

Abstract
Characterizing infectivity as a function of pathogen dose is integral to microbial risk assessment. Dose-response experiments usually administer doses to subjects at one time. Phenomenological models of the resulting data, such as the exponential and the Beta-Poisson models, ignore dose timing and assume independent risks from each pathogen. Real world exposure to pathogens, however, is a sequence of discrete events where concurrent or prior pathogen arrival affects the capacity of immune effectors to engage and kill newly arriving pathogens. We model immune effector and pathogen interactions during the period before infection becomes established in order to capture the dynamics generating dose timing effects. Model analysis reveals an inverse relationship between the time over which exposures accumulate and the risk of infection. Data from one time dose experiments will thus overestimate per pathogen infection risks of real world exposures. For instance, fitting our model to one time dosing data reveals a risk of 0.66 from 313 Cryptosporidium parvum pathogens. When the temporal exposure window is increased 100-fold using the same parameters fitted by our model to the one time dose data, the risk of infection is reduced to 0.09. Confirmation of this risk prediction requires data from experiments administering doses with different timings. Our model demonstrates that dose timing could markedly alter the risks generated by airborne versus fomite transmitted pathogens.
Author Summary
We model the relationship between the temporal patterns of pathogen exposure and infection take off within people. Since different routes of transmission (e.g., airborne versus surface transfer routes) may result in different temporal patterns of exposure, this model helps to better compare the risks of transmission from one person to another through these different routes. Previous models assumed that the risk of infection is the same whether pathogens are inoculated all at once or over one day. Our model, in contrast, captures how one pathogen affects the potential of immunity to keep concurrently or subsequently arriving particles from initiating an infection. Since the pattern of timing of airborne and surface spread pathogen arrivals differ, our model shows that each airborne pathogen could carry less risk than each surface transmitted pathogen. Unfortunately, data to fully fit our model are not currently available. Therefore new experiments will have to be conducted where doses are given across different temporal windows.
Introduction
Microbial risk assessment models are valuable tools for estimating the risks associated with exposures to pathogens in the environment pathogens [1]. Central to this estimate is a dose-response model that predicts the probability of infection given a dose exposure magnitude. In current microbial risk assessment models dose accumulates over time and the probability of infection is based on the total accumulated dose over that period of time [2]–[4]. This assumes that each pathogen particle carries a risk of infection that is independent of when other pathogens have arrived to a host; i.e., three exposures to dose X generate the same total risk as one exposure to a 3× dose. We put forth an alternative dose response model that assumes the current capacity of immune effectors to control an arriving pathogen should be affected by 1) how many effectors are occupied fighting previously or simultaneously arriving pathogens, 2) how many effectors have been depleted in fighting previously arriving pathogens, and 3) how many effector reinforcements have arrived due to usual effector turnover rates or due to a stimulus from prior pathogen exposure.
If dose-timing effects arise from such immune effector dynamics, then infection-risk calculations that do not take these dose-timing effects into account could lead to errors. For example, errors could arise in models of influenza transmission as follows. Pathogens arriving to a host via aerosols do so more frequently but at lower doses than pathogens arriving via hand or fomite mediated inoculations. Models of influenza transmission that do not account for dose-timing effects, such as the model by Atkinson and Wien [4], might misdirect influenza control resources to masks from hand hygiene. Models that assume independent single dose effects will require more extreme cleaning to reduce risks to acceptable levels than models capturing immune effects on dose timing.
Evaluating the potential importance of such dose-timing effects is difficult for two reasons. First, immune control of pathogens is complex; not enough detailed knowledge regarding that complexity is available to provide a high degree of confidence in a-priori causal model predictions. Second, there is almost no direct observational data documenting the presence or absence of dose-timing effects. Although various studies have given pathogen exposure doses over time [5]–[10], only Brachman et al. [11], has been conducted in a manner that allows one to calculate risks for comparable doses administered over different temporal windows.
In this paper we have taken an approach intended to stimulate science that will address both of these issues. We develop a simple model that illustrates the need to generate new data that can describe dose-timing effects while at the same time providing a base upon which to build more realistic models that incorporate more data and theory on immunity. Our model addresses immune control of pathogens between the time pathogens arrive at a host and the time they are either eliminated or have multiplied enough so that an acquired immune response will be needed for control.
We make our model general enough to capture dynamics of pathogen control that might arise from established antibodies and T-cells, macrophages, polymorphonuclear leukocytes, plasma cells, dendritic cells, complement cascades, chemokines, interleukins, interferons, toll like receptors, and other diverse elements affecting immunity. But we lump all these mediators of pathogen control into a highly abstract entity we label as immune effectors. We assume that the dynamic effects of limited immune effector numbers are similar whether the limitation arises from immune effectors being occupied with previously arrived pathogens or from prior consumption of immune effectors in their process of killing pathogens. Therefore we only model the latter source of immune effector limitations. The resulting model is one where any single pathogen always has some chance of initiating an infection but the risk of infection associated with each additional pathogen exposure can markedly increase at higher pathogen doses given over short temporal windows. The exact dynamics of our model will vary as realistic details are added to it. Our goal here is simply to illustrate the importance and inevitability of immune mediated dose-timing effects so as to stimulate further empirical and theoretical work.
The structure of the paper is as follows: in the methods section we describe the Cumulative Dose model and analyze its dynamics. In the results section we use the Cumulative Dose model to fit experimental data assuming a fixed temporal exposure window to simulate the archetypical single dose experiment of dose-response trials. Using the estimated model we show the effect of changing the length of the temporal exposure window. Finally, the conclusions and future research are presented in the discussion section.
Methods
Cumulative Dose Model
The model is based on a stochastic population of individual pathogens and immune effectors. Since the focus of our analysis is how small populations of pathogens either die out or lead to infection initiation, we cannot rely on the mean-field solution provided by the deterministic framework [12]–[14].
The state of the system is defined by the pair ( ) representing the number of immune effectors and the number of
pathogens, in any single host, respectively. The system is defined by the
