

Abstract
Signaling by the platelet-derived growth factor receptor-β (PDGFRβ) is diminished when the PDGFRβ is phosphorylated on seryl residues by G protein-coupled receptor kinase-5 (GRK5), but mechanisms for GRK5 activation by the PDGFRβ remain obscure. We therefore tested whether the PDGFRβ is able to tyrosine-phosphorylate and thereby activate GRK5. Purified GRK5 was tyrosine-phosphorylated by the wild-type PDGFRβ to a stoichiometry of 0.8 mol phosphate/mol GRK5, an extent ∼5 times greater than observed with a Y857F PDGFRβ mutant that fails to phosphorylate exogenous substrates but autophosphorylates and activates Src normally. The degree of PDGFRβ-mediated phosphorylation of GRK5 correlated with GRK5 activity, as assessed by seryl phosphorylation of the PDGFRβ in purified protein preparations, in intact cells expressing a tyrosine-to-phenylalanine GRK5 mutant, and in GRK5 peptide phosphorylation assays. However, tyrosyl phosphorylation of GRK5 was not necessary for GRK5-mediated phosphorylation of the β2-adrenergic receptor, even though β2-adrenergic receptor activation promoted tyrosyl phosphorylation of GRK5 in smooth muscle cells. Phosphorylation of the PDGFRβ by GRK5 in smooth muscle cells or in purified protein preparations reduced PDGFRβ-mediated peptide phosphorylation. In contrast, phosphorylation of GRK5 by the PDGFRβ enhanced the Vmax of GRK5-mediated peptide phosphorylation, by 3.4-fold, without altering the GRK5 KM for peptide. We conclude that GRK5 tyrosyl phosphorylation is required for the activation of GRK5 by the PDGFRβ, but not by the β2-adrenergic receptor, and that by activating GRK5, the PDGFRβ triggers its own desensitization.
The platelet-derived growth factor receptor-β (PDGFRβ) is a receptor protein tyrosine kinase that is critical for fetal development and wound healing, and it is intimately involved in the pathogenesis of atherosclerosis and malignant neoplasia (Heldin and Westermark, 1999). Agonist binding induces PDGFRβ dimerization and subsequent autophosphorylation, a process that creates phosphotyrosyl docking sites for proteins that contain SH2 and phosphotyrosyl-binding domains. Some of these proteins are activated by PDGFRβ-mediated tyrosyl phosphorylation, whereas others are activated as a result of the actions of other proteins that are also associated with the PDGFRβ. Because PDGFRβ signaling can persist even after receptor endocytosis (Wang et al., 2004), regulation of PDGFRβ signaling is critically important to cellular homeostasis.
Desensitization of PDGFRβ signaling can be achieved by a variety of mechanisms, including agonist-induced PDGFRβ seryl phosphorylation mediated by GRKs (Freedman et al., 2002; Hildreth et al., 2004; Wu et al., 2005, 2006) and perhaps casein kinase I (Bioukar et al., 1999), as well as tyrosyl dephosphorylation and PDGFRβ degradation (Heldin and Westermark, 1999). Although GRK2 seems to mediate most agonist-induced seryl phosphorylation and desensitization of the PDGFRβ in fibroblasts (Wu et al., 2005), GRK5 does so in smooth muscle cells (SMCs) (Wu et al., 2006). Mechanisms for GRK-mediated desensitization of the PDGFRβ remain incompletely understood, but they seem to be GRK-specific and involve reduced PDGFRβ autophosphorylation (Hildreth et al., 2004; Wu et al., 2006).
Belonging to a seven-member family of serine/threonine kinases, GRKs share a conserved central catalytic domain flanked by distinct amino- and carboxyl-terminal domains that help target GRKs to receptors and membranes, respectively (Premont and Gainetdinov, 2007). GRKs bind to and phosphorylate agonist-occupied receptors of at least two general types: seven-transmembrane G protein-coupled receptors (Premont and Gainetdinov, 2007), and receptor protein tyrosine kinases that can also activate heterotrimeric G proteins (Freedman et al., 2002). GRK-mediated serine/threonine phosphorylation of receptors leads to the attenuation of certain types of receptor signaling (e.g., through heterotrimeric G proteins or phospholipase Cγ) and potentiation of other types of receptor signaling (e.g., through extracellular signal-regulated kinases or Src) (Wu et al., 2006; DeWire et al., 2007). GRK activity is regulated—in an isoform-specific manner—by mechanisms including serine/threonine phosphorylation by protein kinase C isoforms (Pitcher et al., 1998; Premont and Gainetdinov, 2007) and autophosphorylation (Pronin and Benovic, 1997).
As allosteric enzymes, GRKs are known to be activated by agonist-occupied seven-transmembrane receptors (Premont and Gainetdinov, 2007). However, GRK2 is also activated by tyrosyl phosphorylation of its amino-terminal domain, either by Src (Sarnago et al., 1999; Penela et al., 2001) or the PDGFRβ (Wu et al., 2005). Whether this activation mechanism obtains for GRK5 remains an open question, because GRK2 and GRK5 share only ∼58% sequence similarity (Premont et al., 1994) and belong to distinct GRK phylogenetic subfamilies (Premont and Gainetdinov, 2007). Moreover, GRK2 and GRK5 have demonstrated distinct seven-transmembrane receptor substrate preferences (Gainetdinov et al., 1999; Iwata et al., 2005), distinct phosphorylation sites on specific 7-transmembrane receptors (Fredericks et al., 1996; Hu et al., 2002), and apparently distinct phosphorylation sites on the PDGFRβ (Hildreth et al., 2004; Wu et al., 2006). Further highlighting the differences between GRK2 and GRK5 are the distinct roles these kinases play in recruiting the β-arrestin adaptor protein isoforms to seven-transmembrane receptors (DeWire et al., 2007). Likewise, recruitment of the phosphatase Shp2 to the PDGFRβ is enhanced when the PDGFRβ is phosphorylated by GRK5 and not by GRK2 (Wu et al., 2006). Because of these multiple distinctions between GRK5 and GRK2, we sought to determine 1) whether GRK5-mediated desensitization of PDGFRβ signaling is triggered by PDGFRβ-mediated tyrosyl phosphorylation of GRK5 and 2) how this putative GRK5 tyrosyl phosphorylation affects GRK5 enzymatic activity.
Materials and Methods
Plasmid Constructs. Plasmids encoding the N-terminal Flag-tagged human PDGFRβ (both WT and Y857F mutant) and bovine GRK5, each in pcDNAI (Invitrogen, Carlsbad, CA), were described previously (Wu et al., 2005), as was the plasmid encoding an N-terminal Flag-tagged human β2-adrenergic receptor mutant (β2ART68F,Y132G, Y219A or β2ARTYY) in pcDNA3 (Shenoy et al., 2006). The full-length bovine GRK5 cDNA was subcloned into pcDNA3.1(+) (Invitrogen) using unique EcoRI and XbaI sites that flank the cDNA sequence. Site-directed mutagenesis of GRK5 was performed by using the Expand High Fidelity PCR system (Roche Applied Science, Indianapolis, IN), based on the PCR overlap extension method (Cai et al., 2002), with the following primers (the sense primer, but not the complementary primer, is listed, 5′ to 3′, with mutations underlined): tcggtggcagaattcgaagttactc (Y90F); ttatgaccaagttcctcacccca (Y109F); gagaacattgtcttcagagatttg (Y309F); and gcctgagccccgacttctgg (Y368F). PCR fragments bearing the appropriate mutation were combined in a final amplification step using primers flanking the most 5′ and 3′ mutations. The final product was digested with BamHI and subcloned into the bovine GRK5 plasmid in pcDNA3.1(+). This GRK5 mutant with 4 tyrosine-to-phenylalanine mutations was designated “4YF.” GRK5 constructs containing Y90F and Y109F mutations, or Y309F and Y368F mutations, were named “2YFA” or “2YFB,” respectively.
A chimeric receptor (ChiR) comprising the extracellular domain of the human colony-stimulating factor-1 (CSF-1) receptor (c-fms) and the transmembrane and cytoplasmic domains of the human PDGFRβ was generously provided by Dr. Karen Symes (Symes and Mercola, 1996). To epitope-tag the N terminus of the ChiR, we subcloned the full-length ChiR cDNA into pBlueScript II SK(-) (Stratagene, La Jolla, CA) and used cassette PCR to replace the endogenous signal sequence of the CSF-1 receptor with a hemagglutinin signal sequence followed by the Flag epitope, as described previously (Freedman et al., 2002). We introduced a tyrosine-to-phenylalanine mutation into the ChiR, at residue 857 of the human PDGFRβ cytoplasmic domain, by subcloning an 824-base pair BspE1/SacII fragment from the Y857F PDGFRβ construct we made previously (Wu et al., 2005). The full-length N-terminal Flag-tagged WT and Y857F ChiRs were excised from pBlueScript II SK(-) and subcloned into pcDNA3.1(+). All mutant constructs were subjected to dideoxy sequencing to ensure that no PCR errors were introduced into the amplified fragments.
