

Abstract

In the Rh2(OAc)4-catalyzed amidoglycosylation of glucal 3-carbamates, anomeric stereoselectivity and the extent of competing C3–H oxidation depend on the 4O and 6O protecting groups. Acyclic protection permits high α-anomer selectivity with further improvement in less polar solvents, while electron-withdrawing protecting groups limit C3-oxidized byproducts. Stereocontrol and bifurcation between alkene insertion and C3–H oxidation reflect an interplay of conformational, stereoelectronic, and inductive factors.
2-Amino sugars having a 2,3-cis stereo array include N-acetylmannosamine (ManNAc, 1), which is the biosynthetic precursor of the sialic acids,1 and 2-allosamine, a constituent of the potent chitinase inhibitor allosamidin (2)2 and a useful ligand scaffold (3)3 for asymmetric catalysis. The challenge of stereoselective C2–N bond construction is acute in these systems, and control of anomeric configuration in the preparation of glycoside derivatives is desirable. Synthetic methods based on intermolecular additions to glycals typically place the C2-N group trans to the C3-oxygen substituent.4 Gin’s activated-sulfoxide-mediated acetamidoglycosylation5 of glucals is an exception, producing ManNAc structures, though with N-acetylglucosamine (GlcNAc) byproducts.5c
As an alternative,6 we have used intramolecular nitrogen atom delivery from allal 3-azidoformates,7 allal 3-carbamates,8 and glucal 3-carbamates9 to establish the 2,3-cis relationship. With the 3O-carbamoyl glycals, we extended Du Bois’s C–H amidation method10 to alkene insertion,11,12 a new reaction of allylic carbamates.13 Mechanistic studies14 imply that these conditions produce rhodium nitrenoids having reactivity strikingly analogous to metal carbenoids.15 With iodosobenzene (PhIO)16 instead of PhI(OAc)2 as the oxidant, we achieved in situ glycosylation of alcohols without nucleophilic competition from acetate, an overall amidoglycosylation process.8,9,17
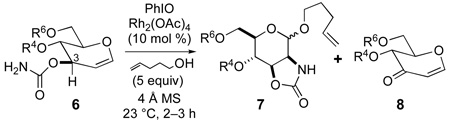
Allal frameworks (e.g., 4, Scheme 1) provided high 1,2-trans selectivity, offering a concise route to β-linked 2-amido allopyranosides as found in allosamidin.7,8 However, in the C3-epimeric series, our one-pot amidoglycosylation process applied to glucal 3-carbamates 6a and 6b, having 4O,6O acetonide or di-tert-butylsilylene protection, gave anomeric mixtures only slighly favoring the 1,2-trans products 7-α and also generated dihydropyranone byproducts 8a and 8b via oxidation at the C3–H bond (Table 1, entries 1 and 5).9 Using 4-penten-1-ol as the acceptor, we were able to stereoconvergently advance either anomer of n-pentenyl glycoside18 7a, but the lack of amidoglycosylation selectivity stymied direct access to α-linked ManNAc derivatives.9
Scheme 1.

β-Selective allal 3-carbamate amidoglycosylation
Table 1.
