

Abstract
The signaling enzyme phospholipase D (PLD) and the lipid second messenger it generates, phosphatidic acid (PA), are implicated in many cell biological processes, including Ras activation, cell spreading, stress fiber formation, chemotaxis, and membrane vesicle trafficking. PLD production of PA is inhibited by the primary alcohol 1-butanol, which has thus been widely employed to identify PLD/PA-driven processes. However, 1-butanol does not always effectively reduce PA accumulation, and its use may result in PLD-independent deleterious effects. Consequently, identification of potent specific small-molecule PLD inhibitors would be an important advance for the field. We examine one such here, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI), which was identified recently in an in vitro chemical screen for PLD2 inhibitors, and show that it rapidly blocks in vivo PA production with subnanomolar potency. We were surprised to find that several biological processes blocked by 1-butanol are not affected by FIPI, suggesting the need for re-evaluation of proposed roles for PLD. However, FIPI does inhibit PLD regulation of F-actin cytoskeleton reorganization, cell spreading, and chemotaxis, indicating potential utility for it as a therapeutic for autoimmunity and cancer metastasis.
The PLD superfamily extends from viruses and bacteria to humans (Jenkins and Frohman, 2005). Mammalian PLDs have been shown to be involved in many cell biological processes, including Golgi budding (Chen et al., 1997; Yang et al., 2008), Ras activation (Zhao et al., 2007), mitochondrial dynamics (Choi et al., 2006), cell spreading (Du and Frohman, 2009), F-actin stress fiber formation (Cross et al., 1996; Kam and Exton, 2001), and dynamin-driven epidermal growth factor receptor endocytosis (Lee et al., 2006). Classic members of the superfamily, such as PLD1 and PLD2 in humans, perform a transphosphatidylation reaction using water to hydrolyze phosphatidylcholine (PC) to generate PA. More divergent family members can use other lipids or even DNA as substrates, or perform synthetic reactions by fusing lipids via a primary hydroxyl group using the transphosphatidylation mechanism (Sung et al., 1997). Primary alcohols, such as 1-butanol, are used preferentially over water by classic PLDs, and cause PLD to generate phosphatidyl (Ptd)-alcohol instead of PA. The presence of as little as 0.1% 1-butanol in cell culture media has been shown to inhibit many of the cell biological processes listed above, from which it has been inferred that these events are driven by PLD (for review, see McDermott et al., 2004).
The mechanism of action of PA is complex. It can function as a membrane anchor to recruit and/or activate proteins that encode specific PA-binding domains, can exert biophysical effects on membranes when the concentration is increased locally because it is a negatively charged lipid, or can be converted to other bioactive lipids such as diacylglycerol or lysophosphatidic acid. Ptd-Butanol (Ptd-But) is thought to be unable to recruit or activate target proteins, to affect membrane structure, or to be able to serve as a substrate to generate diacylglycerol or lysophosphatidic acid. Nonetheless, despite the widespread utilization of 1-butanol over the past 20 years, concerns have been raised as to whether it fully blocks PA production at the concentrations used (Skippen et al., 2002) and whether it and Ptd-But have other effects on cells that extend beyond inhibiting PA production (for review, see Huang et al., 2005; Huang and Frohman, 2007). Furthermore, cellular levels of PA are dictated by convergent synthetic and degradative enzymes that, in addition to the PLD pathway, include de novo synthesis by acylation of glycerol 3-phosphate and phosphorylation of diacylglycerol, and dephosphorylation catalyzed by membrane-bound and soluble phosphatases. Effects of primary alcohols on these enzymes are largely unexplored.
Several other inhibitors of PLD activity have been described including ceramide (Vitale et al., 2001), neomycin (Huang et al., 1999), and natural products (Garcia et al., 2008), but these compounds either sequester the requisite PLD cofactor Ptd-inositol 4,5-bisphosphate (PIP2), work indirectly to inhibit PLD activity, or have many other effects on signaling pathways that complicate their use and interpretation (for review, see Jenkins and Frohman, 2005). A small molecule screen to identify inhibitors of human PLD2 using an in vitro biochemical assay recently identified halopemide, a dopamine receptor antagonist, as a modest inhibitor of PLD2 activity and the analog 5-fluoro-2-indolyl des-chlorohalopemide (FIPI) as being even more potent (Monovich et al., 2007). We show here that FIPI is a potent in vivo inhibitor of both PLD1 and PLD2, setting the stage for a new era of exploration and validation of cell biological roles for mammalian PLD. We provide evidence that supports several proposed functions for PLD, but we also demonstrate a lack of support for others, raising questions about prior studies that relied on primary alcohol-mediated inhibition to define in vivo PLD function.
Materials and Methods
PLD Inhibitor. FIPI and benzyloxycarbonyl-des-chlorohalopemide were synthesized as described previously (compounds 4k and 4g from Monovich et al., 2007) and purified by preparative HPLC (YMC S5 ODS column, 20 × 100 mm; Waters, Inc.) using a gradient of 20% aqueous methanol to 100% methanol with 0.1% trifluoroacetic acid. The compounds were confirmed to have the correct structure (see Monovich et al., 2007 for the FIPI structure) by proton NMR and electrospray ionization mass spectrometer and they gave single, symmetrical peaks on HPLC analysis.
Cell Culture and Transfection. Chinese hamster ovary (CHO) stable cell lines inducibly expressing wild-type PLD1 and PLD2 under the control of tetracycline (Du et al., 2004; Su et al., 2006) were cultured in Ham's F-12 medium containing 10% tetracycline-free fetal bovine serum (FBS) from Clontech (Mountain View, CA). Recombinant protein expression was induced by adding 1 μg/ml doxycycline (Dox) for 24 h. An NIH3T3 cell line inducibly expressing Flag-tagged MitoPLD under the control of mifepristone was induced by addition of 1 nM mifepristone for 24 h. Bone marrow-derived macrophages were isolated from mice as described previously (Zhang et al., 2005). Min6 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 1% 2-mercaptoethanol, l-glutamate, and penicillin/streptomycin. Other cell lines were cultured in Dulbecco's modified Eagle's medium containing 10% FBS.
