

Abstract
Occupational exposure to nickel(Ni), chromium(Cr), and arsenic(As) containing compounds has been associated with lung cancer and other adverse health effects. Their carcinogenic properties may be attributable in part, to activation and/or repression of gene expression induced by changes in the DNA methylation status and histone tail post-translational modifications. Here we show that individual treatment with nickel, chromate, and arsenite all affect the gene activating mark H3K4 methylation. We found that nickel(1 mM), chromate(10 μM), and arsenite(1 μM) significantly increase tri-methyl H3K4 after 24 h exposure in human lung carcinoma A549 cells. Seven days of exposure to lower levels of nickel(50 and 100 μM), chromate(0.5 and 1 μM) or arsenite(0.1 0.5 and 1 μM) also increased tri-methylated H3K4 in A549 cells. This mark still remained elevated and inherited through cell division seven days following removal of 1 μM arsenite. We also demonstrate by dual staining immunofluorescence microscopy that both H3K4 tri-methyl and H3K9 di-methyl marks increase globally after 24 h exposure to each metal treatment in A549 cells. However, the tri-methyl H3K4 and di-methyl H3K9 marks localize in different regions in the nucleus of the cell. Thus, our study provides further evidence that a mechanism(s) of carcinogenicity of nickel, chromate, and arsenite metal compounds may involve alterations of various histone tail modifications that may in turn affect the expression of genes that may cause transformation.
Keywords: Nickel, Chromate, Arsenite, Histone 3 Lysine 4 Methylation
Introduction
The nucleosome, the fundamental subunit of chromatin, is composed of 146 bp of DNA wrapped twice around an octamer of the four core histones (H3, H4, H2A, and H2B) ([Luger et al., 1997], [Kornberg and Thomas, 1974] and [Kornberg and Lorch, 1999]). Post-translational modifications (i.e., acetylation, methylation, phosphorylation, ubiquitination, etc.) of the N- and C-terminal tails of the four core histones play an important role in regulating chromatin biology (Zhang and Reinberg, 2001). The implementation and removal of these post-translational modifications have been shown to be fundamental to the regulation of quite diverse biological processes such as DNA replication, repair and transcription (Kouzarides, 2007).
Methylation of lysine 4 in histone H3 (H3K4) in the promoter region of genes has been linked to transcriptional activation in a variety of eukaryotic species (Martin and Zhang, 2005). H3K4, as well as other lysines in the tails of histone proteins that are subject to methylation, can occur in mono-methylated, di-methylated, and tri-methylated forms ([Schneider et al., 2005], [Santos-Rosa et al., 2002] and [Wood et al., 2007]). High levels of H3K4 tri-methylation are associated with the 5′ regions of active genes and there is a strong positive correlation between this modification, transcription rates, active polymerase II occupancy and histone acetylation (Ruthenburg et al., 2007). However, patterns of H3K4 di-methylation differ between yeast and vertebrate chromatin: in S. cerevisiae, di-methylated H3K4 appears spread throughout genes, peaking toward the middle of the coding region, and is associated with transcriptional “poised” as well as active state; mono-methylation in S. cerevisiae, was found to be most abundant at the 3′ end of genes (Ruthenburg et al., 2007). In vertebrates, the majority of H3K4 di-methylation colocalizes with H3K4 tri-methylation in discrete zones proximate to highly transcribed genes (Ruthenburg et al., 2007). Interestingly, there exists a subset of di-methylated sites in the absence of tri-methylated H3K4 and these regions do not correlate with transcriptional start sites but instead their presence is highly dependent on the cell type tested (Ruthenburg et al., 2007). Epidemiological, cell culture, and animal experimental studies have shown an increased cancer incidence associated with chronic exposure to nickel(II), chromate(VI), and arsenite (III) ([Salnikow and Zhitkovich, 2008], [Oller et al., 1997], [Sunderman, 1984] and [Trott et al., 1995]). For example, occupational exposure to nickel compounds has been associated with lung and nasal cancer, Cr(VI) exposure with a high incidence of lung cancer and a variety of cytotoxic and genotoxic effects, and arsenite exposure with skin, lung, bladder, kidney, and liver cancers ([Costa and Klein, 2006], [National Research Council, 2000] and [Gibb et al., 2000]). Along with occupational exposure, the massive growth of manufacturing activities in industrialized countries, large consumption of nonferrous metals, and the creation of large toxic Superfund sites in the U.S. due to the high volume utilization and poor practices in the disposal of metal-containing waste products are just some of the sources of potential exposure to carcinogenic metals (Salnikow and Zhitkovich, 2008). Although nickel (II), chromate (VI), and arsenite (III) are recognized carcinogenic compounds, their molecular mechanisms of carcinogenesis are not completely understood. Numerous studies have linked the carcinogenic effects of metal compounds to alterations in signaling pathways in the cell, DNA damage via both oxidative and nonoxidative (DNA adducts) mechanisms, and activation or silencing of gene expression through epigenetic mechanisms by inducing changes in DNA methylation and histone tail post-translational modifications.
