

Abstract
Niemann-Pick C disease (NPC) is a fatal, neurovisceral genetic disorder. Cell culture studies showed that NPC1 or NPC2 mutations cause malfunctions in cellular cholesterol trafficking and lead to accumulation of cholesterol and other lipids in the late endo/lysosomes. Previous work showed that neuronal cholesterol accumulation occurs in the brains of young postnatal NPC1-/- mice. Here, to evaluate the potential of partial blockage of cholesterol biosynthesis as a therapy for the NPC disease, we first developed a simple method to monitor the relative rates of lipid biosynthesis in mice brains. We next administered squalene synthase inhibitor (SSI) CP-340868 to young mice. The results show that treating 8-day-old NPC1-/- mice with CP-340868 for six days significantly inhibits cholesterol biosynthesis in the mice brains. It reduces neuronal cholesterol accumulation, reduces GM3 ganglioside accumulation, and diminishes astrogliosis in the brain. These results suggest that neuronal cholesterol accumulation contributes to early pathogenesis in the NPC1-/- mice brains. The SSI treatment also reduced brain galactolipid content, suggesting that blocking endogenous cholesterol synthesis in the young mice brains may disrupt the normal myelin maturation processes. The methods described in the current work have general applicability for lipid metabolism studies in mice brains in various pathophysiological conditions.
Keywords: squalene synthase inhibitor, sterol synthesis in brain, lipid trafficking, neurodegenerative disease, late endosomes/lysosomes
Introduction
Niemann-Pick type C (NPC) disease is a fatal, autosomal recessive neurovisceral disorder. It is characterized by the cellular accumulation of unesterified cholesterol, sphingomyelin, glycolipids, and other lipids within the endosomal/lysosomal system of various tissues including liver, spleen, and the central nervous system (CNS) (reviewed in (Patterson et al., 2001; Chang et al., 2005a)). The disease can be caused by mutations in one of two genetic loci, NPC1 and NPC2. The human NPC1 gene encodes a 1278-aa multi-pass transmembrane protein that contains a sterol-sensing domain (Carstea et al., 1997), which is present in several integral membrane proteins involved in cellular cholesterol homeostasis (Chang et al., 2006). The NPC2 gene encodes a soluble protein known as HE1 (Naureckiene et al., 2000), present in the lysosome. Both NPC1 and NPC2 proteins bind cholesterol (Ohgami et al., 2004; Ko et al., 2003). The NPC2 protein is capable of transferring cholesterol between two membranes, and is believed to be involved in the egress of cholesterol out of the endosomal/lysosomal compartment (Cheruku et al., 2006). Cell culture studies showed that in mutant NPC1 cells, the egress of cholesterol flowing through the late endo/lysosomal pathway from various sources, including LDL-derived cholesterol and a portion of endogenously synthesized sterols (endoSTEROL), is much retarded (Wojtanik and Liscum, 2003; Sugii et al., 2003; Cruz and Chang, 2000).
Mouse models for the NPC disease are available (Chang et al., 2005b; Patterson et al., 2001). These mice show various pathological progressions in the CNS, in manners very similar to those of the human NPC disease. In NPC1-/- mice, at postnatal day (PND) 9, mild abnormalities already become detectable in the corpus callosum, cerebellar white matter, and nerve fibers (Ong et al., 2001). At PND 10, hypomyelination and axonal injury become detectable (Takikita et al., 2004). By PND 22, accumulation of activated astrocytes (i.e., astrogliosis) becomes extensive in the cortex and thalamus regions (Reid et al., 2004); at this stage, Purkinje cells in the cerebellum suffer significant cell loss (13 %), as do astrocytes in the corpus callosum (German et al., 2002). In addition, significant accumulation of gangliosides, mostly GM2 and GM3, has been demonstrated (Zervas et al., 2001a). At the 7th week, severe losses in myelin protein and cholesterol occur (Xie et al., 2000). Death occurs between the 10th and 12th weeks.
In the brain, the cholesterol mass is predominantly associated with the myelin and the plasma membranes of various cellular materials (reviewed in (Dietschy and Turley, 2004)). Most if not all of the cholesterol in the brain is in free, unesterified form. In brains of NPC1-/- mice, the abnormal cellular free cholesterol accumulation cannot be demonstrated by cholesterol mass analysis of isolated brain tissues, but can be demonstrated by staining thin slices of brain sections with a cholesterol-specific fluorescent compound, such as filipin, and viewing under a fluorescent microscope (Zervas et al., 2001a). This is because the amount of cholesterol accumulated within neurons is only a very small fraction of the total cholesterol mass in the CNS. Filipin binds to unesterified cholesterol with high affinity. However, the utility of filipin staining is limited because its fluorescent signal is weak and is subject to rapid photobleaching. Recently, a novel cholesterol stain named BC-theta was developed. This probe is a biotinylated derivative of a bacterial toxin protein (Iwamoto et al., 1997). Similar to filipin, BC-theta also binds to unesterified cholesterol with high affinity. Unlike filipin, BC-theta can be labeled by various avidin/streptavidin derivatives, each with stable fluorescent properties and high sensitivities. BC-theta has been employed to stain cholesterol-rich domains in membranes in intact cells and in vitro (Waheed et al., 2001). Using BC-theta, Reid and colleagues showed that in early postnatal NPC1 mice brains (PND 9), neuronal cholesterol accumulation becomes detectable in various regions of the brain (Reid et al., 2004). In cell culture studies, Karten and colleagues (Karten et al., 2002) showed that intracellular cholesterol accumulation occurred in sympathetic neurons isolated from one-day-old NPC1 mice pups. Treiber-Held and colleagues (Treiber-Held et al., 2003) showed that embryonic neurons (E15-E17) isolated from NPC1 mice brains cultured in a cholesterol-free medium showed massive accumulation of intracellular free cholesterol. Together, these studies show that cholesterol accumulation occurs in the brains of very young mice. In the current work, we first developed a simple method to monitor the relative rates of lipid biosynthesis in mice brains. We then administered a specific squalene synthase inhibitor to partially block endoSTEROL synthesis in mice brains, and examined its effects on neuronal cholesterol accumulation and myelin organization. The methods described here will have applicability for lipid metabolism studies in mice brains in general.
Materials and Methods
Animals, inhibitor injections, tissue preparations, and histochemical stainings
The animal studies were pre-reviewed and approved by the Institutional Animal Use and Care Committee at Dartmouth College, Hanover, New Hampshire: Protocol #11601. Breeding pair of BALB/c NPC1NIH mice was donated by Peter G. Pentchev. Mice were fed with standard mouse diet (Teklad LM-485, Harlan), and bred as NPC+/- heterozygotes. Litters were genotyped via tail snip DNA by PCR method (Loftus et al., 1997). WT and NPC1 mice received daily subcutaneous injections of the squalene synthase inhibitor, CP-340868 (SSI) in saline at 10 μg/g (WT-SSI, NPC-SSI), or saline alone (WT, NPC) once per day for 6 consecutive days starting at postnatal day 8 (ending at postnatal day 14). In some experiments, mice received daily peritoneal injections of lovastatin in vehicle (at 10 μg/g), or received vehicle only as indicated in the figure legend. For histochemical analyses, mice were anesthetized with ether, perfused through their hearts with Dulbecco's phosphate buffered saline (DPBS) without calcium and magnesium, followed by 4 % paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The large tissue sections from the brains were prepared for various staining experiments according to procedures previously described (Reid et al., 2004). BC-theta was prepared as previously described (Waheed et al., 2001) and used at 15 μg/ml. Anti-Calbindin-D-28K (EG-20) rabbit polyclonal antibodies (Sigma) were used to stain Purkinje neurons; anti-glial acidic fibrillary protein (GFAP) rabbit polyclonal antibodies (Sigma) were used to stain reactive astrocytes. After incubation with primaries, slides were washed with 0.5 M Tris containing no FBS, and counterstained with Neurotrace™ fluorescent Nissl Stain (N-21480) 500/525 green (Molecular Probes) according to the manufacturer's literature. Bound BC-theta, GFAP, or Calbindin antibodies were detected with various secondary reagents from Molecular Probes (Eugene, Oregon). Phalloidin conjugated to Alexa 594 for plasma membrane staining and DAPI for nuclei staining were from Molecular Probes.