following set of state transitions:
) representing the number of immune effectors and the number of
pathogens, in any single host, respectively. The system is defined by the
following set of state transitions:
| (1) |
| (2) |
| (3) |
| (4) |
The number of immune effectors  can increase at: 1) a rate
can increase at: 1) a rate  , which models the constant arrival of immune effectors
regardless of the current state of the immunological system; and 2) a rate
, which models the constant arrival of immune effectors
regardless of the current state of the immunological system; and 2) a rate 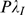 , which models the recruitment of immune effectors in the
presence of pathogens. This term is intended to reflect cytokine induced
recruitment of remote immune effectors to a pathogen invasion site and not
acquired immunity. We assume that the relative endpoints of infection takeoff or
pathogen elimination are reached before an acquired immune effect comes into
play. Immune effectors decrease either at a natural death rate
, which models the recruitment of immune effectors in the
presence of pathogens. This term is intended to reflect cytokine induced
recruitment of remote immune effectors to a pathogen invasion site and not
acquired immunity. We assume that the relative endpoints of infection takeoff or
pathogen elimination are reached before an acquired immune effect comes into
play. Immune effectors decrease either at a natural death rate  , or at a mass-action deactivation rate due to the encounter
with pathogens
, or at a mass-action deactivation rate due to the encounter
with pathogens  .
.
The number of pathogens  can increase by reproduction at a rate
can increase by reproduction at a rate  or by arrival during the inoculation period at a rate
or by arrival during the inoculation period at a rate  . Here
. Here  represents the net reproduction rate that aggregates birth and
death rates. Pathogen numbers decrease due to interaction with immune effectors
as a mass-action deactivation process at the rate
represents the net reproduction rate that aggregates birth and
death rates. Pathogen numbers decrease due to interaction with immune effectors
as a mass-action deactivation process at the rate  .
.
Dynamics of the Cumulative Dose Model
The initial state of the system is set to 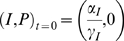 . No chronic low-level exposures or remaining pathogens from
prior exposures are considered. The system starts from the clean state: no
pathogens and the stationary number of immune effectors in the absence of
pathogens. The inoculation process is characterized by the dose of exposure
. No chronic low-level exposures or remaining pathogens from
prior exposures are considered. The system starts from the clean state: no
pathogens and the stationary number of immune effectors in the absence of
pathogens. The inoculation process is characterized by the dose of exposure 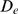 and the temporal exposure length
and the temporal exposure length 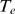 ; i.e., the dose that is composed by
; i.e., the dose that is composed by 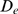 pathogens is inoculated into the host during a period of
pathogens is inoculated into the host during a period of 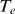 time units. Therefore, the arrival of external pathogens is
modeled as the rate
time units. Therefore, the arrival of external pathogens is
modeled as the rate 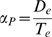 during the inoculation period. Once inoculation has finished
the pathogen arrival rate becomes zero. Thus, the rate
during the inoculation period. Once inoculation has finished
the pathogen arrival rate becomes zero. Thus, the rate 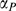 depends on time and is defined as
depends on time and is defined as
 |
During 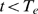 , the pathogens,
, the pathogens,  arrive over a continuous time in the presence of the
immunological response to those pathogens. Once the inoculation has finished,
only the immunological response remains. We set the unit of time to an hour.
That keeps us in the range where we think exposure fluctuations are making a
difference and out of the range where adaptive immune system feedbacks come into
play.
arrive over a continuous time in the presence of the
immunological response to those pathogens. Once the inoculation has finished,
only the immunological response remains. We set the unit of time to an hour.
That keeps us in the range where we think exposure fluctuations are making a
difference and out of the range where adaptive immune system feedbacks come into
play.
Due to stochastic effects and the fate of a relatively small population of
pathogens and immune effectors, the same inoculation dose 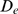 administered in the same time frame
administered in the same time frame 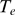 does not necessarily have the same outcome. Each replication
(i.e. run) of the model corresponds to a dose trial on a new subject. All the
numerical results are the average of 104 runs of the Cumulative Dose
model implemented with the Gillespie algorithm [15] using
C. The criteria to stop the simulation is either extinction of
pathogens after the inoculation period (
does not necessarily have the same outcome. Each replication
(i.e. run) of the model corresponds to a dose trial on a new subject. All the
numerical results are the average of 104 runs of the Cumulative Dose
model implemented with the Gillespie algorithm [15] using
C. The criteria to stop the simulation is either extinction of
pathogens after the inoculation period ( ) or pathogens diverging to a very large number,
) or pathogens diverging to a very large number,  , corresponding to no infection and infection respectively. The
probability of infection for a pair
, corresponding to no infection and infection respectively. The
probability of infection for a pair  is the proportion of simulations that diverge to a large
number as opposed to equilibrating to the state of no pathogens.
is the proportion of simulations that diverge to a large
number as opposed to equilibrating to the state of no pathogens.
Figure 1 illustrates the
stochastic process effects on pathogen dynamics given a fixed time of exposure
for different inoculation doses. The main plot in this figure is the time course
of the number of pathogens for 100 independent dose trials given a dose of 60
pathogens administered over one unit of time. The number of pathogens steadily
grow during the inoculation period, from 0 to 1, since the rate of arrival of
pathogens ( ) is much faster than immunological killing of pathogens. Once
the entire dose has been inoculated at
time = 1, the external arrival of pathogens
stop (
) is much faster than immunological killing of pathogens. Once
the entire dose has been inoculated at
time = 1, the external arrival of pathogens
stop ( ) and the immunological response dominates the rest of the
dynamics. In this particular case, the population of pathogens becomes extinct
in 33 cases out of 100, thus, the probability of infection given a dose of 60
pathogens over 1 unit of time is 0.67. Analogously, for a dose of 25 the
probability of infection is 0.02 and for a dose of 90 the probability of
infection is 0.98 (insets of Figure
1).
) and the immunological response dominates the rest of the
dynamics. In this particular case, the population of pathogens becomes extinct
in 33 cases out of 100, thus, the probability of infection given a dose of 60
pathogens over 1 unit of time is 0.67. Analogously, for a dose of 25 the
probability of infection is 0.02 and for a dose of 90 the probability of
infection is 0.98 (insets of Figure
1).
Figure 1. Evolution of the number of pathogens over time for a characteristic
parameter set 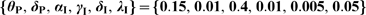 .
.