Many conventional mutagenesis assays have found nickel and arsenite compounds to be nonmutagenic ([Biggart and Costa, 1986], [Klein et al., 1994] and [Kann et al., 2005]). Over the last years, epigenetic mechanisms have been implicated in the actions of both nickel and arsenite’s carcinogenic properties. Studies on the effects of Cr(VI) exposure have also suggested potential epigenetic effects along with mediating DNA damage by inducing oxidative stress and forming stable Cr-DNA adducts (Zhitkovich, 2005). Studies have shown that exposure of cells to nickel salts results in intracellular accumulation of nickel ion, changes in DNA methylation patterns and chromatin condensation, loss of acetylation in all four core histones by inhibiting histone acetyltransferase activity (HAT), and increased global levels of histone H3 lysine 9 (H3K9) methylation by inhibiting the activity of a histone demethylase, with no or very slight inhibition of a G9a methyltransferase activity ([Lee et al., 1995], [Ke et al., 2006], [Chen et al., 2006a] and [Kang et al., 2003]); these changes can result in alterations in gene expression, thus providing a plausible non-genotoxic mechanism to the heritable alterations in gene expression induced by nickel exposure. Cr(VI) was shown to induce genome-wide or gene-specific DNA methylation changes in cultured mammalian cells (Klein et al., 2002) and in plants (Labra et al., 2004). Also, the promoter region of the tumor suppressor gene p16 and the DNA mismatch repair gene (hMLH1) were subject to DNA hypermethylation in tissue samples of chromate exposed workers ([Klein et al., 2002] and [Takahashi et al., 2005]). Numerous reports have suggested that arsenic disrupts DNA methylation. For example, increased DNA hypomethylation of target genes; such as, the metallothienin gene, and transcriptional silencing of tumor suppressor genes induced by DNA hypermethylation in the promoter region of that gene has been reported ([Zhao et al., 1997] and [Marsit et al., 2006]).
Because H3K4 methylation is known to impact the transcriptional machinery and is strongly correlated with transcriptional activation when found in the promoter region of genes, we studied whether nickel, chromate, and arsenite had an effect on the global H3K4 methylation status. Here we report the effect of nickel (II), chromate (VI), and arsenite (III) on H3K4 methylation. We used A549 cells in the study because using human lung cells is relevant to the airborne route of exposure of nickel, chromate, and arsenite containing compounds, and this cell linehas been well established as a model to study the toxic and epigenetic effects of metals([Chen et al., 2006a] and [Mei et al., 2002]). Our study provides further evidence suggesting that alterations in histone modifications contribute to metal-induced aberrant gene expression and carcinogenesis.
Materials and Methods
Chemicals
NiCl2·6H2O, NaAsO2 andNiSO 4 were purchased from Sigma (St. Louis, MO). Potassium chromate (K2CrO4) was obtained from J. T. Baker Chemical Co. (Phillipsburg, NJ).
Cell culture
Cells were grown at 37°C in an incubator with a humidified atmosphere containing 5% CO2. A549 cells were cultured in F-12K medium (Invitrogen, Carlsbad, CA). Normal Human Bronchial Epithelial (NHBE) cells were cultured in Lonza clonetics BEGM Bulletkit (CC-3170) and Reagent Pack (CC-5034). TGR-1 (c-Myc+/+), HO15.19 (c-Myc−/−) and HOmyc3 (HO15.19 with reconstituted c-Myc expression) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen). TGR-1, HO15.19 and HOmyc3 cells are all derivatives of the Rat-1 cell line (Mateyak et al., 1999). All medium was supplemented with 10% fetal bovine serum (FBS, ATLAS Biological, Fort Collins, CO) and 1% penicillin/streptomycin (Grand Island, NY). Cells were passaged at 80–90% confluence by trypsinization. All treatments were administered when cell density reached approximately 70–80% confluence.
Histone extraction
Histones were extracted from the cells as previously described(Chen et al., 2006a). Briefly, cells were grown in 10 cm dishes, were washed with ice-cold PBS and lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, PH 7.4, 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1ug/ml Aprotinin, 1ug/ml leupeptin, 1ug/ml pepstatin, 1 mM Na3VO4, 1 mM NaF) supplemented with a protease inhibitor mixture (Roche Applied Sciences, Indianapolis, IN) for 10 min. The pellet was collected by centrifugation at 10,000 × g for 10 min. The pellet was washed once in 10 mM Tris-Cl and 37 mM EDTA (pH 7.4), and resuspended in 200 μl 0.4 N H2SO4. After 1.5 h incubation on ice, the supernatant was collected by centrifugation at 14,000 × g for 15 min, and mixed with 1 ml cold acetone and kept at −20°C overnight. The histones were collected by centrifugation at 14,000 × g for 15 min. After one wash with acetone, the histones were air dried and suspended in 4 M urea.
