Grenache et al., 2007). For each genotype, mean blood-vessel density ± s.e.m. was determined for five wounds from five separate 6.5-week-old mice. Similar trends were seen in 18-week-old mice.
Cell culture
Mouse keratinocyte cell lines that express or lack α3β1 integrin were derived as previously described (DiPersio et al., 2000a). Keratinocyte growth medium consisted of Eagle's minimum essential medium (BioWhittaker, Walkersville, MD) supplemented with 4% fetal bovine serum (BioWhittaker) from which Ca2+ had been chelated, 0.05 mM CaCl2, 0.04 μg/ml hydrocortisone, 5 μg/ml insulin, 2×10–9 M T3, 10 units/ml interferon-γ (Sigma, St Louis, MO), 10 ng/ml epidermal growth factor, 100 units/ml penicillin, 100 μg/ml streptomycin, and L-glutmamine (Invitrogen Corporation). Keratinocytes were maintained at 33°C, 8% CO2 on tissue-culture dishes coated with 30 μg/ml denatured rat tail collagen (BD Biosciences, Bedford, MA). HUVECS from pooled donors were purchased from VEC Technologies (Troy, NY). Endothelial cell growth medium consisted of M199 medium supplemented with 20% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml heparin and 50 μg/ml of endothelial cell growth supplement (Collaborative Biomedical Products). Cells were maintained at 37°C, 5% CO2 on tissue-culture dishes coated with 0.2% gelatin.
Transwell assays of endothelial-cell migration
Transwell migration assays were performed as described (Meadows et al., 2004). Briefly, transwell tissue-culture inserts (Costar, Corning, NY) with 8 μm pores were coated with 0.2% gelatin. HUVECs were serum-starved for 24 hours, trypsinized and seeded onto top surfaces in serum-free MCDB-131 at 5×104 cells per insert. Lower chambers contained serum-free MCDB-131 medium that had been conditioned by wild-type, α3+ or α3– keratinocytes, in a 70:30 ratio with complete EGM-2 medium (Lonza, Walkersville, MD). Unconditioned medium in the same 70:30 ratio was used to establish baseline cell migration. For some experiments, conditioned medium was collected from 293 cells that overexpress recombinant His-tagged MRP3, and from untransfected 293 cells as a control. After 4 hours of migration, cells were fixed in 3.7% formaldehyde/PBS and stained with 0.5% Crystal Violet. Non-migratory cells on the top surface were removed with cotton swabs and cells that had migrated to the bottom surface were permeabilized with 0.1% Triton X-100 and stained with DAPI (Pierce). Cells were visualized using an Olympus inverted IX70 microscope, and three random 10× fields were collected using a SensiCam digital camera (Cooke, Eugene, OR). The total cell number per field was quantified using ImagePro Plus (Media Cybernetics, Silver Spring, MD).
RT-PCR
Total RNA was isolated from mouse keratinocytes using the Purescript RNA Isolation kit (Gentra Systems, Minneapolis, MN) or Trizol Reagent (Invitrogen Corporation), then reverse transcribed to produce cDNA using the First-Strand cDNA Synthesis kit (Promega). PCR reactions were carried out in PCR REDTaq ready mix (Sigma). PCR primers and conditions for amplication of Mmp9 were previously described (Iyer et al., 2005). PCR primers and conditions for amplication of Mrp3: forward primer, 5′-CCCTTCTTCGATTCAACCATG-3′, reverse primer 5′-CATGTAACACTACAGGACG-3′; 94°C for 30 seconds, 51°C for 30 seconds, 72°C for 1 minute, 18 amplification cycles. PCR primers and conditions for amplication of β-actin: forward primer, 5′-GCCAGGTCATCACTATTGG-3′, reverse primer 5′-AGTAACAGTCCGCCTAGAAGC-3′; 94°C for 30 seconds, 51°C for 30 seconds, 72°C for 1 minute, 18 amplification cycles. PCR primers and conditions for amplication of Vegf: forward primer, 5′-CCGGTGGACATCTTCCAGGAGTA-3′, reverse primer, 5′-CTCACCGCCTCGGCTTGTCACA-3′; 94°C for 30 seconds, 58°C for 30 seconds, 72°C for 1 minute, 26 amplification cycles. Signals were visualized using a Bio-Rad FlourS 2000 and quantified using Quantity One software (Bio-Rad, Hercules, CA).