For transfection, cells were grown on coverslips in 24-well plates and transfected with 0.5 μg of DNA per well using LipofectAMINE Plus (Invitrogen, Carlsbad, CA). Six hours after transfection, the media were replaced with fresh growth medium, and the cells were cultured for another 24 h.
PLD Activity Assay. The in vitro PLD activity assay was performed using an in vitro head-group release assay as described previously (Morris et al., 1997). In vivo PLD activities were determined using transphosphatidylation to measure the accumulation of Ptd-But in intact cells (Hammond et al., 1995; Morris et al., 1997). CHO cells were incubated with FIPI (diluted from a 7.5 mM stock concentration in DMSO) or medium containing a matching concentration of DMSO for 30 min before addition of 0.3% 1-butanol for 30 min. For the time-course study, cells were preincubated with 100 nM FIPI for 5, 10, 15, or 30 min followed by a 10-min incubation with 0.3% 1-butanol. For recovery experiments, cells were incubated with 2 μCi/ml [3H]palmitic acid and then switched to fresh growth medium containing 50 μg/ml cycloheximide. Thirty minutes later, one set of cells was treated with 100 nM FIPI for 30 min followed by three washes with PBS and addition of fresh growth medium containing cycloheximide. The cells were incubated for 1 or 8 h. During the last 30 min, FIPI was added to a second set of cells, after which all of the sets of cells were assayed for PLD activity.
Immunofluorescent Staining. Cells were fixed with 4% paraformaldehyde for 10 min followed by permeabilization with 0.1% Triton X-100. Hemagglutinin-tagged PLD1 and PLD2 proteins were detected by a monoclonal antibody (3F10) followed by secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen). F-actin was stained by phalloidin conjugated with Rhodamine. Other proteins were GFP-tagged and visualized with green fluorescence. For glucose-stimulation assays, the cells were cultured in low-glucose buffer or stimulated with high glucose for 10 to 60 min in the presence of FIPI or vehicle control (DMSO). Images were taken using a Leica TCS SP2 spectral laser scanning confocal microscope with the Leica DMIRE2 inverted platform (Leica Microsystems, Heidelberg, Germany). Fluorescence intensity quantification was performed using ImageJ software (http://rsb.info.nih.gov/ij/).
Western Blotting. Cells were lysed in 1× Laemmli sample buffer and resolved by 10% SDS-PAGE. The blots were blocked with 1% casein in Tris-buffered saline. Western blotting with anti-phospho-ERK and total ERK (Sigma, St. Louis, MO) and other primary antibodies (Cell Signaling Technology, Danvers, MA) was performed as suggested by the suppliers. The blots were developed using secondary antibodies conjugated with Alexa Fluor 680 or IRDye 800 (Rockland Immunochemicals, Gilbertsville, PA). Fluorescent signals were detected with an Odyssey infrared imaging system from LICOR Biosciences (Lincoln, NE).
Cell Migration Assay. HL-60 cells were induced to differentiate using 1.75% DMSO to direct them toward a neutrophil phenotype. Differentiated HL-60 cells (dHL-60) were resuspended at 106/ml in chemotaxis buffer (RPMI 1640 medium + 0.5% bovine serum albumin), and 200 μl of the cell suspension was placed in the inserts of transwell plates that were separated by a 6.5-mm diameter, 5 μm pore membrane from lower wells containing 500 μl of 10 nM fMLP in chemotaxis buffer. The transwell plates were incubated for 1 h at 37°C after which the number of cell that migrated to the lower wells was calculated by placing 10-μl aliquots on a hemocytometer and counting four fields. Each experiment was repeated at least four times.
Cell Spreading. Cell spreading assays were performed as described previously (Du and Frohman, 2009). In brief, CHO cells were suspended by trypsinization, rested in the incubator for 2 h, plated on fibronectin-coated coverslips for the indicated times, and fixed in 4% paraformaldehyde before processing for immunofluorescent staining.
Insulin Secretion and Quantification. Min6 cells cultured in 24-well plates were washed twice in KRBH (4.74 mM KCl, 125 mM NaCl, 5 mM NaHCO3, 1.2 mM MgSO4, 1 mM CaCl2, 25 mM HEPES, 1.2 mM KH2PO4, and 2.5 mM glucose) buffer supplemented with 0.1% bovine serum albumin. All wells were subsequently preincubated in low-glucose KRBH (2.5 mM glucose) buffer for 60 min. The buffer was then completely aspirated and fresh low glucose buffer (control) or low glucose buffer containing 75 or 750 nM FIPI inhibitor was added for an additional 30 min. After this preincubation period, fresh low-glucose KRBH, high-glucose KRBH (20 mM glucose), or high-glucose KRBH buffer containing 75 or 750 nM FIPI inhibitor was added. After 60 min, all samples were collected, and the cells were lysed in ethanol/acid (70% ethanol and 0.18 M HCL) and assayed for total insulin. Secreted and total insulin was measured using an ultrasensitive enzyme-linked immunosorbent assay kit (Mercodia, Winston Salem, NC) and plate reader (Bio-Rad Laboratories, Hercules, CA).
HPLC Tandem Mass Spectrometry Measure of PA. Human embryonic kidney 293 cells were cultured in 35-mm dishes and serum-starved overnight before incubation with vehicle or 750 nM FIPI for 30 min before the addition of 100 ng/ml PMA. Cells were incubated for a further 60 min, washed in ice-cold PBS, and scraped in ice-cold methanol. Lipids were extracted using acidified organic solvents, including [17C]lysophosphatidic acid as an internal recovery standard. Dried lipid extracts were resuspended in methanol, and an aliquot was removed for determination of total lipid phosphorous after wet digestion in perchloric acid. PA species were quantitated by HPLC tandem mass spectrometry using an ABI 4000 Q-Trap Hybrid Triple Quadrupole Linear Ion Trap Mass spectrometer.