Western blot
The protein concentration was determined using Bio-Rad DC (detergent compatible) protein assay (Bio-Rad, Hercules, CA), and 5 μg histones were separated by 15% SDS-PAGE gel and transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad). Immunoblotting was performed using mono-methyl H3K4 (1:5000; Upstate Biotechnology), di-methyl H3K4 (1:5000; Upstate Biotechnology), tri-methyl H3K4 (1:5000; Abcam), and HRP-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The detection was accomplished by chemical fluorescence following an ECL Western blotting protocol (Amersham, Piscataway, NJ). After transfer to PVDF membranes, the gels were stained with Bio-safe Coomassie stain (Bio-Rad) to assess histone loading in each lane.
Immunofluorescence staining
The cells were cultured in Culture slides (BD FalconTM, Bedford, MA), and exposed to various chemicals individually. At selected time intervals, cells were fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.2% Triton X-100 for 5 min. Cells were then quenched with fresh 0.1% sodium borohydride for 5 min and blocked in blocking buffer (10% Goat serum, 1%BSA in PBS). Dual-color immunofluorescence was performed by staining H3K9 di-methylation and H3K4 tri-methylation sequentially. H3K9 di-methylation was stained with 1:2000 di- methylated H3K9 antibody (Abcam, Cambridge, MA) and visualized by Alexa Fluor 594 secondary antibody (red; Molecular Probes). H3K4 tri-methylation was stained with 1:5000 tri-methylated H3K4 antibody (Upstate Biotechnology) and visualized with Alexa Fluor 488 conjugated secondary antibody (green; Molecular Probes). Primary antibodies were diluted in blocking buffer and incubated overnight at 4 °C. The cells were then incubated with Alexa Fluor 488 and Alexa Fluor 594 conjugated secondary antibody (Molecular Probes, Eugene, OR) for 1h and mounted with ProLong Gold Anti-fade Reagent with DAPI (Molecular Probes). The image was visualized and captured by a Leica TSC SP5 confocal microscope.
Statistical Analysis
Two-tailed Student’s t tests were used to determine the significance of differences in the methylation levels between treated samples and controls. Differences were considered significant at a P < 0.05.
Results
Nickel compounds increased H3K4 di- and tri-methylation, but did not affect H3K4 mono-methylation
In order to determine if exposure to nickel compounds results in changes in H3K4 methylation, A549 cells were exposed to 500 μM and 1 mM NiCl2 for 24 h. Global levels of H3K4 methylation were measured using antibodies specific for mono-, di- and tri-methyl H3K4. 24 h exposure to 500 μM and 1 mM NiCl2 resulted in a 2-fold increase in di- methyl H3K4 (Figure 1B), but had no effect on mono-methylation(Figure 1A). Tri-methyl H3K4 was increased 3 fold by 1 mM NiCl2 after 24 h (Figure 1C). Statistical analysis of Figure 1A, 1B and 1C are shown in Figure 1D. Tri-methylated H3K4 was also increased by seven days exposure to 50 μM and 100 μM(Figure 1E). To further confirm that the increase in the levels of H3K4 tri-methylation by nickel compounds was not cell-type specific, Normal Human Bronchial Epithelial (NHBE) cells were treated with 250 μM and 1 mM NiSO4 for 24 h. Exposure of NHBE cells to NiSO4 also resulted in an increase in H3K4 tri-methylation (Figure 1C). Here, we have observed both NiCl2 and NiSO4 increased H3K4 tri-methylation levels in both A549 and NHBE cells.
Figure 1.

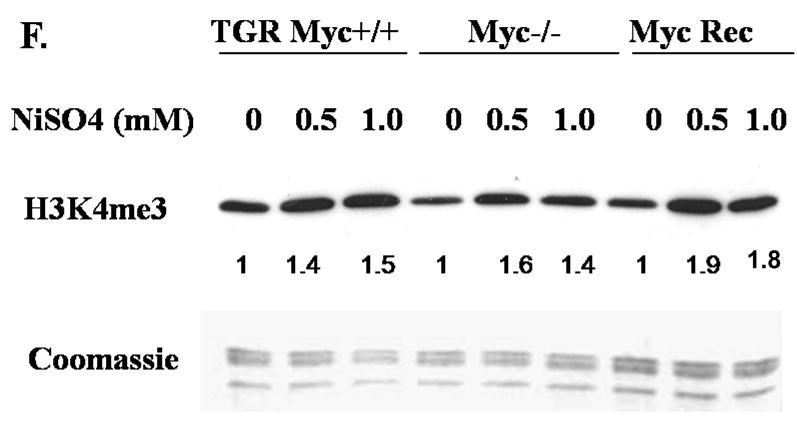
A549 cells were treated with 500 μM and 1 mM of NiCl2 for 24 h (A, B and C), and 50 μM and 100 μM NiCl2 for seven days(E). NHBE cells were treated with 250 μM and 1 mM of NiSO4 for 24 h (C). TGR-1 (c-Myc+/+), HO15.19 (c-Myc−/−) and HOmyc3 (HO15.19 with reconstituted c-Myc expression) cells were treated with 500 μM and 1 mM NiSO4 for 24 h(F). Histones were extracted and immunoblotted with mono-, di- and tri- methyl H3K4 antibodies. H3K4me1, H3K4me2 and H3K4me3 represent H3K4 mono-, di- and tri-methylation respectively. Gels were stained with Coomassie blue to assess histone loading. The numbers below the figure represent the relative intensity of the bands. Shown is a representative experiment but the results were repeated at least two more times. A graph was generated from the quantification of the bands from A, B and C(D). and a students T test determined that there was a *statistically significant change (P < 0.05) compared to control samples.