Slides were treated with Prolong anti-fade from Molecular Probes. Images were collected with a confocal microscope (Bio-Rad MRC-1024) and constructed with LaserSharp software.
Quantitation of neuronal cholesterol accumulation by BC-theta
For quantitating the BC-theta staining signals, the procedure described previously was employed (Reid et al., 2004). For each section, six image scans encompassing all areas of that section were taken, and the sum of the BC-theta fluorescent signals was determined. Sections from WT mice exhibited low levels of BC-theta positive signals, representing background staining of synaptosomal membranes and myelin-associated cholesterol. Therefore, to serve as a baseline, we adjusted the IRIS and GAIN features of the MRC1024 confocal microscope so that WT brain sections exhibited mean fluorescent intensities of 1000 arbitrary units. Once these parameters were set, sections from WT-SSI, NPC1, and NPC1-SSI were scanned and the intensity values measured. To insure that the measured intensity values were proportional to the signal intensity, instrument settings were chosen such that the signals recorded from the brightest samples prepared from the NPC1 mice do not go beyond the full-scale value. To confirm these findings, a second experiment using a second set of brains from WT, WT-SSI, NPC1, and NPC1-SSI mice were examined in the same manner. Neurotrace staining was used to aid in the identification of regions and was used as an internal standard for staining intensities between NPC and WT sections. Measurements of neurotrace fluorescent intensities showed less then 10 % difference in values, indicating that the differences were specific to BC-theta staining.
Sterol and fatty acid synthesis measurements in mice brains
Mice were anesthetized with isoflurane, then mounted onto the Kopf small animal stereotaxic instrument. A sagittal skin incision, about 0.5 cm in length, was made at the top of the head to expose the skull. 3H-acetate (at 20 Cii/mmol), 100 μCi in 3 μl of PBS, was injected into one of the two lateral ventricles of the brain, with a fixed, 5 μl-size Hamilton glass syringe. Bone marks (as described in (Paxinos and Franklin, 2003)) were used as reference points to determine the site of injection. Duration for injection was 1 min. Afterwards, the skin incision was sealed by using mouse glue (3M Vetbond). The mice were kept in the cages at room temp for up to 4 hrs as indicated, and were sacrificed by decapitation with a quick scissor cut. The brain was dissected into five anatomic regions: cortex, thalamus, brain stem, cerebellum and spinal cord (up to10 mm in length below the brain stem). The samples were subjected to lipid analyses using a previously described procedure with minor modifications (Limanek et al., 1978). For sterols analysis, the brain tissues were weighed and homogenized in 0.5 ml of Buffer A (25 mM Tris pH 7.6, 1 mM EDTA) in an Eppendorf tube with a Kontes pellet pestle micro grinder. The homogenates were extracted with 3 ml of chloroform/methanol (2:1 in volume), plus 1 ml of H2O in 13 × 100 mm glass tubes (Fisher Scientific). The samples were centrifuged at 1,000 g in a Beckman TJ-6 centrifuge for 5 min. The top phase was removed and the lower phase was dried to 0.1 ml with N2 blowing. To each sample 3 ml of ethanol/benzene/H2O (80:20:5 in volume) and 0.34 ml of 10 M KOH were added, and the mixture was saponified at 80 °C for 1 hr. The samples were then dried with N2, 1 ml of H2O per sample was added and the samples were vortexed thoroughly, then extracted with 2 ml of petroleum ether 4 times. The collected 8 ml of petroleum ether fraction was reduced in volume with N2 to 3 ml, washed with 1 ml of 3 % NaHCO3 and then 1 ml of H2O. Finally, the 3 ml of petroleum ether fraction was completely dried with N2 stream, dissolved in 80 μl of ethyl acetate, and spotted onto a thin layer chromatography (TLC) plate (J.T. Baker, Si250). The running solvent system was CH2Cl2 (Dichloromethane): ethyl acetate (97:3). The sterol bands were scraped off the TLC and counted, using scintillation fluid for nonaqueous samples (Ecoscint O, from National Diagnostics). This fluid does not cause quenching of lipid samples present in silica. The counting efficiency was 40%. For fatty acids analysis, after the petroleum ether extraction, the 1 ml aqueous phase was acidified with 0.3 ml of 12 M HCl and then extracted with 2 ml of petroleum ether 4 times. The collected 8 ml of petroleum ether fraction was concentrated to 3 ml with N2 blowing, washed with 1 ml of 5 mM HCl and then with 1 ml of H2O. Finally the petroleum ether fraction was completely dried with N2 and dissolved in 80 μl of ethyl acetate and spotted onto TLC plate. The running system was petroleum ether:ether:acetic acid (90:10:1). The fatty acid bands were scraped off the TLC and counted in the same manner as described above. Statistical analyses of results shown in Figs. 3 and 4 were performed with GraphPad Prism Version 4.01, using a two-tailed, unpaired Student's t-test. The difference between two sets of values was considered significant when the p value was less than 0.05. Error bars represent mean + SEM from 4 to 7 mice per group.
Figure 3. Relative sterol synthesis in the young WT and NPC1-/- mice brains.
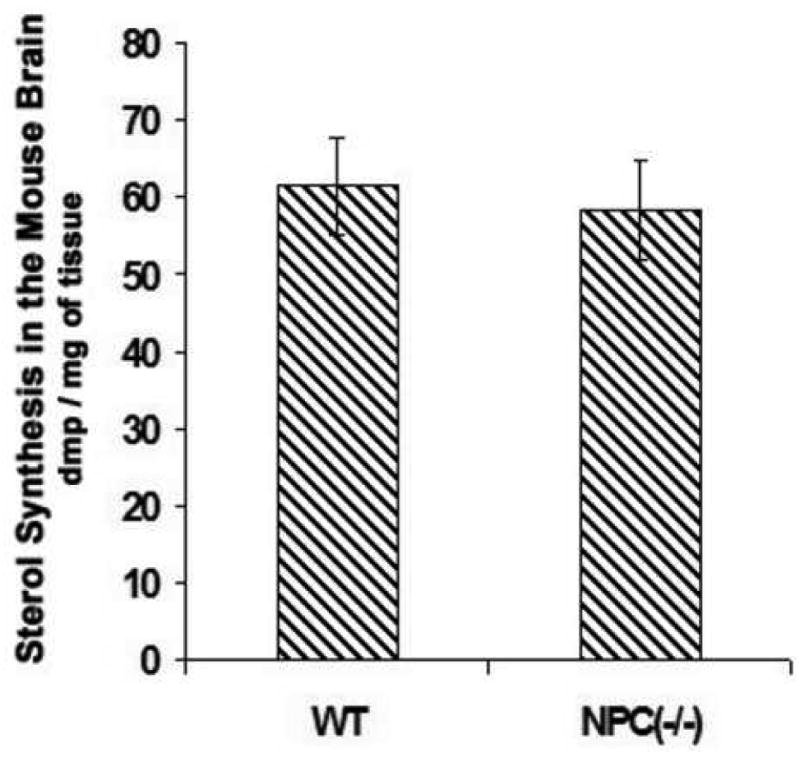
On postnatal day 10, 3 μl of 3H-acetate (100 μCi) per mouse was injected into the lateral ventricle of the brains; 1 hr later mice were sacrificed and the brains were processed for lipid analyses according to procedures described in Materials and Methods. Four mice were used for each group. The results shown are from a single experiment. Each bar represents the mean + SEM. Statistical Analysis showed that there was no significant difference in values between the WT mice and the NPC1-/- mice.
Figure 4. Effect of lovastatin (A, B) or CP-340868 (C, D) on lipid syntheses in the WT mice brains.