Western blot
Keratinocytes were cultured on collagen-coated dishes in the absence of serum for 24 hours. Conditioned medium was collected and cells were lysed in cell lysis buffer (Cell Signaling Technology, Beverly, MA). Protein concentrations were determined using the BCA Protein Assay Kit (Pierce). Samples of equal protein content, or equivalent proportions of culture supernantant, were assayed by immunoblot using rabbit anti-MRP3 or the corresponding pre-immune serum (1:500) (Fassett and Nilsen-Hamilton, 2001), or rabbit anti-keratin (1:1000) (Covance Inc, Princeton, NJ), followed by peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:5000) (Cell Signaling Technology). Chemiluminescence was performed with the SuperSignal Kit (Pierce), and signals were visualized using a Bio-Rad FlourS 2000 and quantified using Quantity One software (Bio-Rad). MRP3 signals were normalized to keratin 14 signals.
shRNA-mediate suppression of MRP3
For use of shRNA to stably suppress MRP3, we used MISSION™ lentiviral shRNA constructs (Sigma) encoding shRNAs that target murine Mrp3, or a non-targeting control shRNA. Lentiviral constructs were transfected into the packaging cell line, 293FT using lipofectamine plus reagent (Invitrogen Corporation). Viral supernatants were added to α3-integrin-expressing keratinocytes for 24 to 48 hours, followed by culture in keratinocyte growth medium containing 10 μM puromycin (MP Biomedicals, Solon, OH) to select populations of stably transduced cells. All experiments and reagents described in this study were approved by the Institutional Biosafety Committee of Albany Medical College. All animal studies were approved by the Institutional Animal Care and Use Committee of Albany Medical College.
We are grateful to Peter Vincent for assistance with statistical analyses and critical reading of the manuscript. We also thank Kevin Pumiglia and Livingston Van De Water for helpful discussions and critical reading of the manuscript, Pierig Lepont, Lee Bendickson and Jelaun Newsome for technical assistance, and Susan Dymecki, Andrew McMahon and Rupert Timple for providing valuable reagents. This research was supported by grants from the National Institutes of Health to C.M.D. (R01CA84238) and from the NIDDK to J.A.K. K.M. was supported by a pre-doctoral training grant from the National Heart Lung and Blood Institute (NIH-T32-HL-07194). Deposited in PMC for release after 6 months.
References
- Bengtson, N. W. and Linzer, D. I. (2000). Inhibition of tumor growth by the antiangiogenic placental hormone, proliferin-related protein. Mol. Endocrinol. 14, 1934-1943. [DOI] [PubMed] [Google Scholar]
- Brakebusch, C., Grose, R., Quondamatteo, F., Ramirez, A., Jorcano, J. L., Pirro, A., Svensson, M., Herken, R., Sasaki, T., Timpl, R. et al. (2000). Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 19, 3990-4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho, R. M., Squarize, C. H., Patel, V., Millar, S. E., Zheng, Y., Molinolo, A. and Gutkind, J. S. (2007). Requirement of Rac1 distinguishes follicular from interfollicular epithelial stem cells. Oncogene 26, 5078-5085. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay, N., Wang, Z., Ashman, L. K., Brady-Kalnay, S. M. and Kreidberg, J. A. (2003). alpha3beta1 integrin-CD151, a component of the cadherin-catenin complex, regulates PTPmu expression and cell-cell adhesion. J. Cell Biol. 163, 1351-1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielowiec, J., Borowiak, M., Morkel, M., Stradal, T., Munz, B., Werner, S., Wehland, J., Birchmeier, C. and Birchmeier, W. (2007). c-Met is essential for wound healing in the skin. J. Cell Biol. 177, 151-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choma, D. P., Pumiglia, K. and DiPersio, C. M. (2004). Integrin α3β1 directs the stabilization of a polarized lamellipodium in epithelial cells through activation of Rac1. J. Cell Sci. 117, 3947-3959. [DOI] [PubMed] [Google Scholar]