The members of the MYC transcription factors have been shown to bind to approximately 15% of genes as well as intergenic regions and globally influence the acetylation and methylation of histones ([Knoepfler et al., 2006] and [Fernandez et al., 2003]). To determine if the nickel-induced increase in tri-methyl H3K4 was dependent upon c-Myc activation, the Rat-1 cell lines (wild type Rat-1 cells: TGR-1, c-Myc knockout Rat-1 cells: HO15.19, and HO15.19 with reconstituted c-Myc expression: HOmyc3) were treated with 500 μM and 1 mM NiSO4 for 24 h. The results show that exposure of wild type Rat-1 cells, c-Myc knockout and c- Myc reconstituted cells with 500 μM and 1 mM NiSO4 for 24 h resulted in an increase in H3K4 tri-methylation (Figure 1F). Interestingly, even though 24 h NiSO4 exposure was able to induce an increase in global tri-methyl H3K4, there was a decrease in the H3K4 tri-methyl mark in the c-Myc knockout Rat-1 cells when compared to wild type or c-myc reconstituted cell lines. Consistent with our results, a group recently published a global reduction of tri-methyl H3K4 12 h following MYC inactivation in an osteosarcoma cell line (Wu et al., 2007). Based on these results, we conclude that c-Myc did not play a significant role in nickel’s induction of H3K4 tri-methylation levels.
Chromate increases tri-methyl H3K4 but has no or little effect on H3K4 mono-and di-methylation
Several studies of chromate induced lung cancer suggested that Cr(VI) may silence tumor suppressor genes by inducing DNA methylation in their promoters (Kornberg and Thomas, 1974). However, very little is known about whether Cr (VI) affects the post-translational modifications of histone tails. Studies in our lab suggest that exposure of A549 cells to potassium dichromate is able to induce global changes in various modifications in histone tails, such as H3K9 di-methylation, and the subsequent silencing of specific tumor suppressor genes whose promoters are subject to the changes in histone tail modifications(Sun et al., 2008).
To examine if exposure to chromate results in changes in H3K4 methylation levels, A549 cells were exposed to 5 uM and 10 uM chromate at 5 μM and 10 μM for 24 h and the levels of H3K4 methylation were measured using antibodies specific for mono-, di- and tri-methyl H3K4. 24 h exposure to chromate resulted in no change on mono-methyl H3K4 (Figure 2A), or a slight increase on di- methyl H3K4(Figure 2B), but a significant increase on tri- methyl H3K4 at the concentration of 10 μM (Figure 2C). Statistical analysis of Figure 2A, 2B and 2C are shown in Figure 2D. Increased tri-methylated H3K4 was also observed in the cells exposed to 0.5 μM and 1 μM chromate for seven days (Figure 2E).
Figure 2.

A549 cells were treated with 5 μM and 10 μM of potassium chromate Cr(VI) for 24 h(A, B and C), and 0.5 μM and 1 μM potassium chromate for seven days(E). Histones were extracted and immunoblotted with mono-, di- and tri- methyl H3K4 antibodies. H3K4me1, H3K4me2 and H3K4me3 represent H3K4 mono-, di- and tri-methylation respectively. Gels were stained with Coomassie blue to assess histone loading in each lane. The numbers below the figure represent the relative intensity of the bands. . Shown is a representative experiment but the results were repeated at least two times. A graph was generated from the quantification of the bands from A, B and C(D). and and a students T test determined that there was a *statistically significant change (P < 0.05) compared to control samples.
Arsenite increases di- and tri-methyl H3K4 but decreases mono-methyl H3K4
In addition to DNA methylation, post-translational modifications in the tails of histone proteins are recognized as epigenetic marks that modulate gene expression. Arsenic’s effect on the DNA methylation status of its target genes suggests that epigenetic mechanisms may play a role in arsenic-induced carcinogenesis. Recently, our group published the first evidence that arsenite can disrupt the epigenetic machinery that regulates histone modifications and found that arsenite can modulate the methylation of histone H3 (Zhou et al., 2008). In order to examine if arsenite modulates the H3K4 methylation state, A549 cells were exposed to 1 μM and 5 μM arsenite for 24 h. After arsenite exposure, H3K4 methylation status was measured using antibodies specific for mono-, di- and tri-methylated H3K4. 24 h exposure to 1 μM and 5 μM arsenite resulted in an increase in di- (Figure 3B) and tri-methyl (Figure 3C) H3K4, but a decrease in mono-methyl H3K4 (Figure 3A). Statistical analysis of Figure 3A, 3B and 3C were are in Figure 3D. The increase of H3K4 di-or tri-methylation at 5 μM is less than that of at 1 μM. This is consistent with a previous assessment that arsenic’s carcinogenic dose-response is non-linear(U.S. EPA, 1997).