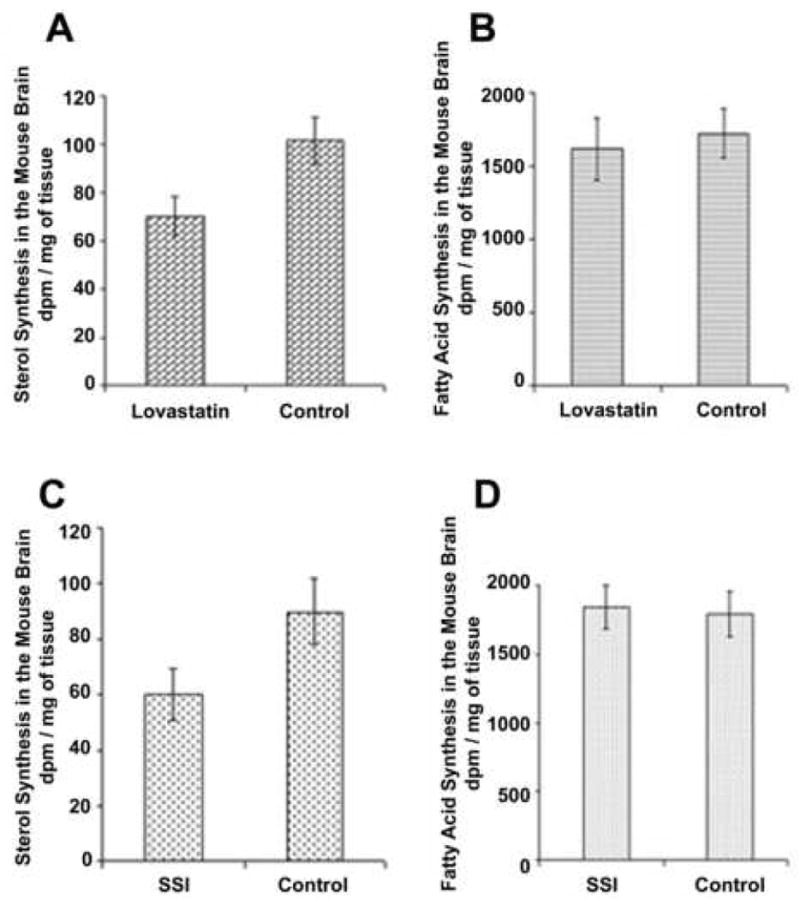
Mice at PND 8 were peritoneally injected with 10 μg of Lovastatin/g of body weight, or were subcutaneously injected with 10 μg of CP-340868 (SSI)/g of body weight. The control mice were injected with the same amount of vehicle only. The injections were given once every day for 8 days. On day 15, 3 μl of 3H-acetate (100 μCi) per mouse was injected into the lateral ventricle of the brains; 1 hr later mice were sacrificed and the brains were processed for lipid analyses according to procedures described in the Materials and Methods section. The results shown are representative of three separate experiments. For each experiment, 9 mice were used for each group. Lovastatin was prepared at 1 mg/ml of lovastatin in mazol PG810 (propylene glycol dicaprylate). SSI was prepared at 1 mg/ml of saline. Each bar represents the mean + SEM. For sterol synthesis values, statistical analysis showed that the difference in values between the SSI-treated or the lovastatin-treated group and the control group was significant. For fatty acid synthesis values, statistical analysis showed that there was no significant difference in values between the SSI-treated or the lovastatin-treated group and the control group.
Analysis of gangliosides, and non-acidic glycolipids in the mice brains
Mice were killed and dissected, and frozen tissues were stored at −80 °C in tight containers prior to analysis. For each mouse, brain analyses were carried out on one cerebrum hemisphere after dissecting out the cerebellum and brainstem. The procedures used for total lipid extraction, separation and purification of main lipid fractions were similar to those used in previous studies (Fujita et al., 1996; Liu et al., 2000; Sleat et al., 2004). In short, total lipids were extracted from a 20 % tissue homogenate in water in chloroform:methanol 1:2 (v/v). For ganglioside studies, part of the extract was desalted and separated into two fractions using 100 mg reverse-phase Bond Elut C18 (Varian) columns. The acidic lipid fraction, eluted with methanol 12:1 (v/v), contained all the gangliosides and was used without further purification. Total sialic acid was measured by the Svennerholm's resorcinol method as described (Fujita et al., 1996). An extract aliquot corresponding to 3 mg tissue was spotted using a Linomat 4 device (Camag) on high-performance thin layer chromatography (HPTLC) silica gel 60 plates (Merck, Darmstadt) developed with chloroform-methanol- 0.2 % CaCl2 55:45:10 (v/v/v) and sprayed with resorcinol-HCl reagent to visualize the sialic acid moiety of individual gangliosides. Densitometric quantification at 580 nm was done using a Camag TLCII scanner equipped with the Cats software. After normalization to the total sialic acid content, individual concentrations were calculated by taking into account the number of sialic acids for each ganglioside. For quantitation of galactosylceramides, the total lipid extract was saponified to discard phosphoglycerides and desalted (Fujita et al., 1996). Suitable aliquots (equivalents of 0.3-0.6 mg wet tissue) were spotted on HPTLC plates together with standards of galactosylceramide from bovine brain in the range of 0.5 to 2 nmol. The plates were developed in chloroform-methanol-water 65:25:4 (by vol.) and sprayed with orcinol-sulphuric acid reagent to visualize hexose-containing compounds. Densitometry was performed at 650 nm as above and quantitation made from the calibrated standards.
Results
Injecting 3H-acetate to the mice brains to monitor relative sterol and fatty acid synthesis rates in mice brains
We aimed at developing a simple method to monitor relative lipid biosynthesis rates in intact mice brains. Labeled acetate (with 14C at C1 and/or C2, or with 3H at C2) has been used extensively by many laboratories to monitor relative rates of lipid syntheses in intact cells or in animals. This method has the intrinsic limitation that the intracellular labeled acetyl CoA pool may undergo variable dilution by the unlabeled acetyl CoA in different tissues under different physiological conditions. This problem has been overcome by employing labeled water, which equilibrates rapidly with unlabeled water within the cell interior, thus eliminating the uncertainties associated with the precursor pool. The labeled water method has the disadvantage that very large amounts of radioactivity are required in order to obtain reliable radioactivity into the sterols. Anderson and Dietschy (Andersen and Dietschy, 1978) compared the labeled water incorporation method and labeled acetate incorporation method for monitoring sterol synthesis in various systemic tissues of rats in vivo, and showed that the labeled acetate method is valid for the purpose of monitoring relative changes in sterol syntheses rates. In brains, another challenging technical limitation exists: Webber and Edmond (Webber and Edmond, 1979) injected individual 14C- labeled lipid precursors, including [3-14C] acetoacetate, [2-14C] acetate, [1-14C] octanoate, etc., subcutaneously to rats, and monitored the incorporation of the 14C label into sterols and fatty acids in the rat brains. The results showed that for most of the precursors employed, the extents of the label incorporated into lipids were low, and the incorporation rates were linear for only 10 to 15 min; afterwards, the incorporation reached certain plateau values. The low yield in incorporation was probably because acetate injected subcutaneously cannot effectively reach the brain; the rapid decline of linearity in incorporation rate was mostly likely due to the rapid aerobic oxidation of labeled acetyl coenzyme A, converting the 14C label into 14CO2, thus halting its utilization for lipid syntheses; however, at various times, the amount of labeled sterols produced from various labeled lipid precursors was a constant fraction of the amount of labeled fatty acids. These results suggested that if injected directly into the CNS, the labeled lipid precursors could be used to monitor the relative rates of sterol and fatty acid syntheses in the animal brains.