Results
FIPI Is a Potent Inhibitor of Classical Mammalian PLD. FIPI was reported to exhibit a 50% inhibitory concentration (IC50) of 20 nM against PLD2 using an unpublished in vitro biochemical assay (Monovich et al., 2007). Using a well established assay system, we have described previously biochemical characterization of recombinant PLD1 and PLD2 (Hammond et al., 1995, 1997; Colley et al., 1997). When separated from cytoplasmic factors, PLD1 requires provision of a stimulator such as ADP-ribosylation factor or Rho small GTPases, or protein kinase C, whereas PLD2 exhibits constitutive activity, as shown in Fig. 1A (“no DMSO” and “0” FIPI concentration). FIPI inhibited both PLD1 and PLD2 in a dose-dependent manner, with 50% loss of activity observed at approximately 25 nM. Thus, consistent with Monovich et al. (2007), FIPI is a potent, concentration-dependent PLD2 inhibitor, and we show here that it inhibits PLD1 equally well under standard in vitro assay conditions.
Fig. 1.

FIPI is a potent in vitro and in vivo PLD inhibitor. A, FIPI blocks PLD activity in vitro. An in vitro headgroup release assay was performed in the presence of increasing amounts of FIPI on PLD protein-containing membrane fractions prepared from Sf9 insect cells infected with baculoviral constructs expressing human PLD1 (Hammond et al., 1995) or mouse PLD2 (Colley et al., 1997). The FIPI was diluted from a 7.5 mM stock in DMSO, and control wells contained matching amounts of DMSO or no DMSO. PLD2 exhibits constitutive activity and was assayed directly. As shown, PLD1 requires a soluble stimulator to exhibit significant activity, and ADP-ribosylation factor was used for that purpose in this assay. The assay was performed in duplicate; values shown are means with a variance between duplicates of less than 5%. B, FIPI blocks PLD activity in vivo. CHO cell lines inducibly expressing PLD1 or PLD2 were assayed for in vivo PLD activity after overnight induction with Dox and a 1-h pretreatment with the indicated concentrations of FIPI or vehicle control (DMSO). Activities are shown in comparison to basal activity (PLD1, no PMA stimulation; PLD2, no Dox induction). Assays were performed in duplicate, and the figure shown represents cumulative results from three independent experiments. C, FIPI blocks translocation of a PA sensor to the plasma membrane. PLD2-inducible CHO cells were transfected with the PA sensor GFP-Spo20-PABD or the mutant GFP-Spo20-PABD-L67R that does not bind PA. PLD2 expression was induced using 24 h of Dox treatment. *, nucleus; arrows, plasma membrane shown in expanded form in inset; arrowheads, endocytic vesicles. Representative cells are shown. Similar results were observed in two independent experiments. D, kinetics of FIPI action. PLD2-expressing CHO cells were preincubated with FIPI for varying lengths of time before being assayed using the in vivo PLD assay. Assays were performed in duplicate, and the figure shown represents cumulative results from three independent experiments. E, FIPI is slowly reversible. PLD-expressing CHO cells were preincubated in cycloheximide to stop new protein synthesis and cultured for an additional 1 or 8 h before assaying in the in vivo assay. One set of cells for each time period was exposed to FIPI at the beginning of the culture period and then washed into fresh medium; the other was exposed at the end of the culture period. Assays were performed in duplicate, and the figure shown represents cumulative results from two independent experiments. F, FIPI does not inhibit MitoPLD. FIPI was added before inducing MitoPLD, which causes mitochondria to aggregate perinuclearly in a PA-dependent manner (Choi et al., 2006). Arrow, mitochondrial cluster, green; F-actin, red. Scale bar, 10 mm. Representative cells shown from two independent experiments.
We next examined FIPI potency in vivo, making use of CHO cell lines that inducibly overexpress PLD1 and PLD2 (Du et al., 2004; Su et al., 2006). Parental CHO cells exhibit very low-to-undetectable levels of PLD activity in nonstimulatory (basal) conditions (Fig. 1B, orange line). In contrast, cells induced to overexpress PLD2 exhibit high levels of activity under basal conditions (Fig. 1B, blue line; average 11.4-fold increase in activity, n = 3). FIPI was added into the cell culture media 1 h before performing an in vivo PLD assay and was found to be a potent inhibitor of PLD2 with an IC50 of 10 nM.
PLD1-overexpressing cell lines exhibit very low levels of PLD activity under basal conditions (Fig. 1B, black diamond; average 1.5-fold increase in activity, n = 3). Stimulation is required to trigger PLD1 activation, for example by using PMA (Fig. 1B, red line; average 18-fold increase in activity over PLD1-overexpressing cells under basal conditions, n = 3) to activate endogenous protein kinase C, which is a direct upstream regulator of PLD1 (for review, see McDermott et al., 2004). PLD1 activity was inhibited even more strongly by FIPI (IC50 = 1 nM). Finally, parental cells stimulated with PMA to activate endogenous PLD1 and/or PLD2 exhibited half-maximal inhibition at 0.5 nM (green line). Thus, FIPI efficiently crosses cell membranes and inhibits PLD1 and PLD2 action on their endogenous substrate in the cytoplasmic environment. These results also indicate that FIPI inhibits both the hydrolytic (Fig. 1A) and transphosphatidylation (Fig. 1B) activities of PLD1 and PLD2. We additionally assayed a second analog, benzyloxycarbonyl-des-chlorohalopemide, which was reported to be much less potent against PLD2 in the in vitro assay (Monovich et al., 2007). We found that it displayed an IC50 of approximately 250 nM against PLD2 in the in vivo assay (data not shown), confirming the essential structure-function relationship reported, although the degree of the difference in efficacy was less than described for the in vitro assay.