Cell Culture and Transfection. Human embryonic kidney (HEK) 293 cells were cultured and transient transfection was performed as described previously (Freedman et al., 2002). HEK cells stably expressing the WT Flag-tagged PDGFRβ (Wu et al., 2005) were used as the source of immunoprecipitated PDGFRβ for peptide phosphorylation assays. HEK cells stably expressing a Flag-tagged human β2-adrenergic receptor mutant (β2ARTYY) were described previously (Shenoy et al., 2006). Cotransfections with PDGFRβ and GRK5 plasmids used 2 μg of PDGFRβ constructs and 8 μg of GRK5 constructs per 100-mm dish of HEK cells. In transfecting GRK5 constructs into the HEK cells stably transfected with the β2ARTYY, we used 2 μg of GRK5 construct plasmid DNA per 100-mm dish of HEK cells. Cell-surface PDGFRβ expression was measured by flow cytometry, as described previously (Wu et al., 2005), to ensure that, within single experiments, PDGFRβ expression among discrete lines of (co-)transfected cells varied by ≤30%. Transfected cell groups that failed to conform to this standard were not used.
Thoracic aortic SMCs were obtained from age-, gender-, and weight-matched, grk5(-/-) and congenic WT mice using the explant outgrowth technique described previously (Wu et al., 2006). SMCs were used only through passage 7. SMCs were transfected with a Nucleofector II (Amaxa AG, Inc., Gaithersburg, MD) according to the manufacturer's instructions. We electroporated 2 × 106 SMCs per cuvette with 2 to 4 μg of plasmid DNA, in solution “L.” Experiments were conducted 48 h after transfection.
Immunoprecipitations and Immunoblotting. Standard procedures for these two assays were described previously (Freedman et al., 2002; Wu et al., 2005, 2006). The following antibodies were used for immunoprecipitations (IPs): rabbit anti-PDGFRβ IgG (Santa Cruz Biotechnology, Santa Cruz, CA) for endogenous PDGFRβ; anti-Flag M2-agarose (Sigma, St. Louis, MO) for N-terminal Flag-tagged PDGFRβ and ChiR; rabbit anti-GRK5 IgG described previously (Premont et al., 1994); and monoclonal anti-GRK5 IgA(κ) U11/6, created as described previously (Oppermann et al., 1996). Antibodies used for immunoblotting (IB) included anti-GRK5 monoclonal A16/17 IgG (Oppermann et al., 1996), rabbit or goat anti-PDGFRβ IgG (Santa Cruz Biotechnology), rabbit anti-phosphoserine IgG (Chemicon, Inc., Temecula, CA), rabbit anti-β2AR or anti-phospho-β2AR (phospho-Ser355/356) (Santa Cruz Biotechnology), and anti-phosphotyrosine pY20 monoclonal IgG (BD Biosciences, San Jose, CA).
Immune Complex Kinase Assays. Recombinant bovine GRK5 was purified from baculovirus-infected Sf9 insect cells, as we reported previously (Premont et al., 1994; Wu et al., 2006). Kinase assays were performed using purified GRK5 and N-terminal Flag-tagged PDGFRβs immunoprecipitated from PDGF-stimulated HEK cells, essentially as described previously (Wu et al., 2005, 2006). Reactions for stoichiometry calculations contained 1.0 μg of GRK5 in a final volume of 30 μl of reaction buffer: 20 mM Tris-HCl (pH 8.0 at 25°C), 2 mM EDTA, 10 mM MgCl2, 1 mM dithiothreitol, and 0.1 mM [γ-32P]ATP (3 × 105 cpm/ml) (PerkinElmer Life and Analytical Sciences, Inc., Waltham, MA). The phosphorylation reactions were incubated at 30°C for 30 min and then stopped by the addition of 2× SDS sample buffer (30 μl). Divided samples were electrophoresed on parallel 4-to-12% SDS-polyacrylamide gels, one of which was dried for autoradiography and one of which was transferred to nitrocellulose for autoradiography and subsequent IB for GRK5, the PDGFRβ, and phosphoserine. Autoradiograms of protein bands and diluted aliquots of [γ-32P]ATP were quantitated with a PhosphorImager (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK). The cpm in GRK5 bands obtained with the dried gels were within 10% of those obtained with the corresponding nitrocellulose blots. The 32P cpm in each GRK5 band were corrected for the amount of GRK5, quantitated by IB. To calculate the stoichiometry of PDGFRβ-mediated GRK5 phosphorylation, we first subtracted from each 32P GRK5 band the 32P cpm obtained with purified GRK5 incubated in the absence of PDGFRβs (but in the presence of agarose beads from mock-transfected HEK cells). To calculate phosphorylation stoichiometry, the resulting difference was divided by the specific activity of ATP used in the experiment and the ∼7 pmol of GRK5 present in each band.
Peptide Phosphorylation Assays. The initial phase of these assays was performed just like immune complex kinase assays (above), with either purified GRK5 or one of two PDGFRβ preparations immunoprecipitated from 100-mm dishes of cells stimulated with 2 nM PDGF-BB (Millipore, Billerica, MA) for 10 min (37°C): N-terminal Flag-tagged PDGFRβs from HEK cells, or endogenous PDGFRβs from mouse SMCs. We also used recombinant human PDGFRβ cytoplasmic domain preparations (Arg561-Leu1106, GenBank accession number NM_002609), purified from Spodoptera frugiperda insect cells (Calbiochem, Inc., San Diego, CA). PDGFRβs immunoprecipitated from multiple dishes were pooled and subsequently aliquoted to multiple phosphorylation reaction tubes. Purified PDGFRβ cytoplasmic domain preparations were used at a concentration of 54 nM. For assays to determine PDGFRβ kinase activity, we added PDGFRβ substrate peptide (MAEEEEYVFIEAKKK) (Baxter et al., 1998) to the standard immune complex kinase assays (final concentration, 100 μM). To measure GRK5 kinase activity, we added GRK5 substrate peptide (RRREEEEESAAA) (Premont et al., 1994) to the standard immune complex kinase assays (final concentration, 1.0 mM) in the presence of 50 nM GRK5. Global Peptides Services (Fort Collins, CO) synthesized trifluoroacetate salts of the PDGFRβ substrate peptide and caveolin scaffolding domain peptides (caveolin-1, DGIWKASFTTFTVTKYWFYR; caveolin-2, DKVWICSHALFEISKYVMYK) (Carman et al., 1999). GRK5 substrate peptide was synthesized by Sigma (also as a trifluoroacetate salt). Stock peptide solutions were made in H2O with 500 μg/ml bovine serum albumin. Because it was used at relatively high concentrations, the GRK5 substrate peptide was dissolved at 25 mM in 100 mM Tris base, to achieve a pH of 7.0; all subsequent dilutions for kinase reactions were made in 100 mM Tris-acetate, pH 7.0, with 0.5 mg/ml bovine serum albumin. Reactions were terminated by the addition of one reaction volume of 150 mM orthophosphoric acid, and 3 × 30-μl aliquots were spotted onto P81 phosphocellulose paper (Whatman, Clifton, NJ). After drying, phosphocellulose was washed 3 × 10 min with 75 mM orthophosphoric acid and subjected to Cerenkov counting. Nonspecific counts were determined from reaction tubes that contained [γ-32P]ATP and kinase(s) without peptides. Nonspecific cpm were subtracted from the total cpm to obtain specific cpm. For kinetic analyses, double-reciprocal plots were compiled and linear regression was performed with GraphPad Prism software. Reactions containing GRK5 as the sole kinase incorporated no specific counts into the PDGFRβ substrate peptide; likewise, reactions containing the PDGFRβ as the sole kinase incorporated no specific counts into the GRK5 substrate peptide.
Caveolin peptide experiments were performed in two sequential steps. First, PDGFRβ-mediated phosphorylation of GRK5 proceeded for 20 min (30°C). (Control reactions that lacked PDGFRβ IPs contained IPs from HEK cells that lacked PDGFRβs.) Second, GRK5 in the reaction supernatant was separated from immunoprecipitated PDGFRβs and subsequently incubated with scaffolding domain peptides from either caveolin-1 or -2 (final concentration, 30 μM) (Carman et al., 1999), or buffer only, for 10 min. Finally, 1 mM GRK5 substrate peptide along with 5 to 10 μCi of [γ-32P]ATP was added to each reaction tube, and reactions proceeded for 15 min (30°C).