In results not shown, we monitored sterol synthesis rates in the brains of 2-week-old mice by peritoneally injecting 3H-acetate (at 200 μCi/mouse). One hr after injection, we sacrificed the mice for lipid analysis. The results showed that labeled acetate was converted to labeled sterols in the brains. However, the total counts per minute were very low, amounting to 7 dpm labeled sterols per mg brain tissue. To increase the dpm of labeled sterols, we injected 3 μl of 3H-acetate (at 100 μCi/3 μl) directly into one of the two lateral ventricles of each mouse brain with a fixed, 5 μl-size Hamilton glass syringe. At various indicated times after injection, the mice were sacrificed for lipid analysis. The results (Fig. 1A) show that at the 1st hr, the brain injection method labeled sterols at 29 dpm per mg tissue. Since we used only 100 μCi acetate per mouse, the results show that acetate injected in the peritoneal cavity cannot effectively reach the CNS, while acetate injected into the lateral ventricle reaches the CNS in a much more efficient manner. Additionally, results in Fig. 1A show that the incorporation of 3H-acetate into labeled brain sterols increased within the first hour to reach a plateau, then decreased slightly by the 4th hr. We next used the brain injection method to measure relative sterol synthesis rates in mice of 1, 2, and 3 weeks of age. The results (Fig. 1B) show that the 2-week-old mice exhibited a twofold increase in sterol synthesis rate when compared to the 1-week-old mice; the increase was largely sustained in the 3-week-old mice. These results are similar to the results of Quan and colleagues (Quan et al., 2003), who used the 3H-water method for labeling lipids, and showed that a 37 % increase in sterol synthesis occurred when the mice age from the 1st week to the 2nd week, and the increase was largely sustained in the 3-week-old mice. We asked whether the water-soluble substance injected through the lateral ventricle might rapidly diffuse outward to different regions of the brain. To address this question, we injected trypan blue, a non-harmful, water-soluble dye, into one of the lateral ventricles of the mice. After the injection, the mice were sacrificed 5 min later. Sagittal sections of the brain were made to examine the dye diffusion pattern in the subarachnoid space. The results (Fig.2) show that the dye does diffuse rapidly in various regions of the mice brain within minutes. Encouraged by these results, we used the brain injection method to monitor the relative sterol synthesis rate and relative fatty acid synthesis rate in various regions of the 2-week-old mice brains. The results show that sterol syntheses occur in various regions but the rates differ. The rates of sterol synthesis rank in the following order: spinal cords > brain stems > cortex, thalamus, and cerebellum. Again, these results are very similar to the results of Quan and colleagues (Quan et al., 2003), who used the 3H-water method for labeling lipids, and showed that in the 2-week-old mice, spinal cords exhibit the highest absolute sterol synthesis rate, followed by brain stems and cerebellum. We also monitored the relative fatty acid synthesis rates in various brain regions. The results show that contrary to the situation for sterol synthesis rates, thalamus exhibits the highest rate in fatty acid synthesis, followed by cortex, brain stems, and cerebellum; the spinal cord exhibits the lowest rate in fatty acid synthesis. The results presented in Fig. 1C, D also show that liver tissue exhibits much lower relative sterol and fatty acid synthesis rates after brain 3H-acetate injection than those in the brain tissues. This result can be attributed to the fact that the labeled acetate injected into the brain can only be partially accessible to the liver after it moves out of the brain and enters the circulation. We next used the new method to compare the relative sterol synthesis in 10-day old WT and NPC1-/- mice brains. The results (Fig. 3) show that despite the NPC1 mutation, the endogenous sterol synthesis is not up-regulated in the young NPC1-/- mice brains. These results are also similar to the early results of Quan and colleagues (Quan et al., 2003), who used the 3H-water method for labeling lipids in mice brains.
Figure 1.
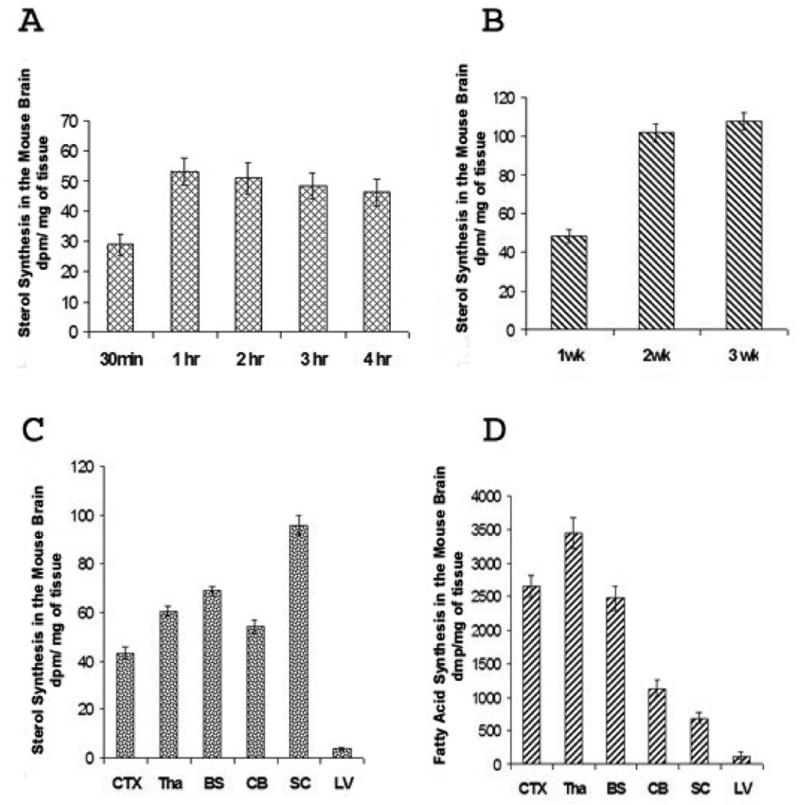
A. Relative sterol synthesis with time in the mice brains. Three μl of 3H-acetate (100 μCi) was injected per mouse into the lateral ventricles of the WT mice brains at 2 weeks of age. At various indicated times after injection, mice were sacrificed and the brains were processed for lipid analysis as described in Materials and Methods. The results shown are the averages of two experiments. For each experiment, five or six mice were used for each time point.
B. Relative sterol synthesis in the brains of mice at 1, 2, or 3 weeks of age. Three μl of 3H-acetate (100 μCi) per mouse was injected into the lateral ventricles of WT mice brains at 1, 2, or 3 weeks of age as indicated. Mice were sacrificed 1 hr after injection for lipid analyses. Six mice were used for each group. The results shown are from a single experiment.
C. Relative sterol syntheses in various regions of the mice brains. Labeled acetates were injected into mice brains as described in Fig. 1B. One hr later, mice were sacrificed; the various anatomical regions of the brains as indicated were isolated and processed for lipid analyses. The results shown are representative of three separate experiments. Eight or nine mice were used for each experiment. CTX: cortex; Tha: thalamus; BS: brain stem; CB: cerebellum; SC: spinal cord; LV: liver.
D. Relative fatty acid syntheses in various regions of the mice brains. Labeled acetates were injected into mice brains as described in Fig. 1B. One hr later, mice were sacrificed; the various anatomical regions of the brains as indicated were isolated and processed for lipid analyses. The results shown are representative of three separate experiments. Eight or nine mice were used for each experiment. CTX: cortex; Tha: thalamus; BS: brain stem; CB: cerebellum; SC: spinal cord; LV: liver.
Figure 2. Diffusion of Trypan blue in the subarachnoid space after injection into lateral ventricle.
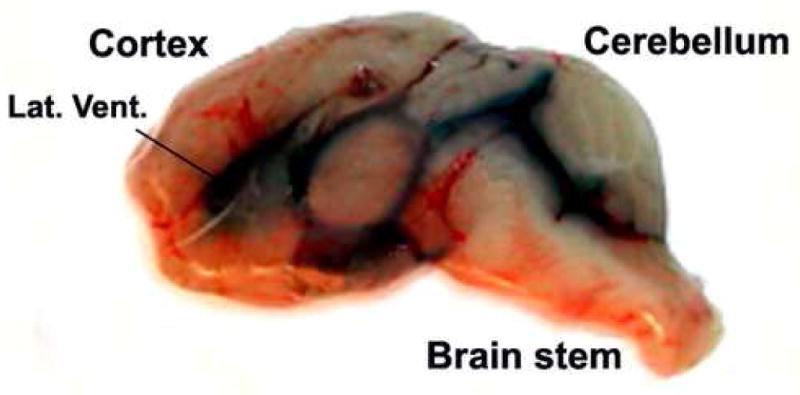
Normal BALB/c mice at one month of age were anesthetized with isoflurane, then mounted on the Kopf small animal sterotaxic instrument. 3 μl of 1 % Trypan blue in PBS was injected into one of the lateral ventricles (indicated as Lat. Vent.) of the brain according to the procedure described in Materials and Methods. The mice were sacrificed 5 min after the injection. Sagittal sections of the brain were made to exam the dye diffusion pattern in the subarachnoid space. The picture taken was within 1 min after the mouse was sacrificed, and is representative of results using 4 mice.