Figure 3.

A549 cells were treated with 1 μM and 5 μM of sodium arsenite for 24 h(A, B and C), and 0.1 μM, 0.5 μM and 1 μM sodium arsenite for seven days(E). Arsenite was removed following seven days exposure to 0.1 μM, 0.5 μM and 1 μM sodium arsenite, and recovered for another seven days (F). Histones were extracted and then immunoblotted with mono-, di- and tri- methyl H3K4 antibodies. H3K4me1, H3K4me2 and H3K4me3 represent H3K4 mono-, di- and tri-methylation respectively. Gels were stained with Coomassie blue to assess histone loading in each lane. The numbers below the figure represent the relative intensity of the bands. . Shown is a representative experiment but the results were repeated at least two times. A graph was generated from the quantification of the bands from A, B and C(D) and a students T test determined that there was a*statistically significant change (P < 0.05) compared to control samples.
Epigenetic effects must be inherited through cell division. A549 cells were exposed to 0.1 μM, 0.5 μM and 1 μM arsenite for 7 days, and the arsenite was removed and the cells were allowed to proliferate in the absence of arsenite for another 7 days. Seven days of exposure to arsenite also increased tri-methylated H3K4 (Figure 3E). At the concentration of 1 μM, the increase was observed even seven days after arsenite was removed from the media (Figure 3F).
Distinct localization of H3K9 di-methylation and H3K4 tri-methylation in nickel, chromate, and arsenite exposed A549 cells
It was of interest that exposure of A549 cells to either nickel, chromate, or arsenite induced H3K4 tri-methyl since this mark in the promoter of genes is associated with gene activation. An increase in global H3K9 di-methylation has also been previously observed with each of these metal treatments. Di-methylation of H3K9 in the promoter region of genes is generally associated with gene silencing (Rice et al., 2003). Because it was unlikely that both the transcriptional silencing and activating signals resided in the promoter of the same gene, we hypothesized that H3K4 tri-methylation and H3K9 di-methylation were positioned in different parts of chromatin. We performed dual immunofluorescence staining of both H3K4 tri-methylation and H3K9 di-methylation and then analyzed their distribution by confocal microscopy. While both H3K4 tri-methylation and H3K9 di-methylation marks were increased 24 h after nickel, chromate, and arsenite treatment, the enrichment pattern for each mark was different. H3K9 di-methylation(red) resided primarily in the periphery of the nucleus, which coincides with the location of heterochromatin, while H3K4 tri-methylation(green) was mainly in the center of the nucleus (euchromatin), though there is some overlapping of the di-methylated H3K9 and tri-methylated H3K4 signal, (Figure 4A, 4B and 4C). The fact that nickel, chromate, and arsenite had differential effects in distinct regions of chromatin suggested that a more detailed analysis of the changes in gene expression pattern after metal treatment will be necessary in order to understand the gene specific changes involved with metal’s epigenetic effects.
Figure 4.
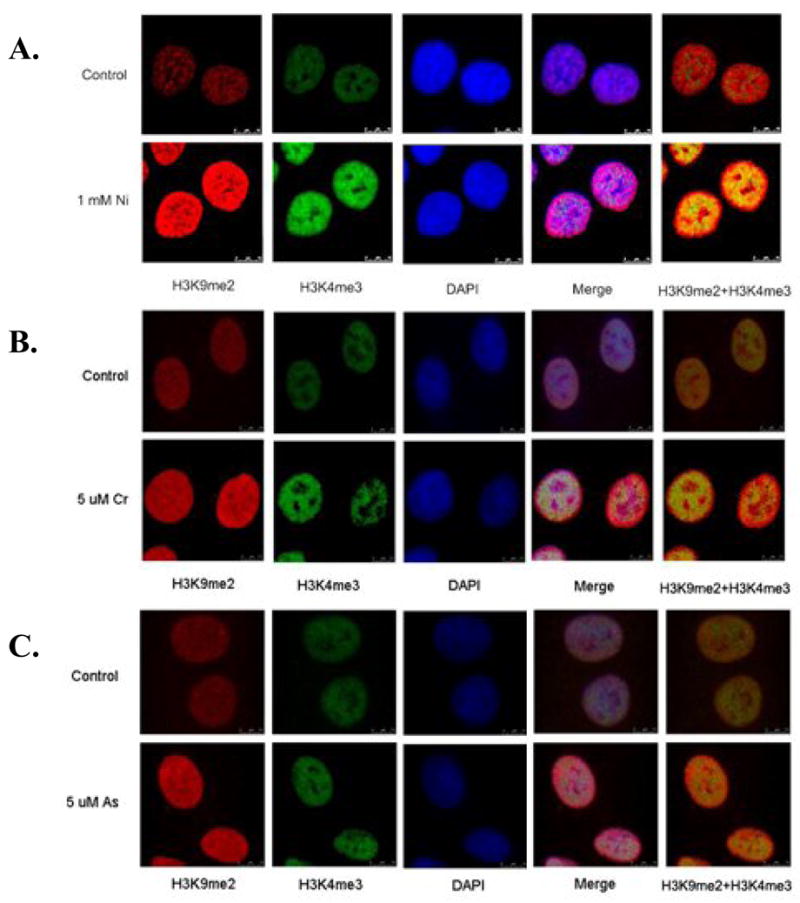
Distinct localization of H3K9 di-methylation (H3K9me2) and H3K4 tri-methylation (H3K4me3) in nickel(A), chromate(B), and arsenite(C)-exposed cells. A549 cells were exposed to 1 mM NiCl2, 5 μM chromate, and 5 μM arsenite for 24 h. After exposure, cells were co-stained with di-methylated H3K9 (red) and tri-methylated H3K4 (green) antibodies. The nucleus was counterstained with DAPI (blue). Merge images show a merge of the red, green and blue staining. The pictures were taken using a confocal microscope.