Protecting group and solvent effects on stereo- and chemoselectivity of glucal 3-carbamate amidoglycosylation
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R6 | R4 | 6 | JH4,H5 in 6a | rxn solvent | 7:8b (%7-α)c | α:βb7 |
| 1d | -CMe2- | a | 9.9e | CH2Cl2 | 3.1:1(40) | 1.3:1 | |
| 2 | a | PhH | 4.3:1(32f) | 1.3:1 | |||
| 3 | a | Hex/PhH 5/1 |
4.9:1(38f) | 2.4:1 | |||
| 4 | a | Hex/PhH 10/1 |
4.7:1(34) | 2.7:1 | |||
| 5d | -SitBu2- | b | 10.0 | CH2Cl2 | 3.2:1 (33) | 2.2:1 | |
| 6 | b | PhH | 3.5:1 (29) | 1.7:1 | |||
| 7 | b | Hex | 3.7:1 (43) | 3.6:1 | |||
| 8 | Bn | Bn | c | 8.8 | CH2Cl2 | 2.9:1 (61) | 12:1 |
| 9 | c | PhH | 2.3:1 (47) | α only | |||
| 10 | c | CH2Cl2/Hex 1/1 | 3.0:1 (54) | 22:1 | |||
| 11 | c | CH2Cl2/PhH 1/1 | 3.0:1 (56) | 23:1 | |||
| 12 | Ac | Ac | d | 7.5 | CH2Cl2 | 15:1 (69) | α only |
| 13 | Ts | Ac | e | 6.7g | CH2Cl2 | 14:1 (69) | α only |
| 14 | -CO- | f | 10.4h | CH2Cl2 | 2.4:1 (32) | 2.9:1 | |
| 15 | TBS | Ac | g | 7.9 | CH2Cl2 | 6.6:1 (68) | 25:1 |
| 16 | Ts | TBS | h | 9.0 | CH2Cl2 | 8.1:1 (67) | 12:1 |
| 17 | TBS | TBS | i | 9.4 | CH2Cl2 | 2.4:1 (46i) | 5.8:1 |
In Hz, CDCl3 unless otherwise noted.
From 1H and 13C NMR of the crude reaction mixture.
Isolated yield of the α anomer.
Data from ref 9.
In DMSO-d6.
NMR yield of the α anomer vs mesitylene as an internal standard.
In C6D6.
In acetone-d6.
Calcd from the total yield (54%) of inseparable anomeric mixture.
Herein we report that proper choice of 4O and 6O protecting groups and solvent enables high levels of stereocontrol and chemoselectivity in amidoglycosylation of glucal 3-carbamates. Our studies also illuminate electronic and conformational aspects of both amidoglycosylation and the competing C3–H oxidation.
For comparison, we began by treating benzylidene-protected allal 3-carbamate 48 under our standard conditions with 4-penten-1-ol as the acceptor (Scheme 1). As in our previous study with other acceptors,8 only β product 5, unaccompanied by dihydropyranone, was observed. The outcome was comparable in the three solvents tested.
By analogy with our earlier results7,8 and Padwa’s studies,11 we attribute high 1,2-trans selectivity to nucleophilic opening of a glycosyl aziridine.4c,17,19 In the glucal series, low anomeric stereocontrol might be due to glycosylation via an oxocarbenium intermediate. Padwa has invoked aziridine-opened zwitterions in reactions of indolyl and benzofuranyl carbamates.11
We evaluated the effect of solvent polarity on anomeric stereoselectivity, beginning with acetonide- and di-tert-butylsilylene-protected glucal 3-carbamates 6a and 6b (Table 1, entries 1–7). There was a modest trend toward higher α selectivity in less polar solvents, which we reasoned favored assistance from the C2 nitrogen versus an oxocarbenium donor. However, with the cyclic 4O,6O protection in 6a and 6b, maintaining N–anomeric contact in the donor entails significant strain.
To better enable an α-selective aziridine donor (C, Figure 2), we removed the stricture of cyclic 4O,6O protection. Dibenzyl-protected 6c20 provided high to complete α selectivity, depending on solvent (Table 1, entries 8–11). In benzene, we detected only the α anomer, but the yield was lowered and we noted chloroform-insoluble material in the crude product that was not present when the reaction solvent was CH2Cl2. Mixed solvents with a less polar component increased α selectivity while better maintaining yields relative to CH2Cl2 alone.
Figure 2.