Tyrosyl Phosphorylation of GRK5 in Intact Cells. After overnight incubation in serum-free medium (Wu et al., 2005), SMCs or HEK cells were preincubated (37°C) in serum-free medium containing either 10 μM AG1295 (Calbiochem) or vehicle [0.1% (v/v) dimethyl sulfoxide] for 20 min and with 50 μM pervanadate (Wu et al., 2005) for 5 min before stimulation with either 2 nM PDGF-BB or 10 μM (-)isoproterenol (Sigma) for 10 min (37°C). Cell lysates were then subjected to parallel IP with antibodies for GRK5 or PDGFRβ. For ChiR experiments, WT SMCs expressing N-terminal Flag-tagged WT or Y857F ChiRs were exposed to serum-free medium containing 50 ng/ml human CSF-1 (Millipore) for 10 min at 37°C in the presence of 50 μM pervanadate as reported previously (Wu et al., 2005). Subsequently, cells were solubilized as described previously (Wu et al., 2005), and lysates were subjected to IP for GRK5 and the ChiR and subsequently to IB for phosphotyrosine and phosphoserine. HEK cells cotransfected with the Flag-tagged PDGFRβ and either the WT or the 4YF GRK5 were assayed 2 days after transfection.
Data Analysis. Independent means were compared with unpaired or paired t tests, depending on the experimental design, with Prism software (GraphPad Software, Inc., San Diego, CA). All p values are two-tailed. The text cites means ± S.D., whereas figures display means ± S.E.
Results
PDGFRβ-Mediated Tyrosyl Phosphorylation of GRK5 Enhances GRK5 Activity. To determine whether the PDGFRβ tyrosine phosphorylates GRK5, we first used purified GRK5 as a substrate for two distinct PDGFRβ constructs: the WT PDGFRβ, and the Y857F PDGFRβ mutant, which fails to phosphorylate exogenous substrates even though it autophosphorylates normally (Baxter et al., 1998) and activates c-Src normally (because it recruits Shp2 normally) (Wu et al., 2005). These N-terminal epitope-tagged PDGFRβ constructs were immunoprecipitated from quiescent or PDGF-stimulated HEK cells. Purified GRK5 was phosphorylated on tyrosyl residues by the WT PDGFRβ in a PDGF-dependent manner. Moreover, the extent of GRK5 tyrosine phosphorylation effected by the WT PDGFRβ was 5-fold greater than that observed with the Y857F PDGFRβ (Fig. 1, A and B). Because the Y857F PDGFRβ autophosphorylates normally and therefore associates normally with PDGFRβ-docking proteins, these data support the inference that GRK5 tyrosyl phosphorylation in these reactions is mediated predominantly by the PDGFRβ itself, rather than by intracellular tyrosine kinases that can associate with the PDGFRβ. To determine the significance of PDGFRβ-mediated tyrosyl phosphorylation of GRK5 in purified protein preparations, we asked whether PDGFRβ-mediated phosphorylation affects GRK5 activity, whether the PDGFRβ phosphorylates GRK5 stoichiometrically, and whether the PDGFRβ phosphorylates GRK5 in intact cells under physiological conditions.
Fig. 1.

The PDGFRβ tyrosine-phosphorylates and activates purified GRK5. HEK 293 cells expressing N-terminal Flag-tagged WT or Y857F mutant PDGFRβs were exposed to medium containing vehicle (-) or 2 nM PDGF-BB (+) for 10 min at 37°C. The cells were then lysed and subjected to PDGFRβ IP. Immune complex kinase assays then proceeded in the absence (-) or presence (+) of 500 nM purified GRK5 for 30 min at 30°C. The PDGFRβ in the pellets and GRK5 in the supernatant were separated and subjected to distinct SDS-PAGE, followed by IB. A, IB of GRK5 were probed first for phosphotyrosine (pTyr) and then for GRK5. The PDGFRβ IB was probed for total PDGFRβ and then either phosphotyrosine (pTyr) or phosphoserine (pSer). Shown are results from a single experiment, representative of three performed. B, band density for pTyr was divided by cognate band density for either GRK5 or PDGFRβ, and these quotients were normalized to those obtained from cells expressing the WT PDGFRβ, to obtain the “percentage of control.” Each ratio was then plotted as the mean ± S.E. from three independent experiments. Compared with the cognate value from the WT PDGFRβ: *, p < 0.05. C, from PDGF-stimulated cells, PDGFRβ pSer band densities were divided by cognate PDGFRβ band densities; each ratio was normalized to that obtained with the WT PDGFRβ in the absence of GRK5 to obtain the “percentage of control.” Each ratio was then plotted as the mean ± S.E. from at least three independent experiments. (Y857F PDGFRβ data in the absence of purified GRK5 are not shown in A.) Compared with control: *, p < 0.05; compared with WT PDGFRβ in the presence of purified GRK5: †, p < 0.05.
As a readout for GRK5 activity, we examined seryl phosphorylation of the PDGFRβ, which we have shown previously to be a substrate for GRK5 (Wu et al., 2006). PDGFRβs immunoprecipitated from PDGF-stimulated HEK cells were phosphorylated on seryl residues, as we have shown previously (Wu et al., 2005, 2006). This seryl phosphorylation can be attributed to intracellular activity of endogenous HEK cell GRKs, occurring before cell solubilization (Hildreth et al., 2004; Wu et al., 2005), and this activity was demonstrably greater on the WT than on the Y857F PDGFRβ (Fig. 1, A and C). Consequent to the addition of purified GRK5, seryl phosphorylation of WT PDGFRβs increased significantly (1.5-fold, p < 0.05; Fig. 1, A and C), but that of Y857F PDGFRβs did not (Fig. 1, A and C). Thus, GRK5-mediated seryl phosphorylation of the PDGFRβ correlated with PDGFRβ-mediated tyrosyl phosphorylation of GRK5. Accordingly, we inferred that PDGFRβ-mediated tyrosyl phosphorylation of GRK5 enhances GRK5 activity.
To test further whether the extent of GRK5 tyrosyl phosphorylation affects GRK5 activity, we created within GRK5 a series of tyrosine-to-phenylalanine mutations designed to reduce PDGFRβ-mediated GRK5 phosphorylation. To predict which sites in GRK5 are targets for the PDGFRβ, we exploited consensus motifs shown to be phosphorylated by the PDGFRβ (Songyang et al., 1995), as well as GRK5's limited homology with GRK2 (Premont et al., 1994). Three GRK5 tyrosyl residues lie in sequences predicted by peptide library studies to constitute sites of PDGFRβ-mediated phosphorylation (Songyang et al., 1995). In addition, GRK5 Tyr90 is the only residue homologous to any of the three GRK2 tyrosyl residues we identified previously as PDGFRβ targets (Wu et al., 2005), and this residue is conserved among GRKs 1 to 6 (Lodowski et al., 2006). Accordingly, we mutated all of these GRK5 tyrosine residues—90, 109, 309, and 368—to phenylalanine, to create a “4YF” GRK5 mutant.
This 4YF GRK5 mutant expressed normally. However, it showed considerably less PDGFRβ-mediated tyrosyl phosphorylation than the WT GRK5, even though GRK5 constructs with corresponding individual tyrosine-to-phenylalanine mutations did not (data not shown). In intact HEK cells, PDGFRβ-mediated phosphorylation of the 4YF GRK5 mutant was only 33 ± 10% of that observed with WT GRK5 (Fig. 2). Along with this reduced tyrosyl phosphorylation, the 4YF GRK5 mutant demonstrated diminished enzymatic activity as well. The magnitude of PDGFRβ seryl phosphorylation effected by the 4YF GRK5 was only 15% of that effected by the WT GRK5 (Fig. 2B). Thus, whether in purified protein preparations or in intact 293 cells, and whether demonstrated by mutations in the PDGFRβ or GRK5 itself, the extent of PDGFRβ-mediated GRK5 tyrosyl phosphorylation correlated with GRK5 activity.
Fig. 2.

GRK5 activity on the PDGFRβ correlates with PDGFRβ-mediated phosphorylation of GRK5. HEK cells were cotransfected with plasmids encoding the N-terminal Flag-tagged PDGFRβ and either no protein (None, empty vector) or one of two GRK5 constructs: WT, or Y90/109/309/368F (4YF). Cells were serum-starved overnight and treated with medium containing vehicle or 2 nM PDGF-BB for 10 min (37°C). Cells were then lysed and subjected to IP for either GRK5 or PDGFRβ, followed by IB. GRK5 blots were probed serially for phosphotyrosine (pTyr) and GRK5, and PDGFRβ blots were probed serially for the PDGFRβ and phosphoserine (pSer). A, results shown are from a single experiment, representative of three performed in duplicate. B, band intensities for pTyr and pSer were normalized to cognate band intensities for GRK5 and the PDGFRβ; the pSer/PDGFRβ quotient for PDGFRβ/empty vector cells was subtracted from each pSer/PDGFRβ quotient to yield values specific for the transfected GRK5 construct. The pTyr/GRK5 and corrected pSer/PDGFRβ values from PDGF-stimulated cells were then normalized to those obtained from cells transfected with the PDGFRβ and WT GRK5, to obtain the “percentage of control.” Plotted are means ± S.E. from three independent experiments performed in duplicate. Compared with cells expressing the PDGFRβ and WT GRK5: *, p < 0.02.