Each line represents an individual replicate with the same parameter set
(100 in total). The fraction of replicates in which the number of
pathogens diverge towards infinity, as opposed to going extinct, is
equivalent to the probability of infection
(pinf) for the dose  (main graph,
(main graph,  and
and  for the insets a) and b) respectively). Temporal
exposure length is fixed at
Te = 1
hour. Probability of infection is 0.67, 0.02 and 0.98 for the main
graph, the inset a), and the inset b) respectively.
for the insets a) and b) respectively). Temporal
exposure length is fixed at
Te = 1
hour. Probability of infection is 0.67, 0.02 and 0.98 for the main
graph, the inset a), and the inset b) respectively.
Temporal Exposure Length
Figure 1 illustrates how the
Cumulative Dose model yields higher probability of infection when the inoculated
dose is increased. The length of time over which the dose is administered,  , also plays a crucial role in the probability of infection. At
one extreme where all the pathogens were inoculated at once (
, also plays a crucial role in the probability of infection. At
one extreme where all the pathogens were inoculated at once ( ), the immune system has no time to react, and the initial
state of the system is
), the immune system has no time to react, and the initial
state of the system is 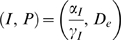 . From this initial state, the immunological response dynamics
determines the fate of the pathogens: either extinction or unbounded growth of
pathogens diverging towards infinity.
. From this initial state, the immunological response dynamics
determines the fate of the pathogens: either extinction or unbounded growth of
pathogens diverging towards infinity.
For  , however, the initial state after all pathogens have been
inoculated (
, however, the initial state after all pathogens have been
inoculated ( ) is not the expected
) is not the expected  , but rather a distribution of probabilities over the space of
possible states. Figure 2
shows the stochastically determined distribution of system states at the point
in Figure 1 where the
exposure time has just ended. It illustrates the effect of different temporal
exposure lengths, ranging from
, but rather a distribution of probabilities over the space of
possible states. Figure 2
shows the stochastically determined distribution of system states at the point
in Figure 1 where the
exposure time has just ended. It illustrates the effect of different temporal
exposure lengths, ranging from  (six minutes) to
Te = 50 hours.
Panel B shows this point of time for the settings in Figure 1. The longer the exposure length, the
larger will be the variance in the distribution of probabilities. Furthermore, a
longer exposure length also affects the average state after inoculation. Both
the pathogen levels and the immune effector levels decrease from the
instantaneous inoculation values as the exposure window length increases. But
the balance between these increasingly favors the immune effectors. Longer
temporal exposure lengths dilute the arrival rate of external pathogens,
(six minutes) to
Te = 50 hours.
Panel B shows this point of time for the settings in Figure 1. The longer the exposure length, the
larger will be the variance in the distribution of probabilities. Furthermore, a
longer exposure length also affects the average state after inoculation. Both
the pathogen levels and the immune effector levels decrease from the
instantaneous inoculation values as the exposure window length increases. But
the balance between these increasingly favors the immune effectors. Longer
temporal exposure lengths dilute the arrival rate of external pathogens,  . Consequently the immunological response has more time to
neutralize the existing pathogens before the arrival of new pathogens. On the
other hand, as the temporal exposure lengths decrease, an increased number of
immune effectors are consumed in killing pathogens, leading to a higher
probability of unbounded growth of pathogens, and thus infection.
. Consequently the immunological response has more time to
neutralize the existing pathogens before the arrival of new pathogens. On the
other hand, as the temporal exposure lengths decrease, an increased number of
immune effectors are consumed in killing pathogens, leading to a higher
probability of unbounded growth of pathogens, and thus infection.
Figure 2. State probability distribution at the end of inoculation ( ) for a dose of
) for a dose of  and temporal exposure length of
and temporal exposure length of 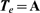 ) 0.1 h, B) 1.0 h, C) 10.0 h and D) 50.0 h.
) 0.1 h, B) 1.0 h, C) 10.0 h and D) 50.0 h.