Model for factors affecting anomeric stereoselectivity and extent of C3–H oxidation
Unfortunately, the dibenzyl protection in 6c did not remedy the chemoselectivity problem. Our mechanistic model (Figure 2) is that amidoglycosylation and C3 oxidation both occur via rhodium nitrenoid conformers A/A’. Du Bois has reported ketone formation in reactions of secondary-alcohol-derived carbamates and tentatively ascribed them to C–H insertion at the α position, followed by fragmentation of the resulting four-membered-ring carbamate.21 In the carbenoid field, Doyle found that the diazoacetate ester of 1-indanol provides 1-indanone and ketene upon treatment with dirhodium(II) catalysts.22 The proposed mechanism involves intramolecular hydride transfer to the rhodium acyl carbenoid.22,23 Another possible path to byproduct 8, involving initial reaction of the enol ether π bond with the hypervalent iodine oxidant, is not consistent with our control experiments.24
The extent of the unwanted oxidation should depend on the electronic characteristics of the C3–H bond. As was found with rhodium carbenoids,25 C–H insertion of rhodium nitrenoids is most favorable for electron-rich C–H bonds.14 Insertion α to oxygen is facile,26 and Parker’s group has described activation of allylic C–H bonds vinylogously α to the ring oxygen of glycal substrates.27 Compain suggested that an anomeric C–H bond is more reactive toward nitrenoid insertion in an axial rather than equatorial position,28 an effect recognized in carbenoid reactions.29 In our glucal 3-carbamate systems, a vinylogous anomeric effect30 may activate the pseudo-axial C3–H bond for insertion (A→8). By contrast, in allal 3-carbamate 4 or in glucal conformer A’ the C3–H bond has the pseudo-equatorial orientation.
Our analysis prompted two substrate-controlled31 approaches for minimizing C3–H oxidation. First, electron-withdrawing 4O and 6O protecting groups might inductively deactivate the C3–H bond toward nitrenoid insertion. Second, shifting the conformational equilibrium from all-equatorial 4H5 (A) toward inverted 5H4 (A’) would diminish stereoelectronic priming of the C3–H bond. The conformational preference in glucals is sensitive to hydroxyl protection,32 and the 4H5/5H4 distribution can affect the stereoselectivity of addition reactions at the glycal alkene.32b Electron-withdrawing substituents at C6, including Br and OTs, along with 3O-ester protection, favor the inverted 5H4 conformation.32 This raised the possibility of simultaneous inductive and conformational deactivation of the C3–H bond, diminishing the formation of byproduct 8. These effects could also improve stereocontrol by stabilizing aziridine donor C relative to B/B’ (half-chair conformers lacking the stereocontrolling element of covalent33 N–C1 attachment) and by favoring the conformation required for forming the aziridine directly (A’→C) without intervention of zwitterion B/B’.
To probe these influences, we synthesized a range of glucal 3-carbamates 6d–i20 for comparison with 6a–c. Analysis of the crude reaction mixtures by 1H and 13C NMR determined anomeric stereoselectivity in products 7 and the proportion of byproducts 8 (see Table 1).34 To estimate conformational effects on the reactivity of the derived rhodium nitrenoids (A/A’), we determined JH4,H5 for each glucal 3-carbamate 6. In various instances, second-order behavior in the 1H NMR spectra (up to 500 MHz) required coupled HSQC techniques for determining JH4,H5.35 Decreasing values of JH4,H5 indicate an increasing proportion of conformationally inverted A’, having the equatorial-equatorial H4–H5 relationship.32
With 4,6-di-O-acetyl-protected 6d and 4-O-acetyl-6-O-tosyl derivative 6e (Table 1, entries 12 and 13), much less of the C3–H-oxidized byproducts formed, compared to dibenzyl-protected 6c. In addition, we detected only the α anomers of 7d and 7e from reactions in CH2Cl2. From the JH4,H5 values, 6d and 6e are both weighted more toward the 5H4 conformation than the dibenzyl-protected 6c.
To better understand conformational and inductive contributions to stereo- and chemoselectivity, we used 6f, having the cyclic-carbonate-locked 4H5 conformation (Table 1, entry 14). Despite the electron-withdrawing character of the 4,6-O-carbonate protection, 6f led to considerable byproduct 8f and a mixture of anomers, supporting conformational flexibility as a requirement for high chemo- and stereoselection.