The contrast between results with WT and 4YF mutant GRK5 supports the inference that the PDGFRβ activates GRK5 by phosphorylating GRK5 on tyrosyl residues before GRK5 can phosphorylate the PDGFRβ on seryl residues. To bolster this inference, we sought to determine that the 4YF GRK5 mutant can, on a substrate distinct from the PDGFRβ, demonstrate catalytic activity comparable with WT GRK5. In this effort, we used a β2-adrenergic receptor mutant that is deficient in G protein coupling (β2ART68F,Y132G,Y219A or β2ARTYY), because this β2ARTYY demonstrates no agonist-induced phosphorylation in HEK cells in the absence of cotransfected GRK5 or GRK6 (and therefore maximizes sensitivity to discern differences in the activity of cotransfected GRKs) (Shenoy et al., 2006). With the agonist-activated β2ARTYY as a substrate, the 4YF GRK5 mutant demonstrated 90 ± 10% of WT GRK5 activity (Fig. 3A), even though GRK5 expression levels in these experiments were somewhat lower than those in our comparable PDGFRβ experiments (Fig. 3B). It is noteworthy that agonist activation of the β2ARTYY did not engender tyrosyl phosphorylation of even WT GRK5 (Fig. 3A). Thus, although the 4YF GRK5 mutant demonstrates impaired catalytic activity on the PDGFRβ (Fig. 2), it demonstrates normal catalytic activity on the β2AR (Fig. 3A). Moreover, it seems that although tyrosyl phosphorylation of GRK5 is necessary for GRK5-mediated phosphorylation of the PDGFRβ (Fig. 2A), it is not necessary for GRK5-mediated phosphorylation of the β2AR (Fig. 3A).
Fig. 3.

GRK5 activity on the β2AR does not require tyrosyl phosphorylation of GRK5. HEK cells stably expressing the N-terminal Flag-tagged β2ARTYY were transiently transfected with plasmids encoding WT GRK5, 4YF GRK5, or no protein (empty vector, None). Cells were stimulated with 10 μM (-)isoproterenol (ISO) for 10 min (37°C). Cell lysates were subjected to parallel IP for either GRK5 or β2ARTYY, followed by SDS-PAGE/IB. A, GRK5 blots were probed sequentially for pTyr and GRK5. Samples from epidermal growth factor-stimulated A431 cells gave abundant bands on the pTyr IB (data not shown). β2ARTYY blots were probed with IgG specific for the β2AR phosphorylated on seryl residues 355 and 356, and then reprobed for the β2AR. Results are from a single experiment representative of five performed. Phospho-β2AR band intensities were divided by cognate β2AR band intensities; the resulting ratios were normalized to those derived from ISO-stimulated cells transfected with WT GRK5, to obtain the “percentage of control.” Data are from five independent experiments. B, 2 μg of cell lysate protein from β2AR (Fig. 3A) and parallel PDGFRβ experiments (Fig. 2) were subjected to SDS-PAGE and IB to compare the expression levels of GRK5. Blots were probed serially for total GRK5 and β-tubulin. Shown are results from a single experiment representative of three performed.
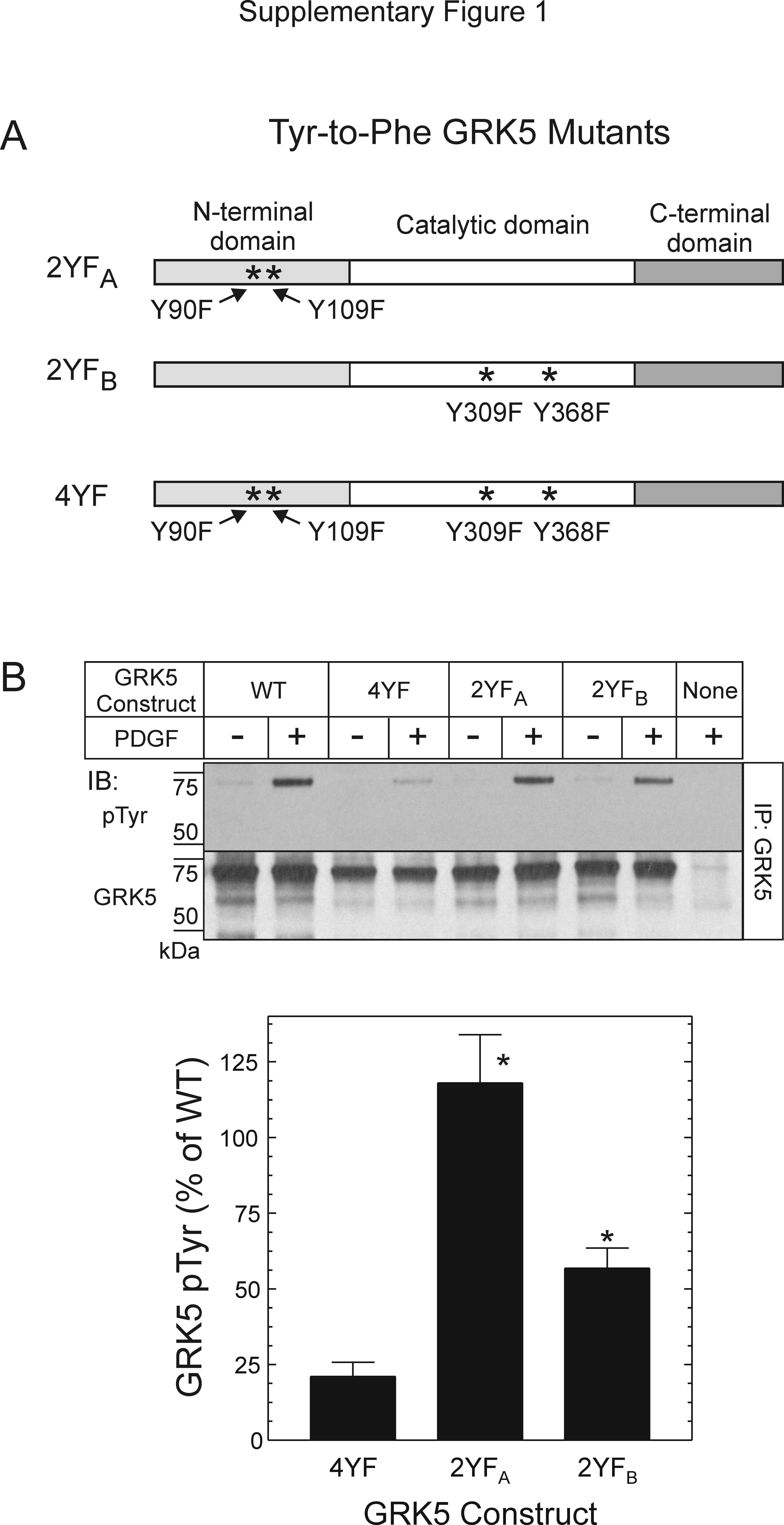
Although the 4YF GRK5 mutant affords insight into mechanisms of PDGFRβ-mediated GRK5 activation, it also begins to elucidate PDGFRβ target sites within GRK5. However, can the PDGFRβ phosphorylate subsets of the tyrosine residues mutated in the 4YF GRK5 mutant? To address this question, we examined GRK5 domain-specific tyrosine-to-phenylalanine mutants that are subsets of the 4YF mutant: Y309F/Y368F GRK5 (with 4YF N-terminal domain residues 90 and 109 restored to tyrosine), and Y90F/Y109F GRK5 (with 4YF catalytic-domain residues 309 and 368 restored to tyrosine) (Supplementary Fig. 1). The Y90F/Y109F GRK5 mutant demonstrated PDGFRβ-mediated tyrosyl phosphorylation comparable with that observed with WT GRK5. In contrast, the Y309F/Y368F GRK5 mutant demonstrated 60 ± 20% of the PDGFRβ-mediated tyrosyl phosphorylation observed with WT GRK5 (Supplementary Fig. 1). Together, these data suggest that 1) the PDGFRβ phosphorylates GRK5 on tyrosyl residues 90 and 109, as well as on tyrosyl residues 309 and 368, and 2) that Tyr90 and/or Tyr109 may inhibit PDGFRβ-mediated GRK5 Tyr phosphorylation, perhaps through hydrogen bonding-mediated steric effects suggested by the GRK6 crystal structure (Lodowski et al., 2006).
The PDGFRβ Tyrosine Phosphorylates GRK5 Stoichiometrically. To determine the stoichiometry of PDGFRβ-mediated tyrosyl phosphorylation of GRK5, we used purified GRK5 and PDGFRβs immunoprecipitated from quiescent or PDGF-stimulated HEK cells, just as in Fig. 1, along with [γ-32P]ATP, so that total protein phosphorylation could be quantitated by PhosphorImager. GRK5 phosphorylation in the presence of the WT PDGFRβ consisted of two components: GRK5 autophosphorylation (Premont et al., 1994) (Fig. 4, lane 5), and PDGFRβ-mediated tyrosyl phosphorylation of GRK5 (Fig. 1). Although PDGFRβ-mediated phosphorylation of GRK5 clearly enhanced the incorporation of 32P into GRK5, it did not alter GRK5 autophosphorylation, as assessed by phosphoserine IB (Fig. 4). Consequently, the amount of 32P incorporated into GRK5 by PDGFRβ-mediated phosphorylation could be obtained by subtracting GRK5 autophosphorylation from total GRK5 phosphorylation observed in the presence of the PDGFRβ. Using this approach, we found that the PDGFRβ tyrosine-phosphorylates GRK5 to a stoichiometry of 0.8 ± 0.2 mol phosphate/mol GRK5. Such a stoichiometry lends credence to the physiological importance of PDGFRβ-mediated tyrosyl phosphorylation of GRK5.