The distribution of probabilities if  would be
would be  given the parameters of the system are
given the parameters of the system are  . The dashed white line is the separatrix of the
deterministic version of the model (see subsection Deterministic
Analysis); if the system were deterministic once inoculation has been
completed, the states that fall below the separatrix would end up in no
infection, and the states above would end up in infection.
. The dashed white line is the separatrix of the
deterministic version of the model (see subsection Deterministic
Analysis); if the system were deterministic once inoculation has been
completed, the states that fall below the separatrix would end up in no
infection, and the states above would end up in infection.
For 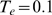 and
and  the average state after inoculation is very close to the ideal
instantaneous inoculation,
the average state after inoculation is very close to the ideal
instantaneous inoculation,  . To better understand the dynamics once inoculation is over,
we included the numerically calculated separatrix as if the system were
deterministic (red-dashed line in Figure 2). Although this separatrix is only truly valid for the
analogous deterministic model, it indicates the probable fate of different
initial states. For the deterministic system, the separatrix separates those
states that go to infection from those that do not (see subsection on
Deterministic Analysis). As temporal exposure length increases, the distribution
of probabilities gravitates towards the space of states that go to no-infection
(below the separatrix).
. To better understand the dynamics once inoculation is over,
we included the numerically calculated separatrix as if the system were
deterministic (red-dashed line in Figure 2). Although this separatrix is only truly valid for the
analogous deterministic model, it indicates the probable fate of different
initial states. For the deterministic system, the separatrix separates those
states that go to infection from those that do not (see subsection on
Deterministic Analysis). As temporal exposure length increases, the distribution
of probabilities gravitates towards the space of states that go to no-infection
(below the separatrix).
Deterministic Analysis
Further understanding of the stochastic dynamics of the Cumulative Dose model can
come from a deterministic description of the system that assumes a continuous
large number of immune effectors and pathogens. We focus our analysis on the
dynamics after the inoculation period, so  is set to 0 and removed from the equations. This analysis on
the deterministic version helps illustrate the interactions between pathogens
and immune effectors that result either in infection or extinction of pathogens.
is set to 0 and removed from the equations. This analysis on
the deterministic version helps illustrate the interactions between pathogens
and immune effectors that result either in infection or extinction of pathogens.
The stochastic system is fully described by a multivariate master equation [16], which can be expanded in a deterministic formulation known as macroscopic law. The deterministic version of the cumulative dose model is as follows,
| (5) |
| (6) |
where 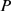 and
and  are continuous variables of the population of pathogens and
immune effectors respectively. The fixed points of the deterministic version of
the cumulative dose model are
are continuous variables of the population of pathogens and
immune effectors respectively. The fixed points of the deterministic version of
the cumulative dose model are  where the pathogen has been eliminated and immune effectors are
in equilibrium and
where the pathogen has been eliminated and immune effectors are
in equilibrium and  where the forces of pathogen growth are balanced by immune
dynamics affecting pathogen death. Note that in the stochastic analyses of this
model as in Figure 1, this
point is never reached. Instead simulations are terminated when growth takes off
toward this point. A simple analysis of the stability of the fixed points
reveals the space of parameters in which the solution is well-defined.
where the forces of pathogen growth are balanced by immune
dynamics affecting pathogen death. Note that in the stochastic analyses of this
model as in Figure 1, this
point is never reached. Instead simulations are terminated when growth takes off
toward this point. A simple analysis of the stability of the fixed points
reveals the space of parameters in which the solution is well-defined.
The point 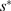 is the equilibrium of no infection—the equilibrium
of the system in the absence of pathogens. When the system gravitates towards
is the equilibrium of no infection—the equilibrium
of the system in the absence of pathogens. When the system gravitates towards  the immunological system prevents pathogens from growing,
resulting in pathogen extinction and therefore no infection.
the immunological system prevents pathogens from growing,
resulting in pathogen extinction and therefore no infection.
To evaluate the stability of the fixed point, we formulate the Jacobian matrix of
the system of equations on  .
.
| (7) |
For a stable equilibrium, both Eigenvalues of the Jacobian matrix need to be
negative, or equivalently, the matrix must have a negative trace and a positive
determinant. For the trace of the Jacobian to be negative the condition  must be true. Since the positive determinant condition,
must be true. Since the positive determinant condition,  , is more restrictive it subsumes the condition for a negative
trace.
, is more restrictive it subsumes the condition for a negative
trace.
The second fixed point  is only well-defined when both
is only well-defined when both  and
and  are positive, since negative number of pathogens and immune
effectors are impossible. The number of pathogens is only positive when
are positive, since negative number of pathogens and immune
effectors are impossible. The number of pathogens is only positive when  . Given the condition of a positive determinant,
. Given the condition of a positive determinant, 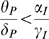 , the sign can only be negative, consequently
, the sign can only be negative, consequently  . Therefore, the system is well defined — i.e. has a
stable equilibrium at no infection and with both fixed points in the positive
quadrant — only when the following condition 8 is met
. Therefore, the system is well defined — i.e. has a
stable equilibrium at no infection and with both fixed points in the positive
quadrant — only when the following condition 8 is met
| (8) |
Once we determine the stability of  we need to characterize the second fixed point
we need to characterize the second fixed point 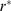 . After some basic algebra, the determinant of the Jacobian
matrix for
. After some basic algebra, the determinant of the Jacobian
matrix for  can be expressed as follows:
can be expressed as follows:  . Given condition 8, both terms are positive, which makes the
determinant negative. As a result the Eigenvalues of the Jacobian are real with
different signs. Therefore,
. Given condition 8, both terms are positive, which makes the
determinant negative. As a result the Eigenvalues of the Jacobian are real with
different signs. Therefore,  is a saddle point as shown in Figure 3.
is a saddle point as shown in Figure 3.
Figure 3. Vector field plot of the deterministic cumulative dose model for a
characteristic parameter set  .
.