The classic in vivo PLD assay used in
Fig. 1, B and D, exploits PLD's
capacity for transphosphatidylation by measuring the production of Ptd-But
when 1-butanol is added into culture medium. To examine the in vivo effect of
FIPI on PLD's hydrolytic activity, which cleaves PC to yield PA, we employed a
means to visualize PA production subcellularly, using a fluorescent PA sensor
consisting of enhanced GFP fused to a 40-amino acid PA-binding domain from the
yeast Spo20 protein (Zeniou-Meyer et al.,
2007). In resting cells, the majority of the sensor localizes to
the nucleus (Fig. 1C). In cells
overexpressing PLD2, the sensor translocates in part to the plasma membrane
and to intracellular membrane vesicles, presumably endosomes, which is where
PLD2 resides (Colley et al.,
1997; Du et al.,
2004). However, no localization of the sensor was observed at
these sites after addition of FIPI to the PLD2-overexpressing cells,
indicating a lack of generation of PA by PLD2. We also examined PA production
by endogenous PLD in human embryonic kidney 293 cells using HPLC tandem mass
spectrometry. Total levels of PA increased 42% upon PMA stimulation for 60 min
(from 32.0 ± 1.9 to 45.5 ± 3.7 pmol/nmol of lipid
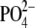 ). In contrast, basal levels of PA
were 17% lower in cells pretreated with 750 nM FIPI for 30 min (26.7 ±
2.1 pmol/nmol of lipid
). In contrast, basal levels of PA
were 17% lower in cells pretreated with 750 nM FIPI for 30 min (26.7 ±
2.1 pmol/nmol of lipid 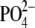 ), and
decreased another 14% upon PMA stimulation (to 23.0 ± 1.4 pmol/nmol of
lipid
), and
decreased another 14% upon PMA stimulation (to 23.0 ± 1.4 pmol/nmol of
lipid 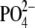 ). Thus, FIPI is efficient at
blocking PMA-stimulated PLD production of PA. Moreover, these data also
suggest that PLD activity is required both to support basal levels of PA and
to compensate for increased rates of PA turnover during signaling events.
). Thus, FIPI is efficient at
blocking PMA-stimulated PLD production of PA. Moreover, these data also
suggest that PLD activity is required both to support basal levels of PA and
to compensate for increased rates of PA turnover during signaling events.
To examine the kinetics of inhibition, FIPI was added to PLD2-overexpressing cells for varying periods of time before beginning the in vivo PLD assay; 15 min was found to suffice to achieve complete inhibition (Fig. 1D). Conversely, we examined the rate at which cells recover PLD activity after being exposed to FIPI (Fig. 1E). PLD2-overexpressing cells were treated with cycloheximide to block new protein production and then cultured with FIPI for 30 min at either the beginning or end of a 1- or 8-h time period before being assayed. Cells exposed to FIPI that were then washed and cultured for 1 h (dark bars) recovered 29% of their PLD2 activity, whereas cells that had an 8-h postexposure period of culture recovered 41% of their activity. Thus, FIPI is not an irreversible suicide inhibitor but neither is it rapidly and completely reversed upon removal of the drug.
Finally, we examined FIPI action on MitoPLD, a highly divergent mammalian PLD superfamily member that uses the classic PLD enzymatic mechanism to hydrolyze the lipid cardiolipin to generate PA (Choi et al., 2006). In wild-type cells, mitochondria exist as both isolated small organelles and an extended tubular network (Fig. 1F). Upon induction of MitoPLD overexpression, the mitochondria aggregate into a single perinuclear cluster (Fig. 1F, arrow) in a PA-dependent manner. FIPI had no effect on this process, indicating that it does not block the catalytic activity of this related but distant family member. We also examined whether FIPI blocks yeast sporulation, a process known to be dependent on the activity of Spo14, the yeast homolog of classic mammalian PLD (Rose et al., 1995), but no inhibition of sporulation was observed (data not shown). Likewise, no inhibition was observed in an in vitro assay (Morris and Smyth, 2007) for autotaxin, a LysoPLD enzyme that hydrolyzes the phosphodiester bond of LysoPC to generate LysoPA but is a member of an unrelated gene family (data not shown). Thus FIPI action seems restricted to PLD1 and PLD2, the classic family members of mammalian PLD.
Because of inherent limitations of the “exogenous substrate” in vitro PLD assay used in Fig. 1A, the mechanism by which FIPI inhibits PLD activity remains to be established. However, because FIPI inhibits both PLD1 and PLD2, the mechanism cannot involve interference with any of the selective activators of PLD1, and because the inhibition is selective for PLD1 and PLD2 over other PLD superfamily enzymes (including Spo14, a yeast classic PLD), a general effect on substrate accessibility can be discounted. These results suggest that FIPI is a direct inhibitor of the PC phosphodiesterase activity of PLD1 and PLD2.
FIPI Does Not Affect PLD Subcellular Localization, PIP2 Availability, the Actin Stress Fiber Network in Resting CHO Cells, or Selected Signaling Events Proximal to PLD Activation. Compounds that affect membrane properties and dislodge PLDs from the bilayer membrane surface, or that sequester PIP2, the required cofactor for mammalian PLD, inhibit PLD activity indirectly. Changes in the actin cytoskeleton can also regulate PLD activity (Lee et al., 2001), and PLD activation has been reported to lie downstream of AKT, p38, and/or ERK activation during signaling events (Varadharaj et al., 2006; Kang et al., 2008). We examined these possibilities for FIPI. The typical localization of PLD1 to peri-nuclear membrane vesicles and PLD2 to the plasma membrane were not affected by exposure to FIPI (Fig. 2A), and FIPI did not decrease PIP2 availability on the plasma membrane in PLD1- and PLD2-overexpressing cells as assessed using an enhanced GFP-fused PIP2 sensor (based on the PH domain of PLCδ; Fig. 2B). FIPI (750 nM) had no visible effect on cortical F-actin and stress fibers in quiescent CHO cells (Fig. 2C) and it did not significantly inhibit AKT or ERK phosphorylation in the human breast cancer cell line MDA-MB231 in response to serum stimulation, even with extended culture (up to 12 h) in inhibitor-containing medium (Fig. 2D). Because the half-life of FIPI in aqueous media is greater than 5 h (Monovich et al., 2007), the lack of effect on signaling pathways over this time frame is not likely to be due to inhibitor degradation. Finally, FIPI did not significantly inhibit p38 or ERK phosphorylation in bone marrow-derived macrophages stimulated with lipopolysaccharide (LPS; Fig. 2E). Taken together, we have not identified any mechanisms other than direct inhibition of the PLD protein catalytic activity that could account for FIPI's inhibitory actions in vivo.
Fig. 2.