- Clark, R. A. F., Lanigan, J. M., DellaPelle, P., Manseau, E., Dvorak, H. F. and Colvin, R. B. (1982). Fibronectin and fibrin provide a provisional matrix for epidermal cell migration during wound reepithelialization. J. Invest. Dermatol. 79, 264-269. [DOI] [PubMed] [Google Scholar]
- Conti, F. J., Rudling, R. J., Robson, A. and Hodivala-Dilke, K. M. (2003). α3β1-integrin regulates hair follicle but not interfollicular morphogenesis in adult epidermis. J. Cell Sci. 116, 2737-2747. [DOI] [PubMed] [Google Scholar]
- Corbacho, A. M., Martinez De La, Escalera, G. and Clapp, C. (2002). Roles of prolactin and related members of the prolactin/growth hormone/placental lactogen family in angiogenesis. J. Endocrinol. 173, 219-238. [DOI] [PubMed] [Google Scholar]
- deHart, G. W., Healy, K. E. and Jones, J. C. (2003). The role of alpha3beta1 integrin in determining the supramolecular organization of laminin-5 in the extracellular matrix of keratinocytes. Exp. Cell Res. 283, 67-79. [DOI] [PubMed] [Google Scholar]
- DiPersio, C. M., Shah, S. and Hynes, R. O. (1995). α3Aβ1 integrin localizes to focal contacts in response to diverse extracellular matrix proteins. J. Cell Sci. 108, 2321-2336. [DOI] [PubMed] [Google Scholar]
- DiPersio, C. M., Hodivala-Dilke, K. M., Jaenisch, R., Kreidberg, J. A. and Hynes, R. O. (1997). α3β1 integrin is required for normal development of the epidermal basement membrane. J. Cell Biol. 137, 729-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPersio, C. M., Shao, M., Di Costanzo, L., Kreidberg, J. A. and Hynes, R. O. (2000a). Mouse keratinocytes immortalized with large T antigen acquire α3β1 integrin-dependent secretion of MMP-9/gelatinase B. J. Cell Sci. 113, 2909-2921. [DOI] [PubMed] [Google Scholar]
- DiPersio, C. M., van der Neut, R., Georges-Labouesse, E., Kreidberg, J. A., Sonnenberg, A. and Hynes, R. O. (2000b). α3β1 and α6β4 integrin receptors for laminin-5 are not essential for epidermal morphogenesis and homeostasis during skin development. J. Cell Sci. 113, 3051-3062. [DOI] [PubMed] [Google Scholar]
- Fassett, J. T. and Nilsen-Hamilton, M. (2001). Mrp3, a mitogen-regulated protein/proliferin gene expressed in wound healing and in hair follicles. Endocrinology 142, 2129-2137. [DOI] [PubMed] [Google Scholar]
- Frank, D. E. and Carter, W. G. (2004). Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J. Cell Sci. 117, 1351-1363. [DOI] [PubMed] [Google Scholar]
- Grenache, D. G., Zhang, Z., Wells, L. E., Santoro, S. A., Davidson, J. M. and Zutter, M. M. (2007). Wound healing in the alpha2beta1 integrin-deficient mouse: altered keratinocyte biology and dysregulated matrix metalloproteinase expression. J. Invest. Dermatol. 127, 455-466. [DOI] [PubMed] [Google Scholar]
- Grinnell, F. (1992). Wound repair, keratinocyte activation and integrin modulation. J. Cell Sci. 101, 1-5. [DOI] [PubMed] [Google Scholar]
- Grose, R., Hutter, C., Bloch, W., Thorey, I., Watt, F. M., Fassler, R., Brakebusch, C. and Werner, S. (2002). A crucial role of beta 1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 129, 2303-2315. [DOI] [PubMed] [Google Scholar]
- Groskopf, J. C., Syu, L. J., Saltiel, A. R. and Linzer, D. I. (1997). Proliferin induces endothelial cell chemotaxis through a G protein-coupled, mitogen-activated protein kinase-dependent pathway. Endocrinology 138, 2835-2840. [DOI] [PubMed] [Google Scholar]
- Hamelers, I. H., Olivo, C., Mertens, A. E., Pegtel, D. M., van der Kammen, R. A., Sonnenberg, A. and Collard, J. G. (2005). The Rac activator Tiam1 is required for α3β1-mediated laminin-5 deposition, cell spreading, and cell migration. J. Cell Biol. 171, 871-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodivala-Dilke, K. M., DiPersio, C. M., Kreidberg, J. A. and Hynes, R. O. (1998). Novel roles for α3β1 integrin as a regulator of cytoskeletal assembly and as a transdominant inhibitor of integrin receptor function in keratinocytes. J. Cell Biol. 142, 1357-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsken, J., Vogel, R., Erdmann, B., Cotsarelis, G. and Birchmeier, W. (2001). beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 105, 533-545. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687. [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (2007). Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 5Suppl. 1, 32-40. [DOI] [PubMed] [Google Scholar]