Fig. 4.

The PDGFRβ tyrosine phosphorylates GRK5 stoichiometrically. PDGFRβs were immunoprecipitated from HEK cells exposed to medium containing vehicle (-) or 2 nM PDGF-BB (+) for 10 min at 37°C. After IP, purified GRK5 was added to PDGFRβ immune complexes, and kinase assays proceeded with [γ-32P]ATP. Samples were resolved by SDS-PAGE and transferred to nitrocellulose, as described under Materials and Methods.A, 32P autoradiograms from a single experiment, representative of four performed, are presented at the top. Bottom, IBs performed on autoradiographed nitrocellulose were probed serially for phosphoserine (pSer), GRK5, and the PDGFRβ; images from a single experiment represent four performed. B, 32P band intensities in lanes from PDGF-stimulated cells were quantitated by PhosphorImager and divided by the cognate IB band densities for the PDGFRβ and GRK5. Reactions were performed with purified GRK5 or the PDGFRβ, in the absence (None) or presence of each other, as in A. Shown are the means ± S.E. of four independent experiments. Compared with values obtained in reactions with a single kinase: *, p < 0.05.
The PDGFRβ Phosphorylates GRK5 under Physiological Conditions. Thus far, data from purified protein preparations and transfected HEK cells have demonstrated that the PDGFRβ mediates tyrosyl phosphorylation and activation of GRK5, and that this activation is required for GRK5-mediated phosphorylation of the PDGFRβ but not the β2AR. To determine whether reciprocal phosphorylation of GRK5 and the PDGFRβ obtains under physiological conditions, we used vascular SMCs, because GRK5 mediates most of the PDGF-induced PDGFRβ seryl phosphorylation in these cells (Wu et al., 2006). Just as we observed in heterologous systems, PDGF treatment of SMCs induced not only tyrosyl phosphorylation of GRK5 and the PDGFRβ but also concomitant seryl phosphorylation of the PDGFRβ (Fig. 5A).
Fig. 5.

Tyrosyl phosphorylation of GRK5 in vascular SMCs results from activation of the PDGFRβ or the β2AR. A, primary mouse aortic SMCs were exposed to serum-free medium lacking (-) or containing (+) 2 nM PDGF-BB for 10 min (37°C). Cell lysates were then subjected to IP for GRK5 or the PDGFRβ, followed by SDS-PAGE and IB. GRK5 blots were probed serially for total GRK5 and phospho-Tyr (pY); PDGFRβ blots were probed for total PDGFRβ, pSer, and pTyr. Shown are results from a single experiment representative of four performed independently. B, SMCs were exposed to serum-free medium containing vehicle (-), PDGF-BB, or ISO as in Figs. 2 and 3, but in the presence or absence of 10 μM AG1295. Cell lysates were subjected to GRK5 IP, and blots were probed serially for total GRK5 and pTyr (top). To verify the efficacy of AG1295, the indicated SMC lysates were also subjected to SDS-PAGE and IB for phospho-PDGFRβ (pY1021) and β-tubulin (bottom). Shown are results from a single experiment representative of three performed.
Does tyrosyl phosphorylation of GRK5 occur consequent to activation of only the PDGFRβ receptor? Or does it occur consequent to activation of other receptors, too? Stimulation of various seven-transmembrane receptors (including β-adrenergic receptors) can engender the activation of Src, a nonreceptor tyrosine kinase that can phosphorylate GRK2 (Fan et al., 2001; Noma et al., 2007), but Src is not known to phosphorylate other GRKs. To test whether activation of the WT β2AR can trigger tyrosyl phosphorylation of GRK5, we stimulated endogenous SMC β2ARs with (-)isoproterenol. GRK5 immunoprecipitated from these cells indeed did demonstrate agonist-dependent tyrosyl phosphorylation—of a magnitude comparable with that observed when we stimulated the SMCs with PDGF (Fig. 5B). Unlike PDGF-induced tyrosyl phosphorylation of GRK5, however, the isoproterenol-stimulated GRK5 tyrosyl phosphorylation was not inhibited by the PDGFRβ tyrosine kinase inhibitor AG1295 (Fig. 5B and data not shown). Thus, the WT β2AR can promote tyrosyl phosphorylation of GRK5 through a kinase (or kinases) distinct from the PDGFRβ. Moreover, although PDGFRβ-mediated tyrosyl phosphorylation of GRK5 seems necessary for GRK5 activity on the PDGFRβ (Figs. 1 and 2), the functional significance of β2AR-promoted GRK5 tyrosyl phosphorylation remains uncertain (Fig. 3).
Although PDGF elicits tyrosyl phosphorylation of GRK5 under physiological conditions in SMCs (Fig. 5), it is not clear whether this tyrosyl phosphorylation in SMCs is mediated by the PDGFRβ itself or by other tyrosine kinase(s) (as is the case for β2AR-promoted GRK5 tyrosyl phosphorylation, Fig. 5B). We therefore sought to contrast the action on SMC GRK5 of the WT and the (phosphorylation-impaired) Y857F PDGFRβs, both of which activate Src equivalently (Wu et al., 2005). To compare equivalent levels of WT and Y857F PDGFRβs in SMCs, however, we needed to circumvent signaling by the endogenous SMC PDGFRβs. To do so, we transfected mouse SMCs with ChiRs composed of two major domains: 1) the transmembrane and cytoplasmic domains of the PDGFRβ (WT or Y857F); and 2) the extracellular domain of the CSF-1 receptor (Symes and Mercola, 1996). These ChiRs generate PDGFRβ-dependent signaling when they are stimulated (i.e., cross-linked) by CSF-1 (Symes and Mercola, 1996), to which the mock-transfected SMCs do not respond by tyrosine-phosphorylating cellular proteins (IB data not shown). Expression levels of these ChiRs in SMCs were less than that of the endogenous PDGFRβs, as determined by membrane IBs for the PDGFRβ cytoplasmic tail (Fig. 6A). Nonetheless, in response to CSF-1, GRK5 was tyrosine-phosphorylated in SMCs that expressed the ChiRs, and this tyrosyl phosphorylation was 2.2 ± 0.1-fold greater with the WT than with the Y857F ChiR (Fig. 6B, p < 0.05). Thus, both with purified protein preparations and in intact SMCs expressing physiological levels of receptor and GRK5, the PDGFRβ cytoplasmic domain itself affected tyrosyl phosphorylation of GRK5.
Fig. 6.

The PDGFRβ tyrosine-phosphorylates GRK5 in vascular SMCs. Mouse aortic SMCs were transfected with an N-terminal Flag-tagged WT or Y857F ChiR comprising the extracellular domain of the human CSF-1 receptor and the transmembrane and cytoplasmic domains of the human PDGFRβ. Cells were stimulated with 50 ng/ml human CSF-1 (Agonist) for 10 min at 37°C. Cell lysates were divided and subjected to IB for the PDGFRβ cytoplasmic domain, to identify the ChiR (A), or IP for either GRK5 or Flag, followed by SDS-PAGE and IB (B). GRK5 IPs were probed sequentially for GRK5 and phosphotyrosine (pY); ChiR IPs were probed for the PDGFRβ cytoplasmic domain. Shown are results from a single experiment representative of three performed.
To ascertain the stoichiometry of PDGFRβ-mediated GRK5 tyrosyl phosphorylation in intact SMCs, we compared GRK5 immunoprecipitated from PDGF-stimulated SMCs with purified GRK5 phosphorylated by the partially purified PDGFRβ. Tyrosyl phosphorylation of GRK5 in intact SMCs was 1.7 ± 0.2-fold greater than in purified protein preparations (Fig. 7, p < 0.05), perhaps because the pervanadate used on intact cells inhibits the action of phosphotyrosine phosphatases [even those that coimmunoprecipitate with the PDGFRβ (Wu et al., 2006), and may therefore affect our immune complex kinase assays]. Taken together, data from intact SMCs and purified protein preparations (Fig. 4) demonstrate the stoichiometry of PDGFRβ-mediated GRK5 tyrosyl phosphorylation to be ∼1 mol/mol.
Fig. 7.

The PDGFRβ phosphorylates GRK5 in SMCs to a stoichiometry greater than that obtained with purified proteins. GRK5 was immunoprecipitated from SMCs without or with PDGF stimulation, as described for Fig. 5A (lanes 1 and 2). Purified GRK5 was phosphorylated by immunoprecipitated PDGFRβs, as described for Fig. 4, except that only nonradioactive ATP was used (lane 3). All GRK5 species were subjected batch-wise to SDS-PAGE and IB; parallel blots were probed for pTyr and GRK5, as indicated. Shown are the results of a single experiment representative of four performed.