Just one electron-withdrawing group at either 4O or 6O, as in 6g and 6h (Table 1, entries 15 and 16), gave better chemoselectivity than either the dibenzyl- or the disilyl-protected carbamates 6c or 6i (cf. entries 8 and 17). The 4O-acetyl group in 6g engendered a higher 5H4 proportion and excellent anomeric selectivity, but was comparable to the 6O-tosyl of 6h in enforcing chemoselection.36 The large silyl groups in 6i (Table 1, entry 17) led to decreased anomeric selectivity relative to the dibenzyl-protected 6c.
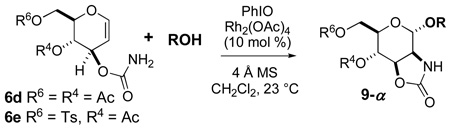
Using stereo- and chemoselective substrates 6d and 6e, we have begun studying reactions with various acceptor alcohols (Table 2). Reducing the amount of acceptor posed a challenge since reactions became sluggish and yields were lower (cf. entries 1 and 2). Reasoning that excess alcohol might be needed to activate CH2Cl2-insoluble iodosobenzene,37 we tested hexafluoroisopropanol (HFIP), a useful solvent in reactions of hypervalent iodine oxidants,38 as an additive (entries 3 and 8). Although yields did not improve much, consumption of starting material was considerably faster with HFIP added, and the weakly nucleophilic additive did not compete as the acceptor. Increasing the concentration of substrate 6 was also helpful, although 0.2 M is close to the upper limit for this initially heterogeneous reaction.
Table 2.
Amidoglycosylation with various acceptor alcohols
 | ||||||
|---|---|---|---|---|---|---|
| entry | 6 | [6] (M) | ROH (equiv) | equiv HFIPa | 9-α(%)b | α:βc |
| 1 | 6d | 0.090 | none | 9da (58) | >20:1 | |
| 2 | 6d | 0.090 | a (2.0) | none | 9da (40) | 12:1 |
| 3 | 6d | 0.091 | a (2.0) | 3.0 | 9da (41) | 7.1:1 |
| 4 | 6d | 0.21 | a (2.0) | none | 9da (47) | 13:1 |
| 5 | 6e | 0.089 | 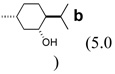 |
none | 9eb (51) | 12:1 |
| 6 | 6e | 0.19 | b (2.1) | none | 9eb (40) | 9.3:1 |
| 7d | 6e | 0.19 | 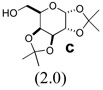 |
none | 9ec (41) | -e- |
| 8 | 6e | 0.19 | c (2.0) | 1.0 | 9ec (44) | -e- |
1,1,1,3,3,3-Hexafluoroisopropanol.
Isolated yield of the α anomer, corrected in entries 1–6 for small amounts of chromatographically inseparable C1–C2 oxidative cleavage byproduct (see Supporting Information for details).
From 1H NMR of the crude reaction mixture.
Starting carbamate 6e (15%) remained.
β Anomer was not detected.
Stereo- and chemoselectivity in the amidoglycosylation of glucal 3-carbamates reflects a confluence of inductive, conformational, and stereoelectronic effects. Acyclic 4O,6O protection enables high anomeric stereocontrol. Electron-withdrawing, 5H4-favoring 4O and 6O groups suppress C3–H oxidation and further enhance stereoselectivity. The net result is direct access to variably protected 2N,3O mannosamine oxazolidinones from glucal 3-carbamates. Studies with other glycal stereoisomers and optimization with a range of acceptors are underway.