Effect of CP-340868 or lovastatin on sterol and fatty acid syntheses in mice brains
Several drugs, including lovastatin and CP-340868, block endogenous cholesterol synthesis. Lovastatin inhibits HMG-CoA reductase activity and blocks the biosynthesis of mevalonate, a key molecule in the isoprenoid biosynthetic pathway. Mevalonate is required for the biosynthesis of cholesterol as well as many other essential non-sterol metabolites (Goldstein and Brown, 1990) that include ubiquinone, dolichol, and prenylation of numerous proteins. In contrast, CP-340868 inhibits squalene synthase activity and blocks the biosynthesis of squalene, which is required for sterols but not for non-sterol metabolites (reviewed in (Tansey and Shechter, 2000)). Several specific squalene synthase inhibitors with different structural types are available. CP-340868 is a potent, reversible, non-competitive squalene synthase inhibitor that shows a high degree of specificity for inhibition of cholesterol formation relative to inhibition of non-sterol polyisoprenoid formation and protein prenylation (Harwood et al., 1997; Pandit et al., 2000). In mice, CP-340868 inhibits sterol synthesis with an ED50 of 0.4 mg/kg, and exhibits an oral bioavailability of 22 %, a plasma half-life of 8.7 hrs, and a peak plasma concentration of 550 ng/ml (35 times its in vitro IC50 for squalene synthase inhibition of 27 nM) after a 10 mg/kg oral dose. CP-340868 is water-soluble and is able to enter the brain interiors of treated animals (results not shown).
We treated the wild-type (WT) mice with lovastatin or with the squalene synthase inhibitor, CP-340868 (SSI). The drugs were administered intraperitoneally (Lovastatin) or subcutaneously (SSI) to the mice once per day at 10 mg/kg, for six consecutive days. At the end of the 6th day, we monitored the effects of the drug on brain sterol synthesis rate and on brain fatty acid synthesis rate. The synthesis rates were monitored by using brain injections of labeled acetate described earlier. The results (Fig. 4A, C) show that both lovastatin and SSI inhibited sterol synthesis in the mice brains by approximately 30 %, while neither drug significantly inhibited fatty acid biosynthesis (Fig. 4B, D). Additional results show that lovastatin inhibited sterol synthesis by approximately 80 %, while SSI inhibited sterol synthesis by approximately 70 % in the mice livers (results not shown). The greater inhibition of hepatic cholesterol synthesis by lovastatin and SSI is not unexpected and is consistent with their greater hepatic relative to brain exposures.
Effect of the 6-day SSI treatment to neuronal cholesterol accumulation and to astrogliosis in young mutant NPC1 mice
We next monitored the effect of the 6-day SSI treatment to very young WT and NPC1 mice. We selected SSI rather than lovastatin for studies evaluating the effects of cholesterol synthesis inhibition on neuronal cholesterol accumulation and astrogliosis since HMG-CoA reductase inhibitors such as lovastatin, in addition to inhibiting cholesterol synthesis, also have the potential to interfere with key polyisoprenoid synthesis and with protein prenylation (Liu et al., 2000), effects that could complicate interpretation of the role of cholesterol synthesis inhibition per se on neuronal cholesterol accumulation and astrogliosis. At the end of the treatment, we examined the neuronal cholesterol accumulation in the brains by BC-theta staining (Reid et al., 2004). The results (Fig. 5A) show that without the SSI, neuronal cholesterol accumulations occur in various brain regions of the NPC1 mice; such accumulations do not occur in the WT mice brains. The SSI treatment clearly reduced neuronal cholesterol accumulation in all regions of the NPC1 mice brains examined. SSI treatment also reduced the extraneuronal cholesterol that accumulates in NPC1 mice, suggesting that SSI treatment may also reduce cholesterol accumulation in extraneuronal, for example glial, cells. We evaluated the relative intensities of the BC-theta staining signals by calculating the relative mean pixel intensities from large collections of fluorescent images taken under confocal microscopy (Reid et al., 2004). The results show that SSI treatment causes a decrease in the BC-theta staining signals by 32 to 44 % in various NPC1 mice brain regions as indicated (Fig. 5B). Additional results show that the six-day SSI treatment also significantly reduced the abnormal cholesterol accumulation in the NPC1 mice livers (Fig. 5C). Previously, we have shown that in NPC1 mice brains, by PND 15, astrocyte activation becomes significant in the thalamus and cortex regions (Reid et al., 2004). To examine the effect of SSI on astrocyte activation, we prepared sections from thalamus and cortex regions of NPC1 mice with or without SSI treatment, and monitored astrocyte activation by GFAP stainings. The results show that at PND 14, without SSI, activated astrocytes appear in the cortex and thalamus regions of the NPC1 mice brains, but not those of the WT mice brains (Fig. 6, 1st and 3rd rows). SSI treatment of NPC1 mice clearly decreased astrocyte activation in these areas (Fig. 6, comparing the 3rd and 4th rows).
Figure 5. The 6-day SSI treatment reduces cholesterol accumulation in the brains and livers of young NPC1-/- mice.
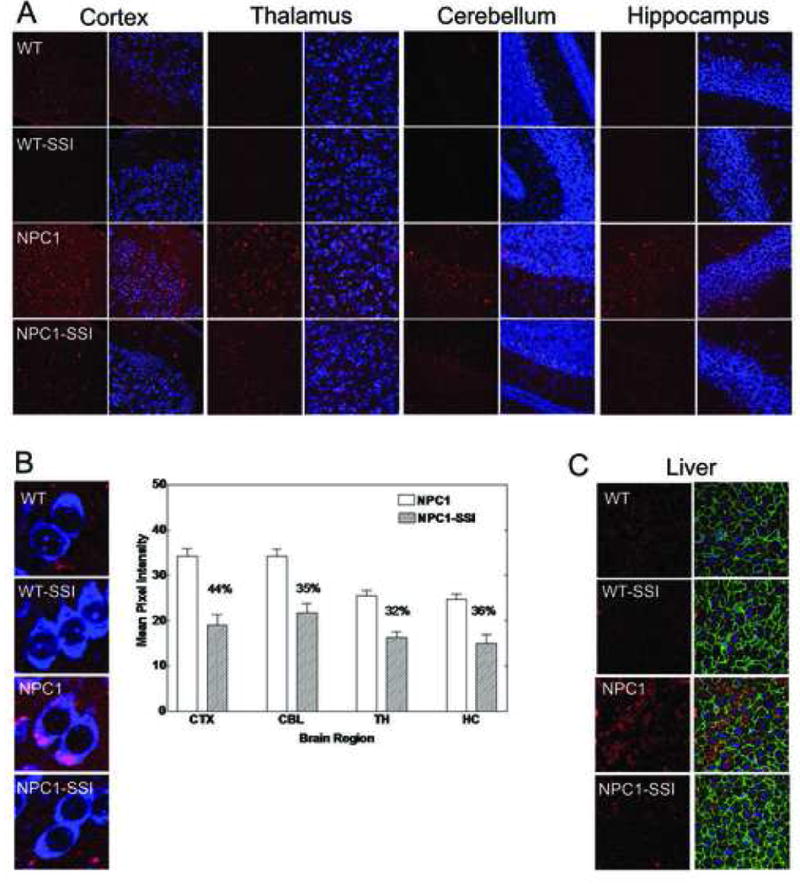
A. Neuronal cholesterol accumulation in WT and NPC1-/- mice brains with or without SSI treatment. WT and NPC1 mice received daily subcutaneous injections of CP-340868 in saline (at 10 μg/g), (WT-SSI; NPC1-SSI) or saline alone (WT; NPC1) once per day for 6 consecutive days starting at postnatal day 8. Two (NPC-/-) mice and 2 age-matched normal sibling mice (WT) were used. Afterwards, the mice (at day 14) were sacrificed and their neuronal cholesterol accumulations were evaluated by the method described previously.16 Briefly, coronal brain sections from the cortex (CTX), thalamus (TH), hippocampus (HC), and cerebellum (CBL) regions were prepared and stained with BC-theta (red) and Neurotrace (blue). Each 7 μm section was examined under a 40X objective using a Bio-Rad MRC1024 confocal microscope. The results are representative of two separate experiments.