- Ito, M., Liu, Y., Yang, Z., Nguyen, J., Liang, F., Morris, R. J. and Cotsarelis, G. (2005). Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med. 11, 1351-1354. [DOI] [PubMed] [Google Scholar]
- Iyer, V., Pumiglia, K. and DiPersio, C. M. (2005). α3β1 integrin regulates MMP-9 mRNA stability in immortalized keratinocytes: a novel mechanism of integrin-mediated MMP gene expression. J. Cell Sci. 118, 1185-1195. [DOI] [PubMed] [Google Scholar]
- Jackson, D., Volpert, O. V., Bouck, N. and Linzer, D. I. (1994). Stimulation and inhibition of angiogenesis by placental proliferin and proliferin-related protein. Science 266, 1581-1584. [DOI] [PubMed] [Google Scholar]
- Kreidberg, J. A., Donovan, M. J., Goldstein, S. L., Rennke, H., Shepherd, K., Jones, R. C. and Jaenisch, R. (1996). Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development 122, 3537-3547. [DOI] [PubMed] [Google Scholar]
- Lamar, J. M., Pumiglia, K. M. and DiPersio, C. M. (2008). An immortalization-dependent switch in integrin function up-regulates MMP-9 to enhance tumor cell invasion. Cancer Res. 68, 7371-7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, V., Lindon, C., Harfe, B. D. and Morgan, B. A. (2005). Distinct stem cell populations regenerate the follicle and interfollicular epidermis. Dev. Cell 9, 855-861. [DOI] [PubMed] [Google Scholar]
- Litjens, S. H., de Pereda, J. M. and Sonnenberg, A. (2006). Current insights into the formation and breakdown of hemidesmosomes. Trends Cell. Biol. 16, 376-383. [DOI] [PubMed] [Google Scholar]
- Liu, Y., Chattopadhyay, N., Qin, S., Szekeres, C., Vasylyeva, T., Mahoney, Z. X., Taglienti, M., Bates, C. M., Chapman, H. A., Miner, J. H. et al. (2009). Coorindate integrin and c-Met signaling regulate Wnt gene expression during epithelial morphogenesis. Development 136, 843-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madlener, M., Parks, W. C. and Werner, S. (1998). Matrix metalloproteinases (MMPs) and their physiological inhibitors (TIMPs) are differentially expressed during excisional skin wound repair. Exp. Cell Res. 242, 201-210. [DOI] [PubMed] [Google Scholar]
- Malyankar, U. M., Rittling, S. R., Connor, A. and Denhardt, D. T. (1994). The mitogen-regulated protein/proliferin transcript is degraded in primary mouse embryo fibroblast but not 3T3 nuclei: altered RNA processing correlates with immortalization. Proc. Natl. Acad. Sci. USA 91, 335-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar, A., Shome, S. G., Lamar, J., Stirling, L., Iyer, V., Pumiglia, K. and DiPersio, C. M. (2004). α3β1 integrin promotes keratinocyte cell survival through activation of a MEK/ERK signaling pathway. J. Cell Sci. 117, 4043-4054. [DOI] [PubMed] [Google Scholar]
- Margadant, C., Raymond, K., Kreft, M., Sachs, N., Janssen, H. and Sonnenberg, A. (2009). Integrin α3β1 inhibits directional migration and wound re-epithelialization in the skin. J. Cell Sci. 122, 278-288. [DOI] [PubMed] [Google Scholar]
- Marinkovich, M. P. (2007). Tumour microenvironment: Laminin 332 in squamous-cell carcinoma. Nat. Rev. Cancer 7, 370-380. [DOI] [PubMed] [Google Scholar]
- Martin, P. (1997). Wound healing-aiming for perfect skin regeneration. Science 276, 75-81. [DOI] [PubMed] [Google Scholar]
- Meadows, K. N., Bryant, P., Vincent, P. A. and Pumiglia, K. M. (2004). Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene 23, 192-200. [DOI] [PubMed] [Google Scholar]