Supplementary Material
Experimental details and compound characterization, including preparation of carbamates 6, determination of JH3,H4 values, and copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

2-Amino sugars with cis-2,3 stereochemistry
Acknowledgment
Dedicated to Prof. Les Lessinger (Barnard College) on the occasion of his retirement and Prof. David R. Williams (Indiana University) on his 60th birthday. We thank the NIH (R15 GM081889-03) for funding and the Undergraduate Science Education Program of HHMI for fellowships. Jessica Anand and Victoria Baranov (Barnard College) did early solvent screens. Simran Buttar and Yetta Levine (Barnard College) prepared several intermediates. We thank Dr. Yasuhiro Itakagi (Columbia University) for mass spectrometric analyses.
References
- 1.Angata T, Varki A. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 2.Berecibar A, Grandjean C, Siriwardena A. Chem. Rev. 1999;99:779–844. doi: 10.1021/cr980033l. [DOI] [PubMed] [Google Scholar]
- 3.Park H, RajanBabu TV. J. Am. Chem. Soc. 2002;124:734–735. doi: 10.1021/ja0172013. [DOI] [PubMed] [Google Scholar]
- 4.(a) Bongat AFG, Demchenko AV. Carbohydr. Res. 2007;342:374–406. doi: 10.1016/j.carres.2006.10.021. [DOI] [PubMed] [Google Scholar]; (b) Griffith DA, Danishefsky SJ. J. Am. Chem. Soc. 1990;112:5811–5819. [Google Scholar]; (c) Dahl R, Finney NS. J. Am. Chem. Soc. 2004;126:8356–8357. doi: 10.1021/ja0319238. [DOI] [PubMed] [Google Scholar]; (d) Leblanc Y, Fitzsimmons BJ, Springer JP, Rokach J. J. Am Chem. Soc. 1989;111:2995–3000. [Google Scholar]; (e) Lemieux RU, Ratcliffe RM. Can. J. Chem. 1979;57:1244–1251. [Google Scholar]; (f) Lafont D, Guilloux P, Descotes G. Carbohydr. Res. 1989;193:61–73. [Google Scholar]; (g) Du Bois J, Tomooka CS, Hong J, Carreira EM. J. Am. Chem. Soc. 1997;119:3179–3180. [Google Scholar]; (h) Guthikonda K, Wehn PM, Caliando BJ, Du Bois J. Tetrahedron. 2006;62:11331–11342. [Google Scholar]
- 5.(a) Liu J, Gin DY. J. Am. Chem. Soc. 2002;124:9789–9797. doi: 10.1021/ja026281n. [DOI] [PubMed] [Google Scholar]; (b) Di Bussolo V, Liu J, Huffman LG, Jr, Gin DY. Angew. Chem. Int. Ed. 2000;39:204–207. doi: 10.1002/(sici)1521-3773(20000103)39:1<204::aid-anie204>3.3.co;2-q. [DOI] [PubMed] [Google Scholar]; (c) Liu J, Di Bussolo V, Gin DY. Tetrahedron Lett. 2003;44:4015–4018. [Google Scholar]
- 6.For a strategy of internally directed glycal aminohydroxylation, see: Nicolaou KC, Baran PS, Zhong Y-L, Vega JA. Angew. Chem. Int. Ed. 2000;39:2525–2529. Nicolaou KC, Baran PS, Zhong Y-L, Barluenga S, Hunt KW, Kranich R, Vega JA. J. Am. Chem. Soc. 2002;124:2233–2244. doi: 10.1021/ja012126h.