FIPI does not alter PLD subcellular localization, access to PIP2, actin stress fibers, or upstream signaling events. A, FIPI does not change the subcellular distribution of PLD. Expression of PLD1 or PLD2 was induced by Dox for 24 h. Cells were treated with 750 nM FIPI for 4 h, fixed, and immunostained for PLD isoforms. B, FIPI does not alter access to PIP2. PLD-induced cells were transfected with the PIP2 sensor PLCδ-PH-GFP, and 24 h later, treated with FIPI for 4 h, fixed, and imaged. C, stress fiber formation in resting CHO cells is not affected by FIPI. CHO cells were treated with FIPI for 4 h, fixed, and stained with Rhodamine-conjugated phalloidin to visualize F-actin. D, FIPI does not affect AKT phosphorylation upon serum stimulation. MDA-MB-231 cells were starved overnight before being pretreated with 750 nM FIPI or vehicle (DMSO) for either 4 or 12 h and then stimulated with serum for 30 min. Cell lysates were analyzed by SDS-PAGE and Western blotting with antibodies against phosphorylated AKT, total AKT, phosphorylated ERK, or total ERK. E, LPS-induced mitogen-activated protein kinase activation is not blocked by FIPI. Bone marrow-derived macrophages were treated with FIPI or vehicle for 4 h, and stimulated with LPS (100 ng/ml) for 10 or 30 min. Cell lysates were analyzed by Western blotting using antibodies against phosphorylated ERK or p38 and total ERK or p38. All results shown are representative of at least three experiments. Scale bar, 10 mm. Representative cells are shown in A to C.
Validation of FIPI as a Specific and Nontoxic PLD Inhibitor through Rescue of PLD2-Driven Suppression of Membrane Ruffling. PLD has been proposed to promote many cell biological processes including Golgi budding, Ras activation, and secretion. However, although demonstrating that FIPI inhibits these processes would be consistent with its action as a PLD inhibitor, off-target or nonspecific toxicity would also constitute a possible explanation for the observed outcomes. To validate FIPI as a specific and nontoxic PLD inhibitor, we chose to test it in a setting in which we could attempt to rescue a cell behavior that is normally suppressed by PLD action.
Stimulation of COS-7 cells with PMA causes dramatic membrane ruffling, which is visualized as concentrated regions of F-actin at the edges and top surfaces of the cells (Fig. 3A, top, arrows). Elevated expression of PLD2 suppresses this ruffling phenotype (Fig. 3A, *). However, culture of the PLD2-overexpressing cells in FIPI restored the ruffling phenotype, as shown in representative cells at the bottom of Fig. 3A and in the quantitation provided in the bar graph.
Fig. 3.

FIPI rescues PLD2-suppressed membrane ruffling and cell spreading. A, hemagglutinin-tagged PLD2 was introduced into COS-7 cells by transient transfection. Cells were serum-starved for 18 h and then treated with 750 nM FIPI. Four hours later, cells were stimulated with 100 ng/ml PMA for 10 min and then fixed with 4% paraformaldehyde. F-actin was visualized using Rhodamine-conjugated phalloidin. *, nucleus; arrows, membrane ruffles. One hundred cells were counted for each condition, and the percentage of cells with ruffling was determined. Three independent experiments were performed with similar results. B, CHO cells in suspension were plated on coverslips with or without a 30-min pretreatment with FIPI. Fifteen minutes after plating, the cells were fixed and stained to visualize F-actin (red) and DNA (green). Scale bar, 10 mm. Representative of two independent experiments.
We have also recently reported a role for PLD2 in early phases of cell spreading (Du and Frohman, 2009). Cell spreading is a critical process that occurs during inflammation and metastasis as cells cease traveling through the vasculature and undergo the morphological changes required to adhere to extracellular matrix. We have shown that the PA generated by PLD2 at the plasma membrane in circulating cells inhibits the enzyme myosin phosphatase, which binds to and is regulated by PA. This leads to increased myosin light chain phosphorylation, myosin activity, and myosin filament formation, which increases peripheral contractile force and hence causes cells to have spherical shapes. Upon attachment, down-regulation of PLD2 activity leads to myosin disassembly, decreased contractile force, and cell spreading. Overexpression of PLD2 prevents spreading, whereas PLD2 RNAi promotes accelerated spreading.
We performed a cell spreading assay in the presence and absence of FIPI. At 15 min after plating suspended spherical CHO cells onto coverslips, the cells had started to spread but were still far from fully flattened (Fig. 3B). In contrast, CHO cells preincubated in FIPI exhibited accelerated spreading (Fig. 3B), phenocopying the result observed for CHO cells stably expressing PLD2 RNAi (Du and Frohman, 2009). Similar results were observed at 7 min after plating, and treatment with FIPI additionally rescued PLD2 overexpression-mediated suppression of cell spreading (data not shown). Taken together, these data demonstrate that FIPI blocks actions of endogenous PLD2 in roles connected to cytoskeletal reorganization and cell trafficking.
Discordance between FIPI- and Alcohol-Mediated Effects on Glucose-Stimulated Insulin Secretion. PLD has been linked to regulated exocytosis in many cell types based on the observation that the signaling-stimulated secretion is inhibited by 1-butanol (for review, see McDermott et al., 2004; Huang and Frohman, 2007). The Min6 pancreatic β-cell line, which releases insulin in response to extracellular elevation of glucose, provides a typical example. Insulin release increases 4- to 5-fold with glucose stimulation (Fig. 4B), and the release is blocked efficiently in a dose-dependent manner by 1-butanol (Fig. 4A). tert-Butanol, the nonprimary isomer of 1-butanol, is commonly used as a control in such assays to reveal potential nonspecific or toxic side effects, because tert-butanol is chemically similar to 1-butanol but cannot be used as a substrate by PLD. As shown in Fig. 4A, tert-butanol has no effect, or a much smaller effect, on insulin release compared with 1-butanol.
Fig. 4.