B. Quantification of neuronal cholesterol accumulation in NPC1-/- mice brains with or without SSI treatment. The results obtained in A were quantified according to the procedures described in Materials and Methods. For each section, the reported mean pixel intensity values are values of the NPC1-/- mouse brain subtracted from values of the WT mouse. For each NPC1-/- mouse brain section, the % reduction in BC-theta signals due to SSI treatment is given. Results represent one of two separate experiments. High magnification images of representative cell types are shown at left.
C. SSI treatment reduces cholesterol accumulation in the livers of NPC1-/- mice. Livers of mice described in A were isolated and processed for staining by BC-theta according to procedure as described in Material and Methods. The green fluorescence represents plasma membrane staining using a phalloidin stain, which binds to plasma membrane F-actin. The blue fluorescence represents nuclear staining using a DAPI stain.
Figure 6. The 6-day SSI treatment significantly reduces astrocyte activation in cortex and thalamus regions of young NPC1-/- mice.
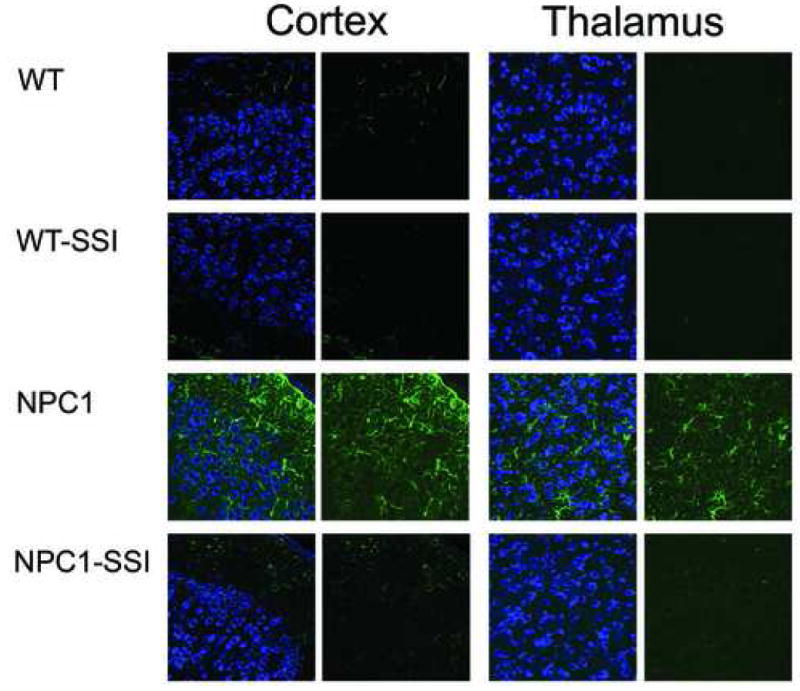
The coronal brain sections of WT and NPC1-/- mice treated with or without SSI as described in Fig. 3 were prepared and stained with neurotrace (left panels) for neurons (blue) and with anti-GFAP antibodies (right panels) for activated astrocytes (green), and viewed under a confocal microscope, according to method described.16 Images are representatives of a large number (more than 30) of photographs obtained.
Effect of the 6-day SSI treatment on brain gangliosides and galactolipids of young NPC1 mice
By mass measurements, the most conspicuous lipid accumulation in the brains of NPC1 mice is that of gangliosides GM2 and GM3 (Liu et al., 2000; Sleat et al., 2004). This is already obvious at the age of 14 PND, as illustrated in Fig. 7 (left top panel, lanes 1 and 6). GM3 and GM2 have been implicated in the pathophysiology of NPC (Zervas et al., 2001b), and therefore, our study on the effect of SSI treatment on brain lipids was primarily focused on gangliosides. There was no change in the concentrations of the major brain gangliosides, including GM1 (Fig. 7, panel A and bar graph; other quantitative data not shown). In WT mice, extremely small amounts of GM3 and GM2 are present. Their concentration was not affected by SSI treatment. On the other hand, in the NPC1 mice, we observed a clear effect of the SSI treatment on the concentration of GM3 ganglioside, with a nearly 50 % decrease in treated animals (Fig. 7, panel A, lanes 4, 5 vs. lane 6, and bar graph). A slight decrease in GM2 ganglioside concentration was also observed, which however remained within the range of variation observed for untreated affected mice (M.T. Vanier, unpublished data) and could thus not be considered as significant. Considering the importance of cholesterol synthesis during brain development and more specifically myelination, the effect of SSI treatment on the most specific myelin lipids, galactosylceramides and sulfatides (galactolipids), was also investigated. Thin-layer chromatographic analysis of the non-acidic lipid fraction (Fig. 7, panel B) disclosed large differences in the content of galactosylceramides between control and NPC1 mice, an already well-documented finding (Liu et al., 2000; Sleat et al., 2004), but also showed marked differences between treated and untreated animals. In the WT mice, SSI treatment significantly reduced the concentration of galactosylceramides with a striking effect on the ceramide moiety containing non-hydroxylated fatty acids. A similar effect also occurred in the NPC1 mice, thus further reducing the already low galactosylceramide content. Other major lipid classes did not appear affected. Galactosylceramides and sulfatides were then purified from the total lipid extract, a chromatographic study obtained using the more specific orcinol staining (panel C) confirmed the initial findings, and further showed that sulfatides were similarly affected. The galactosylceramide concentration of the untreated NPC1 mice was approximately 40 % of that in WT mice. SSI treatment reduced the galactosylceramide content by 45 % in the WT mice, and by 30 % in the NPC1 mutant mice. In untreated animals, both WT or NPC1 mutants, the ceramide moiety with non-hydroxylated fatty acids constituted 35-38 % of total galactosylcearmides, compared with 11-13 % in SSI-treated WT mice and 15-20 % in NPC1 mice.
Figure 7. Lipid profiles in brain tissue from young WT and NPC1 mice treated with SSI.
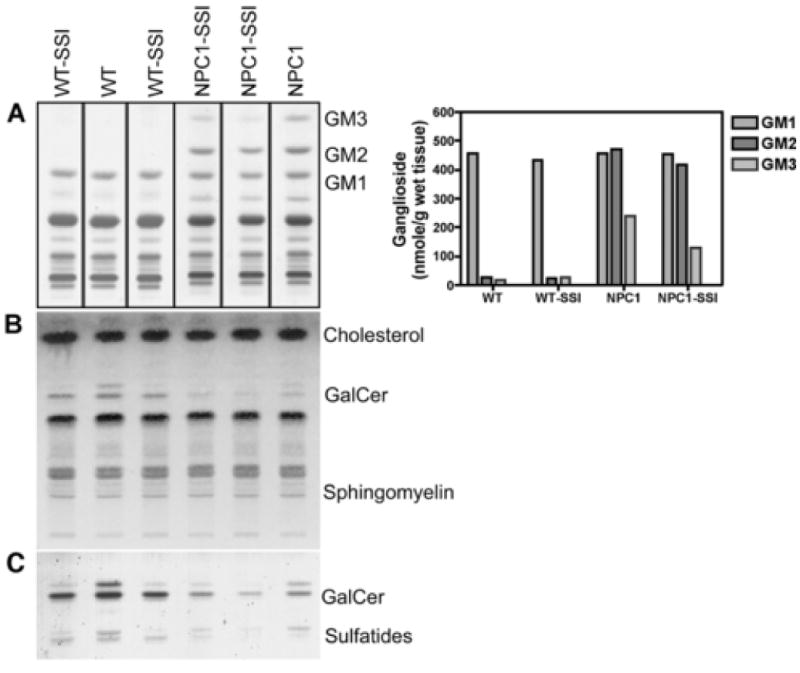
The 6-day SSI treatment was administered as described in Fig. 3. The procedures for lipid extraction, purification and separation, as well as the various chromatographic systems, are described in the Material and Methods section. The brains from one WT mouse without SSI and from one NPC1 mouse without SSI were employed for glycolipid analyses. One of us (M.T.V.) had previously analyzed the glycolipid contents of brains from many WT and NPC1 mice at the same age, and found that the variations of glycolipid contents among individual mice were small. The data reported here for the untreated WT mouse and the untreated NPC1 mouse fall within the expected values. For mice with SSI treatment, the brains from two WT mice and from two NPC1 mice were employed for analyses, and the results were shown individually (in lanes 1, 3, 4, and 5). The values reported in the bar graph are the means of values from the two SSI-treated mice.