- 7.(a) Kan C, Long CM, Paul M, Ring CM, Tully SE, Rojas CM. Org. Lett. 2001;3:381–384. doi: 10.1021/ol0069002. [DOI] [PubMed] [Google Scholar]; (b) Churchill DG, Rojas CM. Tetrahedron Lett. 2002;43:7225–7228. [Google Scholar]
- 8.Levites-Agababa E, Menhaji E, Perlson LN, Rojas CM. Org. Lett. 2002;4:863–865. doi: 10.1021/ol025634k. [DOI] [PubMed] [Google Scholar]
- 9.Bodner R, Marcellino BK, Severino A, Smenton AL, Rojas CM. J. Org. Chem. 2005;70:3988–3996. doi: 10.1021/jo0500129. [DOI] [PubMed] [Google Scholar]
- 10.Espino CG, Du Bois J. Angew. Chem. Int. Ed. 2001;40:598–600. [PubMed] [Google Scholar]
- 11.(a) Padwa A, Stengel T. Org. Lett. 2002;4:2137–2139. doi: 10.1021/ol0259490. [DOI] [PubMed] [Google Scholar]; (b) Padwa A, Flick AC, Leverett CA, Stengel T. J. Org. Chem. 2004;69:6377–6386. doi: 10.1021/jo048990k. [DOI] [PubMed] [Google Scholar]
- 12.Related aziridinations: Wehn PM, Lee J, Du Bois J. 2003;5:4823–4826. doi: 10.1021/ol035776u. Guthikonda K, Du Bois J. J. Am. Chem. Soc. 2002;124:13672–13673. doi: 10.1021/ja028253a. Duran F, Leman L, Ghini A, Burton G, Dauban P, Dodd RH. Org. Lett. 2002;4:2481–2483. doi: 10.1021/ol0200899. Liang J-L, Yuan S-X, Chan PWH, Che C-M. Tetrahedron Lett. 2003;44:5917–5920.
- 13.(a) Knapp S, Yu Y. Org. Lett. 2007;9:1359–1362. doi: 10.1021/ol0702472. [DOI] [PubMed] [Google Scholar]; (b) Donohoe TJ, Johnson PD, Cowley A, Keenan M. J. Am. Chem. Soc. 2002;124:12934–12935. doi: 10.1021/ja0276117. [DOI] [PubMed] [Google Scholar]; (c) Lebel H, Huard K, Lectard S. J. Am. Chem. Soc. 2005;127:14198–14199. doi: 10.1021/ja0552850. [DOI] [PubMed] [Google Scholar]
- 14.Fiori KW, Du Bois J. J. Am. Chem. Soc. 2007;129:562–568. doi: 10.1021/ja0650450. [DOI] [PubMed] [Google Scholar]
- 15.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. New York: Wiley; 1998. [Google Scholar]
- 16.Dauban P, Sanière L, Tarrade A, Dodd RH. J. Am. Chem. Soc. 2001;123:7707–7708. doi: 10.1021/ja010968a. [DOI] [PubMed] [Google Scholar]
- 17.Another Rh-nitrenoid approach to 2-amino glycosides, using glucal sulfamates: Lorpitthaya R, Xie Z-Z, Kuo J-L, Liu X-W. Chem. Eur. J. 2008;14:1561–1570. doi: 10.1002/chem.200701288.
- 18.Fraser-Reid B, Madsen R. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. New York: Marcel Dekker; 1997. pp. 339–356. [Google Scholar]
- 19.Kozlowska-Gramsz E, Descotes G. Tetrahedron Lett. 1981;22:563–566. [Google Scholar]
- 20.See Supporting Information for carbamate syntheses.
- 21. Espino CG, Du Bois J. In: Modern Rhodium-Catalyzed Organic Reactions. Evans PA, editor. Weinheim: Wiley-VCH; 2005. pp. 379–416. See also footnote 17 of Kim M, Mulcahy JV, Espino CG, Du Bois J. Org. Lett. 2006;8:1073–1076. doi: 10.1021/ol052920y.
- 22.Doyle MP, Dyatkin AB, Autry CL. J. Chem. Soc., Perkin Trans. 1995;1:619–622. [Google Scholar]
- 23.Clark JS, Dossetter AG, Wong Y-S, Townsend RJ, Whittingham WG, Russell CA. J. Org. Chem. 2004;69:3886–3898. doi: 10.1021/jo049900e. [DOI] [PubMed] [Google Scholar]
- 24.No reaction of 6b occurred in the absence of Rh(II) catalyst, and non-redox-active Lewis acid catalysts Sm(OTf)3, Ln(OTf)3, or Zn(OTf)2 did not produce byproduct 8b (or any 7b) from 6b. We also noted earlier (ref 8) that tri-O-acetyl-d-glucal does not react under our conditions, though it is a substrate for Kirschning-type oxidation: Kirschning A. Eur. J. Org. Chem. 1998:2267–2274. Compare: Cochran BM, Michael FE. Org. Lett. 2008;10:5039–5042. doi: 10.1021/ol8022165.