1-Butanol inhibits glucose-stimulated insulin release without blocking production of PA, whereas FIPI blocks PA production but does not inhibit insulin release. A, 1-butanol blocks glucose-stimulated insulin release. Min6 cells were assayed for glucose-stimulated insulin release in the presence of increasing amounts of primary or tert-butanol. *, p < 0.05; cumulative results from three independent experiments performed in duplicate. B, Min6 cells were assayed for glucose-stimulated insulin release in the presence of increasing amounts of FIPI added 30 min before the start of the assay. Cumulative results from three independent experiments performed in duplicate. C, Min6 cells were transfected with the GFP-Spo20-PABD PA sensor and maintained in low glucose medium. 24 h later, the cells were pretreated with 750 nM FIPI, 0.5% 1-butanol, or vehicle for 1 h, and then switched to high-glucose medium and fixed after 1 h. Two representative cells are shown for each condition (from two experiments performed with similar results). Arrowheads, plasma membrane. D, quantitation of plasma membrane fluorescence (n = 21, p < 0.001). Scale bar, 10 mm.
We were surprised to observe no inhibition of glucose-stimulated insulin release in the presence of FIPI (Fig. 4B). Likewise, no effect of FIPI was observed on glucose-stimulated insulin release for pancreatic islets (not shown). To address whether FIPI was functioning effectively as a PLD inhibitor for the Min6 cells, we again employed the PA sensor (Fig. 4C). In cells maintained under basal conditions, almost all of the sensor localized to the nucleus rather than to the plasma membrane (Fig. 4C, arrowheads). Upon high glucose stimulation, the PA sensor became easily detected on the plasma membrane. However, no plasma membrane translocation of the PA sensor was detected in high glucose-stimulated cells pretreated with FIPI, indicating that the PLD-mediated production of PA was inhibited (p < 0.01; Fig. 4D). Again we were surprised that 1-butanol treatment did not block the glucose-stimulated plasma membrane translocation of the PA sensor. Taken together, these results suggest that eliminating all PLD activity in Min6 pancreatic β-cells does not affect glucose-stimulated insulin secretion, whereas although 1-butanol exposure does prevent insulin release, it does so through a mechanism other than inhibition of PA production.
FIPI Inhibition of PLD Blunts Chemokine-Stimulated Neutrophil Chemotaxis. HL60 cells differentiated into neutrophils respond to the chemokine fMLP through a signaling pathway involving p38 and ERK phosphorylation that directs them to undergo directional migration. 1-Butanol has been reported to inhibit both the signaling pathways involving p38 (Bechoua and Daniel, 2001) and ERK and the end result of chemotactic movement (Carrigan et al., 2007). Using this model system, we confirmed that 1-butanol does inhibit fMLP-triggered p38 and ERK phosphorylation (81 and 69% decrease, respectively; Fig. 5A); however, consistent with the finding shown in Fig. 2E for other cell types and stimulators, significant inhibition of p38 and ERK phosphorylation was not observed in the presence of FIPI. Nonetheless, FIPI inhibition of PLD did diminish fMLP-directed chemotaxis (p < 0.01, Fig. 5B), validating this role for PLD function and suggesting that PLD regulates chemotaxis via mechanisms distinct from affecting MAKP signaling. Regulation of actin reorganization is a possibility based on the results shown in Fig. 3, because ruffling and cell spreading connect to cell movement.
Fig. 5.

FIPI inhibits neutrophil chemotaxis. A, differentiated HL-60 cells were preincubated with 750 nM FIPI, 0.5% 1-butanol, or DMSO and then stimulated with 1 μM fMLP for 2 min. Cell lysates were analyzed by SDS-PAGE and Western blotting using antibodies against p38, phosphorylated p38, ERK, or phosphorylated ERK. Representative of three experiments performed with similar results. B, differentiated HL-60 cells were treated with 750 nM FIPI for 1 h, resuspended in RPMI 1640 medium-based chemotaxis buffer with FIPI or DMSO, and placed in the upper chamber of 6.5-mm Transwell plates. Bottom wells contained either buffer or 10 nM fMLP. *, p < 0.05; **, p < 0.01, t test. Cumulative data of four independent experiments are presented.
Discussion
Although primary alcohols have long been employed as probes to investigate the role of PLD in cellular processes, concerns have been raised both about potential off-target effects and about lack of complete inhibition of PA production (Skippen et al., 2002; Huang et al., 2005; Huang and Frohman, 2007). Our characterization of FIPI as a selective PLD1 and PLD2 inhibitor provides a novel tool to evaluate the selectivity of 1-butanol for presumptively PLD-regulated processes. Our results support the idea that FIPI is an effective PLD inhibitor both in vitro and in cultured cells but raise and exacerbate concerns about the validity of results obtained using 1-butanol as a probe of PLD function. We show that 1-butanol almost completely blocks glucose-stimulated insulin release (Fig. 4A) but does so without visibly inhibiting production of PA, whereas FIPI blocks PA production but has little effect on insulin release. These data raise the interesting possibility that Ptd-But does not function as an inert lipid, but rather exerts a potent inhibitory effect on the secretory pathway through a mechanism that remains to be defined. Alternatively, the inhibition could ensue from other actions mediated by 1-butanol that are not adequately controlled for through comparison to the actions of tert-butanol.
PLD activity has been linked to regulatory exocytosis using other approaches, such as RNAi-mediated down-regulation of individual isoforms (Waselle et al., 2005). Our findings do not invalidate these reports, because FIPI inhibits both PLD1 and PLD2, and it may well be, for example, that PLD1 facilitates insulin secretion (Waselle et al., 2005), whereas PLD2 opposes it. As well, RNAi-mediated effects take place over hours to days, raising the possibility of secondary effects of the PLD knockdowns, whereas FIPI acts within 15 min, permitting analysis of acute inhibition of PLD activity. Future studies on mice lacking the individual PLD isoforms or using yet-to-be-developed isoform-selective PLD inhibitors will provide clarification. Nonetheless, our findings do suggest that FIPI is a useful tool for reevaluating the many cell biological processes that have been linked to PLD using alcohol-mediated inhibition of PA production or other approaches that inhibit both classic isoforms. In addition to insulin secretion, we also found a lack of evidence that PLD plays roles in activation of ERK, AKT, and p38 in several signaling systems, counter to what has been reported previously. We also saw no effect on F-actin stress fibers in resting CHO cells, which is noteworthy, because there have been several reports that PLD regulates stress fiber formation in the context of LPA signaling or overexpression of activated Rho (Cross et al., 1996; Kam and Exton, 2001). It will interesting to use FIPI to examine the role of PLD in signaling-stimulated stress fiber formation.