Discussion
The choice of the metal exposure doses used for the treatment of A549 cells at 24 h was based on previous cell cytotoxicity studies. 1 mM Ni ions exhibits minimal toxicity in A549 cells after 24 h based on the MTT viability studies (Davidson et al., 2003) and the colony formation assay(Chen et al., 2006b). There was no significant change in the global toxicity in cells treated with less than 10 uM Cr(VI) after 24 h ([Shumilla and Barchowsky, 1999], [Martin et al.,1998]). At concentrations of arsenite at or below 10 μM, no significant changes in cell viability were observed(Talbot et al., 2008). The concentrations of Nickel used in NHBE cells and TGR-1 (c-Myc+/+), HO15.19 (c-Myc−/−) and HOmyc3 (HO15.19 with reconstituted c-Myc expression) cells were comparable to those in A549 cells. The doses used in 7 days exposure were much lower and there was no observable cell death by Ni, Cr and As.
The mechanism of nickel, chromate, and arsenite’s carcinogenesis is unclear but, recent experimental evidence suggests that these metals affect gene expression by an epigenetic mechanism([Chen et al., 2006a], [Sun et al., 2008] and [Zhou et al., 2008]). This report shows that nickel, chromate, and arsenite alter H3K4 methylation states. We find that these metals increase di-and tri-methyl H3K4, while arsenite decreases mono-methyl H3K4. The increase of tri-methylated H3K4 remained through numerous cell divisions after arsenite was removed. Previous work from our group has shown that exposure of cells to these various metals alters other histone post-translational modifications. For example, nickel increased H3K9 di-methylation by inhibiting the demethylating enzyme JHDM2A (Chen et al., 2006a) suggesting that the carcinogenic effects of nickel, chromate, and arsenite may involve aberrant gene expression by epigenetic mechanism(s).
Even though the mechanism by which Cr(VI) affects histone H3K4 methylation is not well understood, we hypothesize that multiple processes may be a target of Cr (VI). For example, our group has observed increased protein and mRNA levels of a histone methyltransferase after chromate exposure of A549 cells (Sun et al., 2008). Also, supplementation with ascorbate, an essential cofactor for histone demethylase activity and the primary reductant required for Cr (VI) conversion to the less toxic Cr(III), can partially reverse the H3K9 di-methylation induced by chromate ([Zhitkovich, 2005] and [Sun et al., 2008]). Therefore, we suggest a mechanism by which hexavalent chromium may target both histone methyltransferase and demethylase activity. Changes in methyltransferase and demethylase activity will alter global histone methylation levels and possibly affect the expression of specific tumor suppressor genes and/or genes that promote carcinogenesis.
Inorganic arsenic exists in the environment in two predominant forms, arsenite [As(III)] or arsenate [As(V)], the former is associated with being more bioreactive and responsible for increasing cancer risks (Barret et al., 1989), however arsenate is converted to arsenite in the body by reductive processes. Since, historically, there has been a lack of animal models to study the cancers associated with inorganic arsenic exposure, the mechanism(s) of arsenic-mediated carcinogenesis remain unclear. The arsenite-induced changes of H3K4 methylation observed in this study are consistent with arsenite’s previous findings that indicate it can activate gene transcription by inducing DNA hypomethylation (Zhao et al., 1997).