PDGFRβ and GRK5 Enzymatic Activities Are Affected Reciprocally by Cross-Phosphorylation. The catalytic activities of GRKs and receptor protein tyrosine kinases are most commonly evaluated in peptide phosphorylation assays, which isolate the effects on enzyme activity from the effects on protein/protein association (Premont et al., 1994; Baxter et al., 1998). Therefore, to determine whether PDGFRβ-mediated GRK5 tyrosyl phosphorylation enhances GRK5 enzymatic activity, and, conversely, whether GRK5-mediated seryl phosphorylation of the PDGFRβ diminishes PDGFRβ enzymatic activity, we used peptide phosphorylation assays with purified protein preparations (Premont et al., 1994; Baxter et al., 1998). GRK5-mediated peptide phosphorylation increased when GRK5 was tyrosyl-phosphorylated, either by the PDGFRβ immunoprecipitated from PDGF-stimulated cells (1.6 ± 0.2-fold) or by the PDGFRβ cytoplasmic domain purified from Sf9 cells (2.3 ± 0.4-fold) (Fig. 8). Thus, PDGFRβ-mediated phosphorylation of GRK5 seemed to increase GRK5 catalytic activity. In contrast, though, GRK5-mediated serine phosphorylation of the PDGFRβ decreased PDGFRβ enzymatic activity by 40 ± 10% (p < 0.05, Fig. 8). Thus, when they phosphorylate each other, or “cross-phosphorylate,” GRK5 and the PDGFRβ engender reciprocal changes in each other's enzymatic activity: GRK5 increases, and the PDGFRβ decreases.
Fig. 8.

Reciprocal regulation of PDGFRβ and GRK5 kinase activities manifests in purified protein preparations. Kinase assays were performed with [γ-32P]ATP as in Fig. 4, with either PDGFRβ substrate peptide or GRK5 substrate peptide for 25 min (30°C), as described under Materials and Methods. The tyrosine kinase was either PDGFRβs immunoprecipitated from PDGF-stimulated 293 cells (PDGFRβ), or the PDGFRβ cytoplasmic domain polypeptide purified from Sf9 cells (PDGFRβ cytodomain). Terminated reactions were spotted onto phosphocellulose and subjected to Cerenkov counting. We normalized cpm from each sample to those obtained with the cognate peptide substrate phosphorylated in the presence of only a single kinase (GRK5 or PDGFRβ); in this way, we obtained kinase activity as a percentage of control. Nonspecific cpm constituted 30 ± 9% of total cpm (see Materials and Methods). Plotted are the means ± S.E. from six independent experiments. Compared with phosphorylation obtained with either GRK5 or the PDGFRβ alone: *, p < 0.05.
Do these enzymatic effects observed with purified proteins also obtain in physiological systems? To address this question, we tested whether PDGFRβ catalytic activity was diminished when, as we have shown previously (Wu et al., 2006), the PDGFRβ is phosphorylated by GRK5 in intact SMCs. We immunoprecipitated PDGFRβs from congenic, PDGF-stimulated SMCs that were (+/+) or (-/-) at the grk5 locus. We then subjected these PDGFRβs to the same sort of peptide phosphorylation assays performed in Fig. 8. In accord with experiments using only purified proteins (Fig. 8), PDGFRβs demonstrated 26 ± 3% (p < 0.05) greater peptide phosphorylation when they were isolated from grk5(-/-) rather than grk5(+/+) SMCs (Fig. 9). Thus, the physiological activity of GRK5 in intact SMCs diminished PDGFRβ catalytic activity.
Fig. 9.

Endogenous GRK5 activity in SMCs reduces PDGFRβ kinase activity. Aortic SMCs of the indicated genotype were serum-starved, challenged with PDGF, solubilized, and then subjected to PDGFRβ IP and subsequent PDGFRβ substrate peptide phosphorylation assay as in Fig. 8. Peptide 32P cpm were normalized to those obtained with PDGF-stimulated WT SMCs, to obtain the percentage of control. Nonspecific cpm constituted 11 ± 6% of total cpm (see Materials and Methods). Plotted are the means ± S.E. from two independent experiments performed in triplicate using two independent pairs of WT and knockout (KO) SMC cell lines expressing equivalent amounts of the PDGFRβ (data not shown). Compared with cognate WT SMCs: *, p < 0.05.
PDGFRβ-Mediated GRK5 Activation Occurs in Noncaveolar Microdomains. GRK5 activity is inhibited when it binds the scaffolding domains of caveolin-1 and -3, but not caveolin-2 (Carman et al., 1999). Because PDGFRβs localize in caveolae after PDGF-induced activation (Matveev and Smart, 2002), we asked whether PDGFRβ-mediated GRK5 tyrosyl phosphorylation (and activation) might relieve caveolin-mediated GRK5 inhibition. We used the caveolin-1 scaffolding domain peptide to address this question, because the formation of caveolae in SMCs requires caveolin-1 (Hardin and Vallejo, 2006). Because PDGFRβ kinase activity, like GRK5, is inhibited by caveolin-1 (Yamamoto et al., 1999), we activated purified GRK5 with the PDGFRβ before testing the effects of the caveolin-1 scaffolding domain on GRK5 activity. GRK5 was separated from the PDGFRβ and then tested for substrate peptide phosphorylation in the absence or presence of the caveolin-1 scaffolding domain. As demonstrated by Carman et al. (1999), the caveolin-1 scaffolding domain peptide inhibited GRK5-mediated peptide phosphorylation by 60 ± 10%, whereas the caveolin-2 scaffolding domain peptide did not (Fig. 10 and data not shown). Furthermore, although PDGFRβ-mediated GRK5 phosphorylation enhanced GRK5 activity by 100 ± 20%, the presence of the caveolin-1 scaffolding domain abrogated this enhancement of GRK5 activity (Fig. 10). Thus, PDGFRβ-mediated tyrosyl phosphorylation of GRK5 does not suffice to overcome inhibition of GRK5 by caveolin-1. Consequently, in light of intact cell data demonstrating that PDGFRβ-mediated tyrosyl phosphorylation of GRK5 enhances GRK5 activity on the PDGFRβ (Fig. 2), these peptide data suggest that the PDGFRβ augments GRK5 activity at an intracellular site (or sites) outside of the caveola.
Fig. 10.

Caveolin-1 abrogates the increase in GRK5 activity resulting from PDGFRβ-mediated tyrosyl phosphorylation. GRK5 substrate peptide phosphorylation assays were performed as in Fig. 8, but in two sequential stages: 1) nonradioactive phosphorylation, in which GRK5 was incubated with immune complexes from PDGFRβ-expressing or -deficient (control) HEK cells; and 2) [γ-32P]ATP phosphorylation, for which GRK5 was separated from immune complexes before combining with GRK5 substrate peptide, in the presence or absence of caveolin-1 (or -2) scaffolding domain peptides, as described under Materials and Methods. GRK5 substrate peptide cpm were normalized to those obtained with GRK5 that had not been prephosphorylated by the PDGFRβ and was not incubated with caveolin peptides (control), to obtain the percentage of control. Shown are the means ± S.E. from four independent experiments. Compared with control: *, p < 0.05. Compared with the cognate reaction conducted without caveolin-1 peptide: †, p < 0.05.
PDGFRβ-Mediated Phosphorylation Augments GRK5 Vmax. To determine mechanisms by which PDGFRβ-mediated phosphorylation increases GRK5 catalytic activity in the absence of the caveolin-1 scaffolding domain, we performed kinetic analyses. The Vmax of GRK5 peptide phosphorylation increased 3.4 ± 0.7-fold (p < 0.05), from 0.5 ± 0.1 to 1.6 ± 0.4 nmol/mg/min, after GRK5 was phosphorylated by the PDGFRβ (Fig. 11). This augmentation in Vmax occurred in the absence of any significant change in GRK5's KM for substrate peptide (0.5 ± 0.1 versus 0.4 ± 0.1 mM, in the presence and absence of the PDGFRβ, respectively) (Fig. 11).
Fig. 11.

GRK5 Vmax value is enhanced by PDGFRβ-mediated tyrosyl phosphorylation of GRK5. Peptide phosphorylation assays were performed (15 min, 30°C) using purified GRK5 (50 nM) and the indicated concentrations of GRK5 substrate peptide in the presence of M2-agarose immunoprecipitates from HEK cells lacking (control, None) or expressing N-terminal Flag-tagged PDGFRβs. Peptide phosphorylation was determined as in Fig. 8, and values were normalized to the maximal kinase-specific 32P counts in the presence of the PDGFRβ (percentage of maximum). Nonspecific CPMs constituted 28 ± 7% of total CPMs (see Materials and Methods). Plotted are the mean peptide phosphorylation values obtained from three independent experiments. Compared with control curve: *, p < 0.01 (two-way analysis of variance).