To avoid overlaps of the vectors they have been normalized. The solid red
lines are the nullclines, the intersections of the nullclines are the
fixed points  (stable pathogen elimination equilibrium) and
(stable pathogen elimination equilibrium) and  (unstable saddle point equilibrium). The dash black
line is the separatrix that separates those configurations that will go
to non-infection equilibrium,
(unstable saddle point equilibrium). The dash black
line is the separatrix that separates those configurations that will go
to non-infection equilibrium,  , and those that will diverge in the number of
pathogens resulting on infection. The separatrix has been calculated
numerically.
, and those that will diverge in the number of
pathogens resulting on infection. The separatrix has been calculated
numerically.
The vector field in Figure 3 illustrates the dynamics of the cumulative dose after the inoculation period. The probability of being in a given state after inoculation is shown in Figure 2. If the system were deterministic then we could anticipate the probability of infection by summing the probability of those states below the separatrix. This does not hold for the stochastic Cumulative Dose model. Nonetheless, the deterministic vector field, shown in Figure 3, serves as an approximate description of what happens in the stochastic model.
For instance, let us take the probability distribution of states when centered at 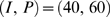 , i.e.,
, i.e.,  and
and 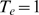 . The typical dynamic results in the decrease in number of
pathogens and immune effectors, gravitating towards the saddle point
. The typical dynamic results in the decrease in number of
pathogens and immune effectors, gravitating towards the saddle point  , from which it will bifurcate to the stable point of
no-infection
, from which it will bifurcate to the stable point of
no-infection 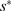 , or an unbounded growth of pathogens. In the case of
, or an unbounded growth of pathogens. In the case of  and
and  , most of the states are already very low in pathogens, and
consequently the number of immune effectors will eradicate the few pathogens
still existing and go to the stable equilibrium of no infection. However, there
is a non-zero probability, albeit small, of being in a state with a large number
of pathogens and a small number of immune effectors. In this case, stochastic
perturbations aside, the pathogens will keep multiplying producing infection in
the host.
, most of the states are already very low in pathogens, and
consequently the number of immune effectors will eradicate the few pathogens
still existing and go to the stable equilibrium of no infection. However, there
is a non-zero probability, albeit small, of being in a state with a large number
of pathogens and a small number of immune effectors. In this case, stochastic
perturbations aside, the pathogens will keep multiplying producing infection in
the host.
Results
Analysis of Exposure Dose Risks
In this section, we fit empirical data on multiple pathogens for the single event inoculation scenario. Next, we extend our analysis to incorporate different temporal exposure windows and patterns of inoculation.
Fitting experimental dose-response data
We selected three different pathogen datasets: 1) poliovirus [17], 2) Cryptosporidium parvum [18] and 3) rotavirus [19]. Analyses of these three datasets are found elsewhere [20].
Several statistical models based on the empirical data have been proposed to describe dose-response data. The most common models are the Exponential model [1]:
| (9) |
where μ is the inoculation dose and r is the per pathogen risk, and the Beta-Poisson model [21]:
| (10) |
where μ is the inoculation dose and α and β are parameters of the beta distribution that describes the host pathogen interaction. Other models such as Log-Logistic and Weibull have been used, but not as commonly.
For parameter estimation we used a classical genetic algorithm [22]. The fitness function of the genetic
algorithm was the mean square error ( ). We fixed the exposure time (
). We fixed the exposure time ( ) of our inoculated dose (
) of our inoculated dose ( ) to 1.0 time units in order to emulate the empirical
dose-response experiments in which the dose is inoculated in a single shot;
i.e., a very short exposure. We present the best fitting curves and discuss
their limitations in the subsection “The Effect of Temporal
Exposure Length”. Then, given these best fitting parameter values,
we present the effect of different temporal exposure windows on the final
probability of infection.
) to 1.0 time units in order to emulate the empirical
dose-response experiments in which the dose is inoculated in a single shot;
i.e., a very short exposure. We present the best fitting curves and discuss
their limitations in the subsection “The Effect of Temporal
Exposure Length”. Then, given these best fitting parameter values,
we present the effect of different temporal exposure windows on the final
probability of infection.
Poliovirus
The first empirical dataset to which we apply the Cumulative Dose model is
Poliovirus type 1 [17]. The cohort for this experiment was 32
2-month-old infants. Inoculation was oral. Figure 4 and Table 1 show the fit alongside a fit to the
Exponential model ( ) according to [18].
) according to [18].
Figure 4. Dose-response curves based on the Exponential Model ( ) and the Cumulative Dose model (
) and the Cumulative Dose model ( ) compared to the experimental dataset for Poliovirus
type 1 (squares).
) compared to the experimental dataset for Poliovirus
type 1 (squares).