Further identification of the specific genes whose expression is affected by nickel, chromate, and arsenite-induced changes in H3K4 methylation status will provide more insight into the epigenetic effects of these metals and help us better understand their cytotoxicity and carcinogenicity. This study, as well as others, raises the question of whether epigenetic mechanism(s) affect other metal compounds’ cytotoxicity/carcinogenicity. In the future, it would be necessary to conduct analysis of potential epigenetic mechanisms of other carcinogenic metals along with identification of the DNA methylation status and mapping post-translational modifications of histones in the promoter regions of their target genes; such as, tumor suppressor and/or genes whose expression is increased following metal exposure.
Acknowledgments
This work was supported by grant numbers ES000260, ES10344, ES014454, ES005512, ES010344 from the National Institutes of Environmental Health Sciences, and grant number CA16087 from the National Cancer Institute.
Footnotes
Conflict of Interest Statement
There are no conflicts of interest for the authors of this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barret JC, Lamb PW, Wang TC, Lee TC. Mechanisms of arsenic induced cell transformation. Biol Trace Elem Res. 1989;21:421–429. doi: 10.1007/BF02917284. [DOI] [PubMed] [Google Scholar]
- Biggart NW, Costa M. Assessment of the uptake and mutagenicity of nickel chloride in salmonella tester strains. Mut Res. 1986;175:209–215. doi: 10.1016/0165-7992(86)90056-4. [DOI] [PubMed] [Google Scholar]
- Chen H, Ke Q, Kluz T, Yan Y, Costa M. Nickel ions increase histone H3 lysine 9 di-methylation and induce transgene silencing. Mol Cell Biol. 2006a;26:3728–3737. doi: 10.1128/MCB.26.10.3728-3737.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Costa M. Effects of soluble nickel on cellular energy metabolism in A549 cells. Exp Biol Med. 2006b;231:1474–1480. doi: 10.1177/153537020623100905. [DOI] [PubMed] [Google Scholar]
- Costa M, Klein CB. Toxicity and carcinogenicity of chromium compounds in humans. Crit Rev Toxicol. 2006;36:155–163. doi: 10.1080/10408440500534032. [DOI] [PubMed] [Google Scholar]
- Davidson T, Salnikow K, Costa M. Hypoxia inducible factor-1 alpha–independent suppression of aryl hydrocarbon receptor–regulated genes by nickel. Mol Pharmacol. 2003;64:1485–1493. doi: 10.1124/mol.64.6.1485. [DOI] [PubMed] [Google Scholar]
- Fernandez P, Frank S, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb HJ, Lees PS, Pinsky PF, Rooney BC. Lung cancer among workers in chromium chemical production. Am J Ind Med. 2000;38:115–126. doi: 10.1002/1097-0274(200008)38:2<115::aid-ajim1>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Kang J, Zhang Y, Chen J, Chen H, Lin C, Wang Q, Ou Y. Nickel-induced histone hypoacetylation: the role of reactive oxygen species. Toxicol Sci. 2003;74:279–286. doi: 10.1093/toxsci/kfg137. [DOI] [PubMed] [Google Scholar]
- Kann S, Estes C, Reichard JF, Huang MY, Sartor MA, Schwemberger S, Chen Y, Dalton TP, Shertzer HG, Xia Y, Puga A. Butylhydroquinone protects cells genetically deficient in glutathione biosynthesis from arsenite-induced apoptosis without significantly changing their prooxidant status. Toxicol Sci. 2005;87:365–384. doi: 10.1093/toxsci/kfi253. [DOI] [PubMed] [Google Scholar]
- Ke Q, Davidson T, Chen H, Kluz T, Costa M. Alterations of histone modifications and transgene silencing by nickel chloride. Carcinogenesis. 2006;27:1481–1488. doi: 10.1093/carcin/bgl004. [DOI] [PubMed] [Google Scholar]
- Klein C, Kargacin B, Su L, Cosentino S, Snow ET, Costa M. Metal mutagenesis in transgenic Chinese hamster cell lines. Environ Health Perspect. 1994;102(Supp 3):63–67. doi: 10.1289/ehp.94102s363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein CB, Su L, Bowser D, Leszczynska J. Chromate-induced epimutations in mammalian cells. Environ Health Perspect. 2002;110(Supp 5):739–743. doi: 10.1289/ehp.02110s5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, MacMahon SB, Eisenman RN. Myc influences global chromatin structure. EMBO J. 2006;25:2723–2734. doi: 10.1038/sj.emboj.7601152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD, Thomas JO. Chromatin structure; oligomers of the histones. Science. 1974;184:865–868. doi: 10.1126/science.184.4139.865. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–290. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Labra M, Grassi F, Imazio S, Di Fabio T, Citterio S, Sgorbati S, Agradi E. Genetic and DNA-methylation changes induced by potassium dichromate in Brassica napus L. Chemosphere. 2004;54:1049–1058. doi: 10.1016/j.chemosphere.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Lee YW, Klein CB, Kargacin B, Salnikow K, Kitahara J, Dowjat K, Zhitkovich A, Christie NT, Costa M. Carcinogenic nickel silences gene expression by chromatin condensation and DNA methylation: a new model for epigenetic carcinogens. Mol Cell Biol. 1995;15:2547–2557. doi: 10.1128/mcb.15.5.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Marsit CJ, Karagas M, Danaee H, Liu M, Andrew A, Schned A, Nelson HH, Kelsey KT. Carcinogen Exposure and gene promoter hypermethylation in bladder cancer. Carcinogenesis. 2006;27:112–116. doi: 10.1093/carcin/bgi172. [DOI] [PubMed] [Google Scholar]