- 25.Wang P, Adams J. J. Am. Chem. Soc. 1994;116:3296–3305. [Google Scholar]
- 26.Fiori KW, Fleming JJ, Du Bois J. Angew. Chem. Int. Ed. 2004;43:4349–4352. doi: 10.1002/anie.200460791. [DOI] [PubMed] [Google Scholar]
- 27.Parker KA, Chang W. Org. Lett. 2005;7:1785–1788. doi: 10.1021/ol050356l. [DOI] [PubMed] [Google Scholar]
- 28.(a) Toumieux S, Compain P, Martin OR. Tetrahedron Lett. 2005;46:4731–4735. [Google Scholar]; (b) Toumieux S, Compain P, Martin OR, Selkti M. Org. Lett. 2006;8:4493–4496. doi: 10.1021/ol061649x. [DOI] [PubMed] [Google Scholar]
- 29.(a) Sulikowski GA, Lee S. Tetrahedron Lett. 1999;40:8035–8038. [Google Scholar]; (b) Wardrop DJ, Zhang W, Fritz J. Org. Lett. 2002;4:489–492. doi: 10.1021/ol016975l. [DOI] [PubMed] [Google Scholar]
- 30.Curran DP, Suh Y-G. Carbohydr. Res. 1987;171:161–191. doi: 10.1016/s0008-6215(00)90885-1. [DOI] [PubMed] [Google Scholar]
- 31.An alternative would be catalyst-controlled chemoselectivity. In our systems, of various Rh(II) carboxylates examined, only Rh2(OAc)4 and Du Bois’s Rh2(esp)2 provided complete conversion, and neither catalyst was satisfactory in limiting C3–H oxidation. Catalyst control of chemoselectivity in rhodium nitrenoid and carbenoid insertions, respectively: Hayes CJ, Beavis PW, Humphries LA. Chem. Commun. 2006:4501–4502. doi: 10.1039/b611662k. Davies HML, Coleman MG, Ventura DL. Org. Lett. 2007;9:4971–4974. doi: 10.1021/ol702218w.
- 32.(a) Thiem J, Ossowski P. J. Carbohydr. Chem. 1984;3:287–313. [Google Scholar]; (b) Roush WR, Sebesta DP, Bennett CE. Tetrahedron. 1997;53:8825–8836. [Google Scholar]
- 33.Crich D, Vinogradova O. J. Am. Chem. Soc. 2007;129:11756–11765. doi: 10.1021/ja0730258. [DOI] [PubMed] [Google Scholar]
- 34.Since dihydropyranones 8 are Michael acceptors for alcohols under basic or acidic conditions (for example: Mann B, Pitts D, Koviach J. J. Carbohydr. Chem. 2005;24:161–168. ), we verified their stability under the reaction conditions. See Supporting Information for details.
- 35. Simova S. Magn. Reson. Chem. 1998;36:505–510. See Supporting Information for details of JH4,H5 determinations.
- 36.van Roeckel CAA, Beetz T, van Aelst SF. Tetrahedron. 1984;40:4097–4107. [Google Scholar]
- 37.Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J. Am. Chem. Soc. 2008;130:343–350. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]
- 38.(a) Kita Y, Takada T, Gyoten M, Tohma H, Zenk MH, Eichhorn J. J. Org. Chem. 1996;61:5857–5864. [Google Scholar]; (b) Huang X, Shao N, Palani A, Aslanian R. Tetrahedron Lett. 2007;48:1967–1971. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details and compound characterization, including preparation of carbamates 6, determination of JH3,H4 values, and copies of NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.