The estimated parameters are  for the Exponential model [3] and
for the Exponential model [3] and 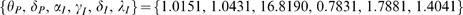 for the Cumulative Dose model.
for the Cumulative Dose model.
Table 1. Probability of infection from experimental data for Polivirus type 1 ( ) compared to the probability of infection based on the
Exponential model (
) compared to the probability of infection based on the
Exponential model (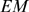 ) and the Cumulative Dose model (
) and the Cumulative Dose model ( ).
).
| Dose | No. of subjects | No. Infected | Fraction Infected 
|

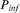
|


|
| 7.0 | 1 | 0 | 0.0 | 0.0617 | 0.0 |
| 16.0 | 2 | 0 | 0.0 | 0.1355 | 0.0 |
| 27.0 | 2 | 0 | 0.0 | 0.2178 | 0.0062 |
| 42.0 | 1 | 0 | 0.0 | 0.3176 | 0.0831 |
| 50.0 | 6 | 3 | 0.50 | 0.3656 | 0.1840 |
| 55.0 | 3 | 1 | 0.333 | 0.3938 | 0.2582 |
| 65.0 | 6 | 0 | 0.0 | 0.4465 | 0.4523 |
| 80.0 | 1 | 1 | 1.0 | 0.5171 | 0.6992 |
| 90.0 | 4 | 3 | 0.75 | 0.5591 | 0.8189 |
| 160.0 | 3 | 3 | 1.0 | 0.7668 | 0.999 |
| 210.0 | 2 | 2 | 1.0 | 0.8521 | 1.0 |
| 280.0 | 1 | 1 | 1.0 | 0.9218 | 1.0 |
The estimated parameters are  for the Exponential model [3] and
for the Exponential model [3] and  for the Cumulative Dose model.
for the Cumulative Dose model.
Cryptosporidium
The cohort for the Cryptosporidium parvum study [18] was
35 healthy subjects (12 men and 17 women, age range between 20 and 45 years).
The strain was an isolate from a calf and the inoculums were orally administered
via capsules. Figure 5 and
Table 2 show the fit
alongside a fit to the Exponential model ( ) according to [20].
) according to [20].
Figure 5. Dose-response curves based on the Exponential Model (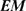 ) and the Cumulative Dose model (
) and the Cumulative Dose model ( ) compared to the experimental dataset for
Cryptosporidium parvum (squares).
) compared to the experimental dataset for
Cryptosporidium parvum (squares).

The estimated parameters are 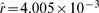 for the Exponential model [30] and
for the Exponential model [30] and  for the Cumulative Dose model.
for the Cumulative Dose model.
Table 2. Probability of infection from experimental data for
Cryptosporidium parvum (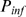 ) compared to the probability of infection predicted by
the Exponential model (EM) and the Cumulative Dose (CD) model.
) compared to the probability of infection predicted by
the Exponential model (EM) and the Cumulative Dose (CD) model.
| Dose | No. of subjects | No. Infected | Fraction Infected 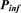
|
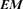

|

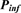
|
| 34 | 5 | 1 | 0.2 | 0.1273 | 0.0848 |
| 108 | 8 | 3 | 0.375 | 0.3511 | 0.3173 |
| 313 | 3 | 2 | 0.6667 | 0.7145 | 0.7421 |
| 504 | 6 | 5 | 0.8333 | 0.8671 | 0.9065 |
| 1129 | 2 | 2 | 1.0 | 0.9891 | 0.9972 |
| 11460 | 3 | 3 | 1.0 | 1.0 | 1.0 |
| 113900 | 1 | 1 | 1.0 | 1.0 | 1.0 |
| 1139000 | 1 | 1 | 1.0 | 1.0 | 1.0 |
The estimated parameters are  for the Exponential model [30]
and
for the Exponential model [30]
and  for the Cumulative Dose model.
for the Cumulative Dose model.
Rotavirus
Finally, we tested the Cumulative Dose model against a dataset for Rotavirus [19].
The cohort for rotavirus was 62 adult males, 18 to 45 years old. The inoculation
was oral. Unlike the previous dose-response empirical datasets, neither the
Cumulative Dose model nor the Exponential model produce a good fit. The
Beta-Poisson model ( ) was statistically a better fit than the Exponential model
[20].
) was statistically a better fit than the Exponential model
[20].
Both the Exponential and the Cumulative Dose model increase too rapidly in relation to the probability of infection of 1; i.e. these models cannot maintain a non-zero or non-one probability of infection for a dose range of several orders of magnitude. Conversely, the Beta-Poisson model does not suffer from this limitation since its convergence to 1 is slower, providing a wider range of variance (Figure 6 and Table 3).
Figure 6. Dose-response curves based on the Exponential Model ( ), the Beta-Poisson model (
), the Beta-Poisson model ( ) and the Cumulative Dose model (
) and the Cumulative Dose model ( ) compared to the experimental dataset for Rotavirus
(squares).
) compared to the experimental dataset for Rotavirus
(squares).