Discussion
This study identifies GRK5 as a novel substrate for the PDGFRβ, both in intact cells and in preparations of purified proteins. PDGFRβ-mediated phosphorylation significantly enhanced GRK5's Vmax, assessed as seryl phosphorylation of either the PDGFRβ or a model peptide substrate. By phosphorylating and thereby activating GRK5, the PDGFRβ triggers GRK5-mediated PDGFRβ phosphorylation that results in PDGFRβ deactivation, which manifests in this study as diminished PDGFRβ catalytic activity—a previously unappreciated mechanism for GRK5-mediated PDGFRβ desensitization (Wu et al., 2006). PDGFRβ-mediated tyrosyl phosphorylation of GRK5 seems necessary for GRK5 activity on the PDGFRβ, even though tyrosyl phosphorylation of GRK5 seems unnecessary for GRK5 activity on the β2AR. Thus, the PDGFRβ phosphorylates GRK5, and GRK5 subsequently phosphorylates the PDGFRβ in a receptor-specific reciprocal feedback loop that regulates the activities of both kinases.
The reciprocity of regulation between the PDGFRβ and GRK5, or their activation/deactivation cycle, mirrors other reciprocal regulatory mechanisms affecting not only the PDGFRβ but also other receptor protein tyrosine kinases. Although the PDGFRβ and EGFR activate the phosphatase Shp2 directly and indirectly, respectively (Neel et al., 2003), Shp2 deactivates specific receptor-triggered signaling pathways by dephosphorylating the PDGFRβ (Klinghoffer and Kazlauskas, 1995; Wu et al., 2006) or the EGFR-phosphorylated adaptor protein Gab1 (Zhang et al., 2002). In particular cell lines, the EGFR (Countaway et al., 1992) and insulin receptor (Kayali et al., 1998) activate phospholipase Cγ, consequently elevate intracellular [Ca2+] and activate calcium/calmodulin kinase II and protein kinase C isoforms, which subsequently phosphorylate and deactivate the EGFR (Countaway et al., 1992) and insulin receptor (Takayama et al., 1988), respectively.
The dependence of GRK5 activity on tyrosyl phosphorylation is illustrated both by studies using a phosphorylation-deficient PDGFRβ mutant and studies using a GRK5 mutant lacking four target sites phosphorylated by the PDGFRβ. In this regard, GRK5 activation seems quite similar to GRK2 activation, which also requires tyrosyl phosphorylation by either the PDGFRβ (Wu et al., 2005) or Src (Fan et al., 2001). However, PDGFRβ-mediated activation of GRK5 may involve phosphorylation-dependent intramolecular interactions both distinct from and shared with GRK2. Approximately 50% of PDGFRβ-mediated GRK2 tyrosyl phosphorylation occurs on the three tyrosyl residues phosphorylated by c-Src (Wu et al., 2005). Of these residues, the two most N terminal have no homologs in GRK5; phosphorylation of these GRK2 residues (Tyr13 and Tyr86) could conceivably affect the interaction between the N terminus of the GRK2 RGS homology domain and the GRK2 pleckstrin homology domain—the latter of which is lacking in GRK5 (Lodowski et al., 2006). The third GRK2 tyrosyl residue phosphorylated by the PDGFRβ is conserved in GRK5 (Tyr90), and, according to the GRK2 and GRK6 crystal structures, lies immediately adjacent to the RGS homology domain region that interfaces with the catalytic domain large lobe (Lodowski et al., 2006). Consequently, phosphorylation at Tyr90 could engender activation of both GRK2 and GRK5 by altering the intramolecular interactions that, in the absence of allosteric activation, maintain the GRK catalytic domain in an “open,” and therefore inactive, conformation (Lodowski et al., 2006). Such a mechanism could explain our observation that PDGFRβ-mediated phosphorylation of GRK5 increases GRK5's Vmax in peptide phosphorylation assays. Alternatively, phosphorylation of Tyr90 in GRK5 may disrupt the homodimerization that is presumed to occur between members of the GRK5 (but not GRK2) family of GRKs (Lodowski et al., 2006). There are GRK2 homologs for two of the three remaining GRK5 tyrosyl residues we identified as PDGFRβ targets (Tyr109 and Tyr309). It is entirely possible that these sites are also PDGFRβ-phosphorylated in GRK2, because the known GRK2 target sites for PDGFRβ-mediated phosphorylation account for only ∼50% of the total PDGFRβ-mediated phosphorylation (Wu et al., 2005).
In intact SMCs and HEK cells, GRK5 activity enhances PDGF-induced association of the PDGFRβ with Shp2, a phosphatase that deactivates PDGFRβ signaling, in part (Wu et al., 2006). Knockdown of Shp2 attenuates GRK5-mediated desensitization of PDGFRβ/PLCγ signaling; therefore, Shp2 seems a likely effector of GRK5-initiated PDGFRβ desensitization (Wu et al., 2006). However, our current work adds another dimension to our evolving understanding of GRK5-mediated PDGFRβ desensitization, by demonstrating that GRK5-mediated phosphorylation of the PDGFRβ directly reduces the catalytic activity of the PDGFRβ on even a peptide substrate. In this way, the effects of GRK5 on the PDGFRβ mirror the effects of GRKs on heptahelical receptors' ability to activate heterotrimeric G proteins: GRK-mediated receptor phosphorylation impairs receptor-mediated G protein activation somewhat, even in preparations of purified proteins, and results in recruitment to the receptors of the accessory β-arrestin proteins, which severely reduce receptor-mediated G protein activation (Pitcher et al., 1998).
The failure of PDGFRβ-mediated GRK5 phosphorylation to relieve caveolin-1-mediated GRK5 inhibition should inform our models for PDGFRβ and GRK5 regulation. PDGF-activated PDGFRβs on the plasma membrane would seem most likely to tyrosine-phosphorylate and activate GRK5 (which resides predominantly on the plasma membrane) (Premont and Gainetdinov, 2007), before the PDGFRβs translocate to caveolae (Matveev and Smart, 2002)—where both the PDGFRβs (Yamamoto et al., 1999) and GRK5 (Carman et al., 1999) are inhibited by caveolin-1. Alternatively, activated PDGFRβs could also phosphorylate and activate GRK5 after they translocate to clathrin-coated pits (Nilsson et al., 1983), or even after internalization in coated vesicles (Kapeller et al., 1993)—because PDGFRβs continue to signal intracellularly (Wang et al., 2004). It is in these noncaveolar compartments, devoid of caveolin-1 (Nabi and Le, 2003), that GRK5-mediated phosphorylation of the PDGFRβ would presumably have the greatest effect on PDGFRβ signal transduction.
We have demonstrated that tyrosyl phosphorylation of GRK5 occurs after activation of not only the PDGFRβ but also the β2AR. It is remarkable that agonist-induced tyrosyl phosphorylation of GRK5 seems to have receptor-dependent, functionally distinct consequences for GRK5 activity. Tyrosyl phosphorylation of GRK5 mediated by the PDGFRβ is required for GRK5 activity on the PDGFRβ, but β2AR-promoted tyrosyl phosphorylation of GRK5—mediated by a kinase distinct from the PDGFRβ—is clearly not required for GRK5 activity on the β2AR, at least under the conditions tested in our studies. This distinction may result, in part, from the superior efficacy of the β2AR (compared with the PDGFRβ) in GRK5 allosteric activation, which may obviate tyrosyl phosphorylation-dependent GRK5 activation. Nonetheless, the increase in GRK5 Vmax effected by PDGFRβ-mediated phosphorylation could be expected to enhance GRK5 activity broadly, on substrates including 7-transmembrane receptors. It remains to be determined how GRK5 function is affected when GRK5 is tyrosine-phosphorylated after seven-transmembrane receptor activation.
Supplementary Material
Acknowledgments
We thank W. Darrell Capel for help with anti-GRK5 antiserum.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL77185, HL73005, HL80525]; the National Institutes of Health National Institute on Drug Abuse [Grant DA016347]; and the American Heart Association, Mid-Atlantic Affiliate [Grant-in-Aid 0655464U and Postdoctoral Fellowship 0625403U].