Panel A: Ganglioside patterns. The bands corresponding to gangliosides GM3, GM2 and GM1 are indicated. The adjacent bar graph reports the concentration of these three gangliosides, expressed as nmol ganglioside / g wet weight of tissue.
Panel B: Chromatographic profiles of total non-acidic lipids (chloroform:methanol 1:2 eluate from Bond-Elut C18 column). An aliquot corresponding to 1 mg tissue was applied to the plate. GalCer: galacotsylceramides.
Panel C: Galactolipids profiles (galactosylceramides and sulfatides). Galactolipids were purified as described in Material and Methods. An aliquot corresponding to 0.6 mg tissue was applied to the plate. For both lipids, the upper band corresponds to a ceramide moiety containing “normal”, unhydroxylated fatty acids, the lower band corresponds to a ceramide moiety with hydroxylated fatty acids.
Discussion
In the current work, we first describe a new injection method to monitor relative sterol and fatty acid syntheses in the brain. We then treat WT and NPC1 mice with the squalene synthase inhibitor CP-340868 (SSI) to examine the effect of blocking sterol synthesis in the NPC1 mice brains. The results show that a six-day SSI treatment of young NPC1 mice significantly diminishes neuronal and possibly also glial cell cholesterol accumulation in various regions. The methods described in our current work can be broadly applied to study the potential relationship between cholesterol synthesis and various neurodegenerative diseases, including Alzheimer's disease.
Previously, we have shown that in the young NPC1 mice brains neuronal cholesterol accumulation can be observed as early as day 9, while activated astrocytes (astrogliosis) begin to appear in the thalamus and cortex by day 15. In general, astrogliosis occurs in response to brain injury. Our current results show that the six-day SSI treatment significantly slows astrocyte activation in NPC1 mice. These results support the hypothesis that in the NPC1 mice, neuronal cholesterol accumulation contributes to the early pathogenesis, resulting in brain injury and leading to astrogliosis; treating mice with SSI attenuates the neuronal injury caused by neuronal cholesterol accumulation. Exactly how neuronal cholesterol accumulation induces neuronal injury is presently unknown. Abnormal accumulation of cholesterol within the endo/lysosomal compartments are believed to cause these compartments to malfunction in various membrane trafficking activities (discussed in (Chang et al., 2005a)). It is possible that the membrane trafficking activities in neurons are particularly prone to the abnormal buildup of cholesterol in the late endo/lysosomes.
When examined in various cell culture systems, in addition to cholesterol accumulation, mutant NPC cells also accumulate large amounts of complex glycosphingolipids, particularly GM2 and GM3 (reviewed in (Patterson et al., 2001)). The relationship between the cellular glycosphingolipid accumulation and cellular cholesterol accumulation is complex (discussed in (Chang et al., 2005b)). In mutant NPC1 cells, Reagan and colleagues showed that cholesterol accumulation inhibits lysosomal sphingomyelinase activity (Reagan et al., 2000); Salvioli and colleagues (Salvioli et al., 2004) showed that cholesterol accumulation inhibits two additional lysosomal enzymes involved in the degradation of glycosphingolipids. These two works provide a plausible mechanism to explain why cellular cholesterol accumulation may lead to glycosphingolipid accumulation in the lysosomes. The results of our current work, demonstrating that the six-day SSI treatment diminishes the GM3 content in the treated NPC1 mice brains, support the hypothesis that in NPC1 mice brains, elevated neuronal cholesterol accumulation results in elevated GM3 levels by delaying the normal degradation of GM3 in neurons and/or other neuronal cells. On the other hand, why SSI administration decreases GM3 but not GM2 in the treated NPC1 mice brains is not clear. It is possible that the elevated GM3 level may mainly be present in activated microglial cells and astrocytes, but not in neurons, of the NPC1 mice brains. Further investigations are required to clarify this issue.
Regarding the potential use of SSI as a drug therapy to treat NPC disease: in our current work, we studied the effect of SSI in very young mutant NPC mice (starting at PND 8, which is presumably equivalent to many weeks preterm in humans). In humans, most patients with NPC disease are not diagnosed until childhood. Thus, determination of the potential of SSI as a therapy for human NPC must await the results of treating older mice with SSI. Our study demonstrated the beneficial effects of SSI on cholesterol and ganglioside accumulation in NPC brains. We also showed that the six-day SSI treatment caused significant reductions in the GalCer and sulfatides contents in the brains, suggesting that partial inhibition of sterol synthesis in the brain could potentially cause disturbances in the myelin maturation process. The conclusion that SSI may affect the myelin maturation process is only a tentative one; the presumed effect of SSI is inferred from reduced galactolipid contents in treated mice brains (Fig. 7). In the future, in order to fully document the SSI effects on myelination, additional morphological evidence is needed. Nevertheless, our results are consistent with the result of Saher and colleagues (Saher et al., 2005), who demonstrated that mice with a conditional mutation of the squalene synthase have a severely perturbed myelin maturation process.
NPC is a fatal disease; currently, it has no cure. In animal models, numerous potential drug therapies have been attempted. Thus far, two different experimental drug therapies have shown partial efficacy in slowing down the disease progression (reviewed in (Patterson and Platt, 2004)). The first is the use of the compound NB-DNJ. This compound inhibits several enzymes involved in the glycolipid synthesis pathway, and has shown efficacy in experimental models for certain glycolipid storage diseases (Cox et al., 2000). In mutant NPC animals, NB-DNJ slows down disease progression and prolongs life spans by approximately 25 % (Zervas et al., 2001b). The second is to treat the mutant NPC1 mice with a neurosteroid called allopregnanolone (Griffin et al., 2004), or with the mirror image of allopregnanolone called ent-allopregnanolone (Langmade et al., 2006). Allopregnanolone or ent-allopregnanolone delays the onset of clinical neurological symptoms, partially prevents Purkinje cell death, reduces GM2 and GM3 accumulation in the brain, and doubles the lifespan of the treated NPC mice. In addition, co-administration of the compound T0901317, a synthetic oxysterol ligand that activates genes in cholesterol catabolism, potentiated the beneficial effects of the neurosteroids (Langmade et al., 2006). It is not yet clear whether or not these drugs reduce neuronal cholesterol accumulation in treated animals; none of these drugs alone was able to completely cure the disease. A complete cure of the NPC disease may require the use of a combination drug therapy. As shown in our current work, SSI reduces early neuronal cholesterol accumulation and abrogates astrogliosis, but it also inhibits the normal myelination process. The SSI therapy may be worthwhile considering as part of the combination therapy for the NPC disease and other neurodegenerative diseases that involve neuronal cholesterol accumulation as a causative agent, if its effect on myelin maturation process can be avoided/minimized.