Thus far, we have identified no other pathways directly inhibited by FIPI. FIPI inhibits the catalytic activity of classic PLD isoforms and seems to do so without causing changes in the localization of the proteins, access to the required cofactor PIP2, or the actin cytoskeleton. Nonetheless, future studies will be required to establish the mechanism by which FIPI inhibits PLD1 and PLD2 activity and to further evaluate its specificity for other signaling pathways. It is intriguing that FIPI exhibits greater potency in vivo (as low as 0.5 nM IC50) than it does in the standard in vitro PLD assay (25 nM IC50). It should be noted that the in vitro assay is performed using positively curved synthetic liposomes containing only a few types of lipids and no proteins, whereas PLD functions in vivo on a planar or slightly negatively curved membrane surface composed of a complex set of lipids and proteins. The latter environment may be less hospitable for PLD enzymatic action, allowing the inhibitor to be visibly effective at a lower concentration. Further investigation using broken cell in vitro assays and other approaches may yield novel insights into PLD mechanisms of action in the cellular setting.
We also show here, using FIPI, that PLD inhibition blunts chemotaxis and blocks effects on cytoskeletal reorganization and elements of cell spreading that are regulated by PLD2. PLD has long been of interest in the context of immune responses (for review, see Huang and Frohman, 2007), but these outcomes are particularly intriguing because PLD has been receiving increasing attention in the cancer field for the past several years. PLD1 and -2 gene expression, protein level, and activity are up-regulated in numerous tumor types (for review, see Huang and Frohman, 2007), and promote mTor activity and block cancer cell apoptosis (Chen et al., 2005). PLD1 facilitates matrix metalloproteinase release (Williger et al., 1999), whereas PLD2 affects cell spreading (Du and Frohman, 2009), proliferation (Zhao et al., 2007), and migration, and PLD2 mutations have been identified in breast cancer (Wood et al., 2007). FIPI is derived from halopemide, a therapeutic for neuroscience applications, and FIPI pharmacokinetics suggest that it has a half-life and bioavailability in vivo that make it a usable candidate for animal studies on metastasis (Monovich et al., 2007). It is likely that FIPI analogs with greater potency or other beneficial characteristics can be generated, and this current study suggests that they will be of potential utility as in vivo agents to examine physiological roles for PLD and to determine whether inhibiting PLD could be useful in the cancer therapeutic setting.
Acknowledgments
We thank Novartis and Dr. Lauren Monovich for providing starting samples of 5-fluoro-2-indolyl des-clorohalopemide.
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM071520, GM071475, GM50388, T32-GM008444]; the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL50040]; the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant F31-DK082280]; the National Institutes of Health National Center for Research Resources [Grants S10-RR024598, P20-RR021954]; the Turner Foundation; an American Heart Association Postdoctoral Fellowship; and the Carol M. Baldwin Breast Cancer Research Fund.
ABBREVIATIONS: PLD, phospholipase D; PC, phosphatidylcholine; PA, phosphatidic acid; Ptd-But, phosphatidyl butanol; PIP2, phosphatidyl inositol 4,5-bisphosphate; FIPI, 5-fluoro-2-indolyl des-chlorohalopemide; HPLC, high-performance liquid chromatography; CHO, Chinese hamster ovary; FBS, fetal bovine serum; Dox, doxycycline; GFP, green fluorescent protein; DMSO, dimethyl sulfoxide; PAGE, polyacrylamide gel electrophoresis; ERK, extracellular signal-regulated kinase; DMSO, dimethyl sulfoxide; KRBH, Krebs-Ringer bicarbonate HEPES buffer; PMA, phorbol 12-myristate 13-acetate; RNAi, RNA interference.
References
- Bechoua S and Daniel LW (2001) Phospholipase D is required in the signaling pathway leading to p38 MAPK activation in neutrophil-like HL-60 cells, stimulated by N-formyl-methionyl-leucyl-phenylalanine. J Biol Chem 276 31752-31759. [DOI] [PubMed] [Google Scholar]
- Carrigan SO, Pink DB, and Stadnyk AW (2007) Neutrophil transepithelial migration in response to the chemoattractant fMLP but not C5a is phospholipase D-dependent and related to the use of CD11b/CD18. J Leukoc Biol 82 1575-1584. [DOI] [PubMed] [Google Scholar]
- Chen Y, Rodrik V, and Foster DA (2005) Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene 24 672-679. [DOI] [PubMed] [Google Scholar]
- Chen YG, Siddhanta A, Austin CD, Hammond SM, Sung TC, Frohman MA, Morris AJ, and Shields D (1997) Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J Cell Biol 138 495-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Huang P, Jenkins GM, Chan DC, Schiller J, and Frohman MA (2006) A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat Cell Biol 8 1255-1262. [DOI] [PubMed] [Google Scholar]
- Colley WC, Sung TC, Roll R, Jenco J, Hammond SM, Altshuller Y, Bar-Sagi D, Morris AJ, and Frohman MA (1997) Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr Biol 7 191-201. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Roberts S, Ridley AJ, Hodgkin MN, Stewart A, Claesson-Welsh L, and Wakelam MJO (1996) Stimulation of actin stress fibre formation mediated by activation of phospholipase D. Curr Biol 6 588-597. [DOI] [PubMed] [Google Scholar]
- Du G and Frohman MA (2009) A lipid-signaled myosin phosphatase surge disperses cortical contractile force early in cell spreading. Mol Biol Cell doi: 10.1091/mbc.E08-06-0555 [DOI] [PMC free article] [PubMed]