The estimated parameters are  for the Exponential model,
for the Exponential model, 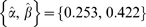 for the Beta-Poisson model [31] and
for the Beta-Poisson model [31] and 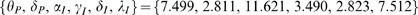 for the Cumulative Dose model.
for the Cumulative Dose model.
Table 3. Probability of infection from experimental data for Rotavirus (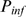 ) compared to the the Exponential, Beta-Poisson and
Cumulative Dose models.
) compared to the the Exponential, Beta-Poisson and
Cumulative Dose models.
| Dose | No. of subjects | No. Infected | Fraction Infected 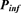
|

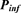
|
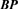

|

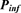
|
| 9×10−3 | 5 | 0 | 0.0 | 0.0009 | 0.0053 | <0.001 (*) |
| 9×10−2 | 7 | 0 | 0.0 | 0.009 | 0.0477 | 0.0053 |
| 9×10−1 | 7 | 1 | 0.1428 | 0.0861 | 0.2509 | 0.0740 |
| 9 | 11 | 8 | 0.7273 | 0.5934 | 0.5442 | 0.6175 |
| 9×101 | 7 | 6 | 0.8571 | 0.9999 | 0.7428 | 0.9999 |
| 9×102 | 8 | 7 | 0.875 | 1.0 | 0.8562 | 1.0 |
| 9×103 | 7 | 5 | 0.7143 | 1.0 | 0.9197 | 1.0 |
| 9×104 | 3 | 3 | 1.0 | 1.0 | 0.9551 | 1.0 |
The estimated parameters are 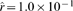 for the Exponential model,
for the Exponential model,  for the Beta-Poisson model [31]
and
for the Beta-Poisson model [31]
and 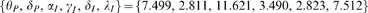 for the Cumulative Dose model. (*) The
dose in the original trial was administered in concentrations, to
work with discrete pathogens as required by the Cumulative Dose
model, we assumed that the concentration of
9×10−2 is equivalent to 9
pathogens. As a consequence the concentration of
9×10−3 could not be tested since
it is a fraction of a pathogen. The probability of infection for a
single pathogen is 10−3. This assumption is
only required by the Cumulative Dose model.
for the Cumulative Dose model. (*) The
dose in the original trial was administered in concentrations, to
work with discrete pathogens as required by the Cumulative Dose
model, we assumed that the concentration of
9×10−2 is equivalent to 9
pathogens. As a consequence the concentration of
9×10−3 could not be tested since
it is a fraction of a pathogen. The probability of infection for a
single pathogen is 10−3. This assumption is
only required by the Cumulative Dose model.
A possible explanation of the poor fit of the Cumulative Dose model is the high degree of acquired immunity to Rotavirus and the changing serotype profile circulating within populations [23]. Unlike the polio virus study, the rotavirus cohort consisting of adults (18–45 years old), is likely to have been exposed multiple times to various rotavirus serotypes [24]. Such heterogeneity in susceptibility flattens out dose response curves beyond what can be captured by exponential dose response models or this Cumulative Dose response model.
The Effect of Temporal Exposure Length
In the previous subsections we fixed temporal exposure length,  , to 1 hour, and assume that this is the time corresponding to
the single shot inoculation, analogous to existing experimental dose-response
trials. In this section, we present simulations for a range of different
temporal exposure lengths, illustrating how longer times affect the dose
response curve. The model is set to the parameters that provided an optimal fit
for a temporal exposure length of
, to 1 hour, and assume that this is the time corresponding to
the single shot inoculation, analogous to existing experimental dose-response
trials. In this section, we present simulations for a range of different
temporal exposure lengths, illustrating how longer times affect the dose
response curve. The model is set to the parameters that provided an optimal fit
for a temporal exposure length of  .
.
Figure 7 shows the
dose-response curves for Poliovirus type 1 for different lengths of exposure for
the estimated parameters used in Figure 4 to fit the experimental data for the condition
Te = 1.0:  . As the exposure length increases, the probability of
infection decreases dramatically. Therefore, assuming that the unit of time is
one hour, and this is the equivalent for a dose that is administered in a single
shot, the probability of infection generated by the Cumulative Dose model for a
dose of
. As the exposure length increases, the probability of
infection decreases dramatically. Therefore, assuming that the unit of time is
one hour, and this is the equivalent for a dose that is administered in a single
shot, the probability of infection generated by the Cumulative Dose model for a
dose of  of 90 pathogens administered in one hour is 0.82. If the dose
were administered not in one hour, but uniformly over ten hours the probability
of infection would be 0.18. If the dose were administered over fifty hours the
probability of infection would be reduced to 0.0001. To obtain the same
probability of infection for a ten hours inoculation period instead of one, we
would require a dose of 139 pathogens instead of 90.
of 90 pathogens administered in one hour is 0.82. If the dose
were administered not in one hour, but uniformly over ten hours the probability
of infection would be 0.18. If the dose were administered over fifty hours the
probability of infection would be reduced to 0.0001. To obtain the same
probability of infection for a ten hours inoculation period instead of one, we
would require a dose of 139 pathogens instead of 90.
Figure 7. Predicted effects of varying exposure times (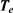 ) when inoculated with Poliovirus type 1.
) when inoculated with Poliovirus type 1.