Acknowledgments
We thank Nathan P. Watson for instruction on using the Kopf small animal stereotaxic instrument. We thank Nobutaka Ohgami, Yoshio Yamauchi, and Elena Y. Bryleva for helpful discussions, and Helina H. Josephson for careful preparation of this manuscript. This work is supported by NIH grants R01 HL 036709 and R21 NS 050716 to T.Y.C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen JM, Dietschy JM. Relative importance of high and low density lipoproteins in the regulation of cholesterol synthesis in the adrenal gland, ovary, and testis of the rat. J Biol Chem. 1978;253:9024–32. [PubMed] [Google Scholar]
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ, Dwyer NK, Neufeld EB, Chang TY, Liscum L, Strauss JF, III, Ohno K, Zeigler M, Carmi R, Sokol J, Markie D, O'Neill RR, van Diggelen OP, Elleder M, Patterson MC, Brady RO, Vanier MT, Pentchev PG, Tagle DA. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:228–31. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol Sensing, Trafficking, and Esterification. Annu Rev Cell Dev Biol. 2006;22:129–57. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005a;280:20917–20. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CCY. The Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005b;280:20917–20. doi: 10.1074/jbc.R400040200. [DOI] [PubMed] [Google Scholar]
- Cheruku SR, Xu Z, Dutia R, Lobel P, Storch J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J Biol Chem. 2006;281:31594–604. doi: 10.1074/jbc.M602765200. [DOI] [PubMed] [Google Scholar]
- Cox T, Lachmann R, Hollak C, Aerts J, van Weely S, Hrebicek M, Platt F, Butters TD, Dwek RA, Moyses C, Gow I, Elstein D, Zimran A. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–5. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Chang TY. Fate of Endogenously Synthesized Cholesterol in Niemann-Pick Type C1 Cells. J Biol Chem. 2000;275:41309–16. doi: 10.1074/jbc.M008272200. [DOI] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–97. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- Fujita N, Suzuki K, Vanier MT, Popko B, Maeda N, Klein A, Henseler M, Sandhoff K, Nakayasu H. Targeted disruption of the mouse sphingolipid activator protein gene: a complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum Mol Genet. 1996;5:711–25. doi: 10.1093/hmg/5.6.711. [DOI] [PubMed] [Google Scholar]
- German DC, Liang CL, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–50. doi: 10.1016/s0306-4522(01)00517-6. [DOI] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–30. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–11. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- Harwood HJ, Jr, Barbacci-Tobin EG, Petras SF, Lindsey S, Pellarin LD. 3-(4-Chlorophenyl)-2-(4-diethylaminoethoxyphenyl)-a-pentenonitrile monohydrogen citrate and related analogs. Reversible, competitive, first half-reaction squalene synthetase inhibitors. Biochem Pharm. 1997;53:839–64. doi: 10.1016/s0006-2952(96)00892-1. [DOI] [PubMed] [Google Scholar]
- Iwamoto M, Morita I, Fukuda M, Murota S, Ando S, Ohno-Iwashita Y. A biotinylated perfringolysin O derivative: a new probe for detection of cell surface cholesterol. Biochim Biophys Acta. 1997;1327:222–30. doi: 10.1016/s0005-2736(97)00061-8. [DOI] [PubMed] [Google Scholar]
- Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem. 2002;83:1154–63. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- Ko DC, Binkley J, Sidow A, Scott MP. The integrity of a cholesterol-binding pocket in Niemann-Pick C2 protein is necessary to control lysosome cholesterol levels. Proc Natl Acad Sci USA. 2003;100:2518–25. doi: 10.1073/pnas.0530027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci USA. 2006;103:13807–12. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limanek JS, Chin J, Chang TY. Mammalian Cell Mutant Requiring Cholesterol and Unsaturated Fatty Acid for Growth. Proc Natl Acad Sci USA. 1978;75:5452–6. doi: 10.1073/pnas.75.11.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu YP, Wada R, Neufeld EB, Mullin KA, Howard AC, Pentchev PG, Vanier MT, Suzuki K, Proia RL. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum Mol Genet. 2000;9:1087–92. doi: 10.1093/hmg/9.7.1087. [DOI] [PubMed] [Google Scholar]
- Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–5. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- Ohgami N, Ko DC, Thomas M, Scott MP, Chang CC, Chang TY. Binding between the Niemann-Pick C1 protein and a photoactivatable cholesterol analog requires a functional sterol-sensing domain. Proc Natl Acad Sci USA. 2004;101:12473–8. doi: 10.1073/pnas.0405255101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong WY, Kumar U, Switzer RC, Sidhu A, Suresh G, Hu CY, Patel SC. Neurodegeneration in Niemann-Pick Type C Disease Mice. Exp Brain Res. 2001;141:218–31. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ., Jr Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J Biol Chem. 2000;275:30610–7. doi: 10.1074/jbc.M004132200. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Platt F. Therapy of Niemann-Pick disease, type C. Biochim Biophys Acta. 2004;1685:77–82. doi: 10.1016/j.bbalip.2004.08.013. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, Blanchette-Mackie JE, Pentchev PG. Niemann-Pick disease type C: A lipid trafficking disorder. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York, New York: 2001. pp. 3611–33. [Google Scholar]
- Paxinos G, Franklin KBJ. The Mouse Brain in Stereotaxic Coordinates, Compact 2nd Edition. second. Academic Press; San Diego, California: 2003. [Google Scholar]
- Quan G, Xie C, Dietschy JM, Turley SD. Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res Dev Brain Res. 2003;146:87–98. doi: 10.1016/j.devbrainres.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Reagan JW, Jr, Hubbert ML, Shelness GS. Posttranslational regulation of acid sphingomyelinase in Niemann-Pick type C1 fibroblasts and free cholesterol-enriched Chinese hamster ovary cells. J Biol Chem. 2000;275:38104–10. doi: 10.1074/jbc.M005296200. [DOI] [PubMed] [Google Scholar]
- Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang TY. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res. 2004;45:582–91. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- Saher G, Brugger B, Lappe-Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S, Nave KA. High cholesterol level is essential for myelin membrane growth. Nat Neurosci. 2005;8:468–75. doi: 10.1038/nn1426. [DOI] [PubMed] [Google Scholar]
- Salvioli R, Scarpa S, Ciaffoni F, Tatti M, Ramoni C, Vanier MT, Vaccaro AM. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann-Pick disease type C. J Biol Chem. 2004;279:17674–80. doi: 10.1074/jbc.M313517200. [DOI] [PubMed] [Google Scholar]
- Sleat DE, Wiseman JA, El-Banna M, Price SM, Verot L, Shen MM, Tint GS, Vanier MT, Walkley SU, Lobel P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc Natl Acad Sci USA. 2004;101:5886–91. doi: 10.1073/pnas.0308456101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugii S, Reid PC, Ohgami N, Du H, Chang TY. Distinct endosomal compartments in early trafficking of low density lipoprotein-derived cholesterol. J Biol Chem. 2003;278:27180–9. doi: 10.1074/jbc.M300542200. [DOI] [PubMed] [Google Scholar]
- Takikita S, Fukuda T, Mohri I, Yagi T, Suzuki K. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J Neuropathol Exp Neurol. 2004;63:660–73. doi: 10.1093/jnen/63.6.660. [DOI] [PubMed] [Google Scholar]
- Tansey TR, Shechter I. Structure and regulation of mammalian squalene synthase. Biochim Biophys Acta. 2000;1529:49–62. doi: 10.1016/s1388-1981(00)00137-2. [DOI] [PubMed] [Google Scholar]
- Treiber-Held S, Distl R, Meske V, Albert F, Ohm TG. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. J Pathol. 2003;200:95–103. doi: 10.1002/path.1345. [DOI] [PubMed] [Google Scholar]
- Waheed AA, Shimada Y, Heijnen HF, Nakamura M, Inomata M, Hayashi M, Iwashita S, Slot JW, Ohno-Iwashita Y. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts) Proc Natl Acad Sci USA. 2001;98:4926–31. doi: 10.1073/pnas.091090798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber RJ, Edmond J. The in vivo utilization of acetoacetate, D-(-)-3-hydroxybutyrate, and glucose for lipid synthesis in brain in the 18-day-old rat. Evidence for an acetyl-CoA bypass for sterol synthesis. J Biol Chem. 1979;254:3912–20. [PubMed] [Google Scholar]
- Wojtanik KM, Liscum L. The transport of LDL-derived cholesterol to the plasma membrane is defective in NPC1 cells. J Biol Chem. 2003;278:14850–6. doi: 10.1074/jbc.M300488200. [DOI] [PubMed] [Google Scholar]
- Xie C, Burns DK, Turley SD, Dietschy JM. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J Neuropathol Exp Neurol. 2000;59:1106–17. doi: 10.1093/jnen/59.12.1106. [DOI] [PubMed] [Google Scholar]
- Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick Disease Type C Accumulate Gangliosides as Well as Unesterified Cholesterol and Undergo Dendritic and Axonal Alterations. J Neuropathol Exp Neurol. 2001a;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- Zervas M, Somers KL, Thrall MA, Walkley SU. Critical Role of Glycosphingolipids in Niemann-Pick Disease Type C. Curr Biol. 2001b;11:1283–7. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]