- Du G, Huang P, Liang BT, and Frohman MA (2004) Phospholipase D2 localizes to the plasma membrane and regulates angiotensin II receptor endocytosis. Mol Biol Cell 15 1024-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia A, Zheng Y, Zhao C, Toschi A, Fan J, Shraibman N, Brown HA, Bar-Sagi D, Foster DA, and Arbiser JL (2008) Honokiol suppresses survival signals mediated by Ras-dependent phospholipase D activity in human cancer cells. Clin Cancer Res 14 4267-4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Altshuller YM, Sung TC, Rudge SA, Rose K, Engebrecht J, Morris AJ, and Frohman MA (1995) Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J Biol Chem 270 29640-29643. [DOI] [PubMed] [Google Scholar]
- Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, and Morris AJ (1997) Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4,5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-α. J Biol Chem 272 3860-3868. [DOI] [PubMed] [Google Scholar]
- Huang P, Altshuller YM, Chunqiu Hou J, Pessin JE, and Frohman MA (2005) Insulin-stimulated plasma membrane fusion of Glut4 glucose transporter-containing vesicles is regulated by phospholipase D1. Mol Biol Cell 16 2614-2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P and Frohman MA (2007) The potential for phospholipase D as a new therapeutic target. Expert Opin Ther Targets 11 707-716. [DOI] [PubMed] [Google Scholar]
- Huang Y, Qureshi IA and Chen H (1999) Effects of phosphatidylinositol 4,5-bisphosphate and neomycin on phospholipase D: kinetic studies. Mol Cell Biochem 197 195-201. [DOI] [PubMed] [Google Scholar]
- Jenkins GM and Frohman MA (2005) Phospholipase D: a lipid centric review. Cell Mol Life Sci 62 2305-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam Y and Exton JH (2001) Phospholipase D activity is required for actin stress fiber formation in fibroblasts. Mol Cell Biol 21 4055-4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DW, Park MH, Lee YJ, Kim HS, Kwon TK, Park WS, and Min do S (2008) Phorbol ester up-regulates phospholipase D1 but not phospholipase D2 expression through a PKC/Ras/ERK/NFκB-dependent pathway and enhances matrix metalloproteinase-9 secretion in colon cancer cells. J Biol Chem 283 4094-4104. [DOI] [PubMed] [Google Scholar]
- Lee CS, Kim IS, Park JB, Lee MN, Lee HY, Suh PG, and Ryu SH (2006) The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nat Cell Biol 8 477-484. [DOI] [PubMed] [Google Scholar]
- Lee S, Park JB, Kim JH, Kim Y, Kim JH, Shin KJ, Lee JS, Ha SH, Suh PG, and Ryu SH (2001) Actin directly interacts with phospholipase D, inhibiting its activity. J Biol Chem 276 28252-28260. [DOI] [PubMed] [Google Scholar]
- McDermott M, Wakelam MJ, and Morris AJ (2004) Phospholipase D. Biochem Cell Biol 82 225-253. [DOI] [PubMed] [Google Scholar]
- Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du Z, LaSala D, Boyar W, et al. (2007) Optimization of halopemide for phospholipase D2 inhibition. Bioorg Med Chem Lett 17 2310-2311. [DOI] [PubMed] [Google Scholar]
- Morris AJ, Frohman MA, and Engebrecht J (1997) Measurement of phospholipase D activity. Anal Biochem 252 1-9. [DOI] [PubMed] [Google Scholar]
- Morris AJ and Smyth SS (2007) Measurement of autotaxin/lysophospholipase D activity. Methods Enzymol 434 89-104. [DOI] [PubMed] [Google Scholar]
- Rose K, Rudge SA, Frohman MA, Morris AJ, and Engebrecht J (1995) Phospholipase D signaling is essential for meiosis. Proc Natl Acad Sci U S A 92 12151-12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skippen A, Jones DH, Morgan CP, Li M, and Cockcroft S (2002) Mechanism of ADP ribosylation factor-stimulated phosphatidylinositol 4,5-bisphosphate synthesis in HL60 cells. J Biol Chem 277 5823-5831. [DOI] [PubMed] [Google Scholar]
- Su W, Chardin P, Yamazaki M, Kanaho Y, and Du G (2006) RhoA-mediated Phospholipase D1 signaling is not required for the formation of stress fibers and focal adhesions. Cell Signal 18 469-478. [DOI] [PubMed] [Google Scholar]
- Sung TC, Roper RL, Zhang Y, Rudge SA, Temel R, Hammond SM, Morris AJ, Moss B, Engebrecht J, and Frohman MA (1997) Mutagenesis of phospholipase D defines a superfamily including a trans-Golgi viral protein required for poxvirus pathogenicity. EMBO J 16 4519-4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadharaj S, Steinhour E, Hunter MG, Watkins T, Baran CP, Magalang U, Kuppusamy P, Zweier JL, Marsh CB, Natarajan V, et al. (2006) Vitamin C-induced activation of phospholipase D in lung microvascular endothelial cells: regulation by MAP kinases. Cell Signal 18 1396-1407. [DOI] [PubMed] [Google Scholar]
- Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, and Bader MF (2001) Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J 20 2424-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waselle L, Gerona RR, Vitale N, Martin TF, Bader MF, and Regazzi R (2005) Role of phosphoinositide signaling in the control of insulin exocytosis. Mol Endocrinol 19 3097-3106. [DOI] [PubMed] [Google Scholar]
- Williger BT, Ho WT, and Exton JH (1999) Phospholipase D mediates matrix metalloproteinase-9 secretion in phorbol ester-stimulated human fibrosarcoma cells. J Biol Chem 274 735-738. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318 1108-1113. [DOI] [PubMed] [Google Scholar]
- Yang JS, Gad H, Lee S, Mironov A, Zhang L, Beznoussenko GV, Valente C, Turacchio G, Bonsra AN, Du G, et al. (2008) COPI vesicle fission: a role for phosphatidic acid and insight into Golgi maintenance. Nat Cell Biol 10 1146-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeniou-Meyer M, Zabari N, Ashery U, Chasserot-Golaz S, Haeberle AM, Demais V, Bailly Y, Gottfried I, Nakanishi H, Neiman AM, et al. (2007) Phospholipase D1 production of phosphatidic acid at the plasma membrane promotes exocytosis of large dense-core granules at a late stage. J Biol Chem 282 21746-21757. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ting AT, Marcu KB, and Bliska JB (2005) Inhibition of MAPK and NF-kappa B pathways is necessary for rapid apoptosis in macrophages infected with Yersinia. J Immunol 174 7939-7949. [DOI] [PubMed] [Google Scholar]
- Zhao C, Du G, Skowronek K, Frohman MA, and Bar-Sagi D (2007) Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat Cell Biol 9 707-712. [DOI] [PubMed] [Google Scholar]