ABBREVIATIONS: PDGFRβ, platelet-derived growth factor receptor-β; β2AR, β2-adrenergic receptor; ChiR, chimeric colony stimulating factor-1/platelet-derived growth factor-β receptor; CSF-1, colony stimulating factor-1; EGFR, epidermal growth factor receptor; GRK, (heterotrimeric) G protein-coupled receptor kinase; IB, immunoblot; IP, immunoprecipitation; ISO, (-)isoproterenol; PDGF, platelet-derived growth factor; SMC, smooth muscle cell; WT, wild-type; Y4F, G protein-coupled receptor kinase-5 mutant with four tyrosine-to-phenylalanine point mutations; PCR, polymerase chain reaction; HEK, human embryonic kidney; PAGE, polyacrylamide gel electrophoresis; AG1295, 6,7-dimethyl-2-phenylquinoxaline.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
References
- Baxter RM, Secrist JP, Vaillancourt RR, and Kazlauskas A (1998) Full activation of the platelet-derived growth factor β-receptor kinase involves multiple events. J Biol Chem 273 17050-17055. [DOI] [PubMed] [Google Scholar]
- Bioukar EB, Marricco NC, Zuo D, and Larose L (1999) Serine phosphorylation of the ligand-activated β-platelet-derived growth factor receptor by casein kinase I-γ2 inhibits the receptor's autophosphorylating activity. J Biol Chem 274 21457-21463. [DOI] [PubMed] [Google Scholar]
- Cai X, Zhang K, and Lytton J (2002) A novel topology and redox regulation of the rat brain K+-dependent Na+/Ca2+ exchanger, NCKX2. J Biol Chem 277 48923-48930. [DOI] [PubMed] [Google Scholar]
- Carman CV, Lisanti MP, and Benovic JL (1999) Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem 274 8858-8864. [DOI] [PubMed] [Google Scholar]
- Countaway JL, Nairn AC, and Davis RJ (1992) Mechanism of desensitization of the epidermal growth factor receptor protein-tyrosine kinase. J Biol Chem 267 1129-1140. [PubMed] [Google Scholar]
- DeWire SM, Ahn S, Lefkowitz RJ, and Shenoy SK (2007) β-Arrestins and cell signaling. Annu Rev Physiol 69 483-510. [DOI] [PubMed] [Google Scholar]
- Fan G, Shumay E, Malbon CC, and Wang H (2001) c-Src tyrosine kinase binds the β2-adrenergic receptor via phospho-Tyr-350, phosphorylates G-protein-linked receptor kinase 2, and mediates agonist-induced receptor desensitization. J Biol Chem 276 13240-13247. [DOI] [PubMed] [Google Scholar]
- Fredericks ZL, Pitcher JA, and Lefkowitz RJ (1996) Identification of the G protein-coupled receptor kinase phosphorylation sites in the human β2-adrenergic receptor. J Biol Chem 271 13796-13803. [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Kim LK, Murray JP, Exum ST, Brian L, Wu JH, and Peppel K (2002) Phosphorylation of the platelet-derived growth factor receptor-β and epidermal growth factor receptor by G protein-coupled receptor kinase-2. Mechanisms for selectivity of desensitization. J Biol Chem 277 48261-48269. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Bohn LM, Walker JK, Laporte SA, Macrae AD, Caron MG, Lefkowitz RJ, and Premont RT (1999) Muscarinic supersensitivity and impaired receptor desensitization in G protein-coupled receptor kinase 5-deficient mice. Neuron 24 1029-1036. [DOI] [PubMed] [Google Scholar]
- Hardin CD and Vallejo J (2006) Caveolins in vascular smooth muscle: form organizing function. Cardiovasc Res 69 808-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH and Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79 1283-1316. [DOI] [PubMed] [Google Scholar]
- Hildreth KL, Wu JH, Barak LS, Exum ST, Kim LK, Peppel K, and Freedman NJ (2004) Phosphorylation of the platelet-derived growth factor receptor-β by G protein-coupled receptor kinase-2 reduces receptor signaling and interaction with the Na+/H+ exchanger regulatory factor. J Biol Chem 279 41775-41782. [DOI] [PubMed] [Google Scholar]
- Hu LA, Chen W, Premont RT, Cong M, and Lefkowitz RJ (2002) G protein-coupled receptor kinase 5 regulates β1-adrenergic receptor association with PSD-95. J Biol Chem 277 1607-1613. [DOI] [PubMed] [Google Scholar]
- Iwata K, Luo J, Penn RB, and Benovic JL (2005) Bimodal regulation of the human H1 histamine receptor by G protein-coupled receptor kinase-2. J Biol Chem 280 2197-2204. [DOI] [PubMed] [Google Scholar]
- Kapeller R, Chakrabarti R, Cantley L, Fay F, and Corvera S (1993) Internalization of activated platelet-derived growth factor receptor-phosphatidylinositol-3′ kinase complexes: potential interactions with the microtubule cytoskeleton. Mol Cell Biol 13 6052-6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayali AG, Eichhorn J, Haruta T, Morris AJ, Nelson JG, Vollenweider P, Olefsky JM, and Webster NJ (1998) Association of the insulin receptor with phospholipase C-γ (PLCγ) in 3T3-L1 adipocytes suggests a role for PLCγ in metabolic signaling by insulin. J Biol Chem 273 13808-13818. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA and Kazlauskas A (1995) Identification of a putative Syp substrate, the PDGF β receptor. J Biol Chem 270 22208-22217. [DOI] [PubMed] [Google Scholar]
- Lodowski DT, Tesmer VM, Benovic JL, and Tesmer JJ (2006) The structure of G protein-coupled receptor kinase (GRK)-6 defines a second lineage of GRKs. J Biol Chem 281 16785-16793. [DOI] [PubMed] [Google Scholar]
- Matveev SV and Smart EJ (2002) Heterologous desensitization of EGF receptors and PDGF receptors by sequestration in caveolae. Am J Physiol Cell Physiol 282 C935-C946. [DOI] [PubMed] [Google Scholar]
- Nabi IR and Le PU (2003) Caveolae/raft-dependent endocytosis. J Cell Biol 161 673-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel BG, Gu H, and Pao L (2003) The `Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci 28 284-293. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Thyberg J, Heldin CH, Westermark B, and Wasteson A (1983) Surface binding and internalization of platelet-derived growth factor in human fibroblasts. Proc Natl Acad Sci U S A 80 5592-5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, et al. (2007) Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 117 2445-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann M, Diversé-Pierluissi M, Drazner MH, Dyer SL, Freedman NJ, Peppel KC, and Lefkowitz RJ (1996) Monoclonal antibodies reveal receptor specificity among G-protein-coupled receptor kinases. Proc Natl Acad Sci U S A 93 7649-7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Elorza A, Sarnago S, and Mayor F Jr (2001) Beta-arrestin- and c-Src-dependent degradation of G-protein-coupled receptor kinase 2. EMBO J 20 5129-5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, and Lefkowitz RJ (1998) G protein-coupled receptor kinases. Annu Rev Biochem 67 653-692. [DOI] [PubMed] [Google Scholar]
- Premont RT and Gainetdinov RR (2007) Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol 69 511-534. [DOI] [PubMed] [Google Scholar]
- Premont RT, Koch WJ, Inglese J, and Lefkowitz RJ (1994) Identification, purification, and characterization of GRK5, a member of the family of G protein-coupled receptor kinases. J Biol Chem 269 6832-6841. [PubMed] [Google Scholar]
- Pronin AN and Benovic JL (1997) Regulation of the G protein-coupled receptor kinase GRK5 by protein kinase C. J Biol Chem 272 3806-3812. [DOI] [PubMed] [Google Scholar]
- Sarnago S, Elorza A, and Mayor F Jr (1999) Agonist-dependent phosphorylation of the G protein-coupled receptor kinase 2 (GRK2) by Src tyrosine kinase. J Biol Chem 274 34411-34416. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, and Lefkowitz RJ (2006) β-arrestin-dependent, G protein-independent ERK1/2 activation by the β2-adrenergic receptor. J Biol Chem 281 1261-1273. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Carraway KL 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, and Eng C (1995) Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature 373 536-539. [DOI] [PubMed] [Google Scholar]
- Symes K and Mercola M (1996) Embryonic mesoderm cells spread in response to platelet-derived growth factor and signaling by phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A 93 9641-9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S, White MF, and Kahn CR (1988) Phorbol ester-induced serine phosphorylation of the insulin receptor decreases its tyrosine kinase activity. J Biol Chem 263 3440-3447. [PubMed] [Google Scholar]
- Wang Y, Pennock SD, Chen X, Kazlauskas A, and Wang Z (2004) Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J Biol Chem 279 8038-8046. [DOI] [PubMed] [Google Scholar]
- Wu JH, Goswami R, Cai X, Exum ST, Huang X, Zhang L, Brian L, Premont RT, Peppel K, and Freedman NJ (2006) Regulation of the platelet-derived growth factor receptor-β by G protein-coupled receptor kinase-5 in vascular smooth muscle cells involves the phosphatase Shp2. J Biol Chem 281 37758-37772. [DOI] [PubMed] [Google Scholar]
- Wu JH, Goswami R, Kim LK, Miller WE, Peppel K, and Freedman NJ (2005) The platelet-derived growth factor receptor-β phosphorylates and activates G protein-coupled receptor kinase-2. A mechanism for feedback inhibition. J Biol Chem 280 31027-31035. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Toya Y, Jensen RA, and Ishikawa Y (1999) Caveolin is an inhibitor of platelet-derived growth factor receptor signaling. Exp Cell Res 247 380-388. [DOI] [PubMed] [Google Scholar]
- Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, and Neel BG (2002) Receptor-specific regulation of phosphatidylinositol 3′-kinase activation by the protein tyrosine phosphatase Shp2. Mol Cell Biol 22 4062-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}