- Michaels, J. t., Dobryansky, M., Galiano, R. D., Bhatt, K. A., Ashinoff, R., Ceradini, D. J. and Gurtner, G. C. (2005). Topical vascular endothelial growth factor reverses delayed wound healing secondary to angiogenesis inhibitor administration. Wound Repair Regen. 13, 506-512. [DOI] [PubMed] [Google Scholar]
- Nelson, J. T., Rosenzweig, N. and Nilsen-Hamilton, M. (1995). Characterization of the mitogen-regulated protein (proliferin) receptor. Endocrinology 136, 283-288. [DOI] [PubMed] [Google Scholar]
- Nguyen, B. P., Ryan, M. C., Gil, S. G. and Carter, W. G. (2000). Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr. Opin. Cell Biol. 12, 554-562. [DOI] [PubMed] [Google Scholar]
- Parfett, C. L., Hamilton, R. T., Howell, B. W., Edwards, D. R., Nilsen-Hamilton, M. and Denhardt, D. T. (1985). Characterization of a cDNA clone encoding murine mitogen-regulated protein: regulation of mRNA levels in mortal and immortal cell lines. Mol. Cell. Biol. 5, 3289-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilcher, B. K., Dumin, J. A., Sudbeck, B. D., Krane, S. M., Welgus, H. G. and Parks, W. C. (1997). The activity of collagenase-1 is required for keratinocyte migration on a type I collagen matrix. J. Cell Biol. 137, 1445-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan, S., Bauer, C., Mundschau, G., Li, Q. and Fuchs, E. (2000). Conditional ablation of beta1 integrin in skin: severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J. Cell Biol. 150, 1149-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, L. E., Conti, F. J., Silva, R., Robinson, S. D., Iyer, V., Rudling, R., Cross, B., Nye, E., Hart, I. R., Dipersio, C. M. et al. (2008). alpha3beta1 integrin-controlled Smad7 regulates reepithelialization during wound healing in mice. J. Clin. Invest. 118, 965-974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, M. C., Lee, K., Miyashita, Y. and Carter, W. G. (1999). Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol. 145, 1309-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salo, T., Makela, M., Kylmaniemi, M., Autio-Harmainen, H. and Larjava, H. (1994). Expression of matrix metalloproteinase-2 and -9 during early human wound healing. Lab. Invest. 70, 176-182. [PubMed] [Google Scholar]
- Santoro, M. M. and Gaudino, G. (2005). Cellular and molecular facets of keratinocyte reepithelization during wound healing. Exp. Cell Res. 304, 274-286. [DOI] [PubMed] [Google Scholar]
- Silva, R., D'Amico, G., Hodivala-Dilke, K. M. and Reynolds, L. E. (2008). Integrins: the keys to unlocking angiogenesis. Arterioscler. Thromb. Vasc. Biol. 28, 1703-1713. [DOI] [PubMed] [Google Scholar]
- Singer, A. J. and Clark, R. A. (1999). Cutaneous wound healing. N. Engl. J. Med. 341, 738-746. [DOI] [PubMed] [Google Scholar]
- Soares, M. J., Alam, S. M., Duckworth, M. L., Horseman, N. D., Konno, T., Linzer, D. I., Maltais, L. J., Nilsen-Hamilton, M., Shiota, K., Smith, J. R. et al. (2007). A standardized nomenclature for the mouse and rat prolactin superfamilies. Mamm. Genome 18, 154-156. [DOI] [PubMed] [Google Scholar]
- Toft, D. J., Rosenberg, S. B., Bergers, G., Volpert, O. and Linzer, D. I. (2001). Reactivation of proliferin gene expression is associated with increased angiogenesis in a cell culture model of fibrosarcoma tumor progression. Proc. Natl. Acad. Sci. USA 98, 13055-13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpert, O., Jackson, D., Bouck, N. and Linzer, D. I. (1996). The insulin-like growth factor II/mannose 6-phosphate receptor is required for proliferin-induced angiogenesis. Endocrinology 137, 3871-3876. [DOI] [PubMed] [Google Scholar]
- Watt, F. M. (2002). Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 21, 3919-3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweers, M. C., Davidson, J. M., Pozzi, A., Hallinger, R., Janz, K., Quondamatteo, F., Leutgeb, B., Krieg, T. and Eckes, B. (2007). Integrin alpha2beta1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J. Invest. Dermatol. 127, 467-478. [DOI] [PubMed] [Google Scholar]