- Martin BD, Schoenhard JA, Sugden KD. Hypervalent chromium mimics reactive oxygen species as measured by the oxidantsensitive dyes 29,79-dichlorofluorescin and dihydrorhodamine. Chem Res Toxicol. 1998;11:1402–1410. doi: 10.1021/tx9801559. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Sedivy JM. c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol Cell Biol. 1999;19:4672–4683. doi: 10.1128/mcb.19.7.4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei N, Kunugita N, Hirano T, Kasai H. Acute arsenite-induced 8-hydroxyguanine is associated with inhibition of repair activity in cultured human cells. Biochem Biophys Res Commun. 2002;297:924–930. doi: 10.1016/s0006-291x(02)02309-4. [DOI] [PubMed] [Google Scholar]
- National Research Council. National Research Council Report: Arsenic in the Drinking Water. Washington, DC: National Academy Press; 2000. [Google Scholar]
- Oller AR, Costa M, Oberdorster G. Carcinogenicity assessment of selected nickel compounds. Toxicol Appl Pharmacol. 1997;143:152–166. doi: 10.1006/taap.1996.8075. [DOI] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- Ruthenburg A, Allis C, Wysocka J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Molecular Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, Arsenic, and Chromium. Chem Res Toxicol. 2008;21(1):28 – 44. doi: 10.1021/tx700198a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. Molecular regulation of histone H3 tri-methylation by COMPASS and the regulation of gene expression. Mol Cell. 2005;19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Shumilla JA, Barchowsky A. Inhibition of Protein Synthesis by Chromium(VI) Differentially Affects Expression of Urokinase and Its Receptor in Human Type II Pneumocytes. Toxicol Appl Pharmacol. 1999;158:288–295. doi: 10.1006/taap.1999.8704. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhou X, Chen H, Li Q, Costa M. Modulation of histone methylation by hexavalent chromium; Proceedings of the 99th Annual Meeting of the American Association for Cancer Research; 2008 Apr 12–16; San Diego, CA. Philadelphia (PA): AACR; 2008. Abstract 6. [Google Scholar]
- Sunderman FW., Jr Carcinogenicity of nickel compounds in animals. IARC Sci Publ. 1984;53:127–142. [PubMed] [Google Scholar]
- Takahashi Y, Kondo K, Hirose T, Nakagawa H, Tsuyuguchi M, Hashimoto M, Sano T, Ochiai A, Monden Y. Microsatellite instability and protein expression of the DNA mismatch repair gene, hMLH1, of lung cancer in chromate-exposed workers. Mol Carcinog. 2005;42:150–158. doi: 10.1002/mc.20073. [DOI] [PubMed] [Google Scholar]
- Talbot S, Nelson R, Self WT. Arsenic trioxide and auranofin inhibit selenoprotein synthesis: implications for chemotherapy for acute promyelocytic leukaemia. Br J Pharmacol. 2008;154:940–948. doi: 10.1038/bjp.2008.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott DA, Cuthbert AP, Overell RW, Russo I, Newbold RF. Mechanisms involved in the immortalization of mammalian cells by ionizing radiation and chemical carcinogens. Carcinogenesis. 1995;16:193–204. doi: 10.1093/carcin/16.2.193. [DOI] [PubMed] [Google Scholar]
- U.S. EPA. Report on the Expert Panel on Arsenic Carcinogenicity: Review and Workshop. Prepared by Eastern Research Group, Inc. for National Center for Environmental Assessment. U.S. Environmental Protection Agency; Washington, DC: 1997. [Google Scholar]
- Wood A, Shukla A, Schneider J, Lee JS, Stanton JD, Dzuiba T, Swanson SK, Florens L, Washburn MP, Wyrick J, Bhaumik SR, Shilatifard A. Ctk complex-mediated regulation of histone methylation by COMPASS. Mol Cell Biol. 2007;27:709–720. doi: 10.1128/MCB.01627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Riggelen J, Yetil A, Fan A, Bachireddy P, Felscher D. Cellular senescence is an important mechanism of tumor regression upon c-myc inactivation. PNAS. 2007;104:13028–13033. doi: 10.1073/pnas.0701953104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc Natl Acad Sci U S A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhitkovich A. Importance of chromium-DNA adducts in mutagenicity and toxicity of chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- Zhou X, Sun H, Ellen T, Chen H, Costa M. Arsenite alters global histone H3 methylation. Carcinogenesis. 2008;29(9):1831–1836. doi: 10.1093/carcin/bgn063. [DOI] [PMC free article] [PubMed] [Google Scholar]