Parameters are defined as stated in Figure 4.
Because data on the impact of temporal patterns of inoculation are currently not
available, a model with dose-time dependence such as ours is not identifiable
[25]; i.e., the model can be fit to existing single
dose empirical data with many different parameters sets. For example, in Figure 8 we show model
simulation results for Cryptosporidium parvum for two different
parameter sets. Both parameters sets have a similar fit to the
Cryptosporidium parvum dataset when  (mean square error using
(mean square error using  and
and 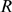 is 3.5×10−3 and
9.7×10−3 respectively). For values of
is 3.5×10−3 and
9.7×10−3 respectively). For values of 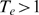 , however, the dose response relationships of the two parameter
sets diverge. Parameter set
, however, the dose response relationships of the two parameter
sets diverge. Parameter set  is much less sensitive to exposure time than
is much less sensitive to exposure time than 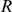 due its slower dynamics. Using parameter set R, pathogens
proliferate faster, are being eliminated by each immune effector more quickly,
are recruiting fewer immune effectors, and are eliminating immune effectors at a
slower rate. On the other hand, using parameter set R, the natural rate of
turnover of immune effectors is more rapid. We cannot argue at this point which
is the most plausible configuration since identifiability cannot be resolved
without data from dosing trials for different exposure lengths.
due its slower dynamics. Using parameter set R, pathogens
proliferate faster, are being eliminated by each immune effector more quickly,
are recruiting fewer immune effectors, and are eliminating immune effectors at a
slower rate. On the other hand, using parameter set R, the natural rate of
turnover of immune effectors is more rapid. We cannot argue at this point which
is the most plausible configuration since identifiability cannot be resolved
without data from dosing trials for different exposure lengths.
Figure 8. Predicted effects of varying exposure times ( ) when inoculated with Cryptosporidium
parvum.
) when inoculated with Cryptosporidium
parvum.

The top graph comes from simulations using the parameter set defined in
Figure 5. The
bottom graph comes simulations using the parameter set 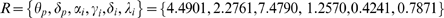 .
.
The Effect of Dosing Patterns over the Exposure Window
In this section we relax the assumption that pathogens are inoculated at a fixed rate. We allow variation both in dose magnitude and length of exposure time, in order to capture a more realistic exposure scenario.
The temporal pattern of inoculation of pathogens within a host depends both on the behavior of the host and the contamination of the environment the host interacts with. For instance, a susceptible host in a venue contaminated with influenza will be exposed to pathogens from air and fomites. However, the temporal patterns of exposure for these two modes of transmission are different. The host is likely to receive a small dose with every breath when breathing contaminated air. In fomite mediated transmission, however, the touching of a mucous membrane with contaminated fingers, for example, is likely to transmit a larger but less frequent dose.
To illustrate this effect we devised an experiment where both the total
inoculated dose 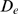 and the exposure time length
and the exposure time length  are fixed. The only parameter that varies is the number of
inoculation events,
are fixed. The only parameter that varies is the number of
inoculation events, 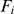 , which ranges from 1 to the total dose
, which ranges from 1 to the total dose  . Consequently, once the number of inoculations events is
determined, the dose inoculated in each event is
. Consequently, once the number of inoculations events is
determined, the dose inoculated in each event is 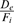 and the rate at which inoculation occur is
and the rate at which inoculation occur is 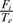 .
.
Figure 9 shows the results of
this experiment where the same parameter sets are used as in Figure 8. The pathogen is
Cryptosporidium parvum, and the same two different
parameters sets, S and R, are used to inform
the cumulative dose model. The total dose inoculated is set to  and the temporal exposure length is set to
Te = 120.0
hours.
and the temporal exposure length is set to
Te = 120.0
hours.
Figure 9. Predicted effects of different temporal patterns of exposure when inoculated with Cryptosporidium parvum.

The main figure displays the probability of infection as function of the
number of inoculation events. The line with circular markers comes from
simulation results using the parameter set defined in Figure 5, and the line
with square markers comes from simulation results using the parameter
set 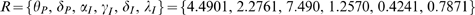 The insets below demonstrate three temporal patterns
for three different patterns of inoculation events:
A = 1,
B = 4 and
C = 50 events respectively. The solid
line represents one instance of the 5000 replicas used in the
experiment. The dashed line represents the average of dose inoculated
over time.
The insets below demonstrate three temporal patterns
for three different patterns of inoculation events:
A = 1,
B = 4 and
C = 50 events respectively. The solid
line represents one instance of the 5000 replicas used in the
experiment. The dashed line represents the average of dose inoculated
over time.
For both parameter sets S and R we observe the same behavior: infectivity decreases as the frequency or number of inoculations events increases. The temporal pattern more likely to be associated with fomite transmission (low frequency and high dose, Figure 9.B) is more likely to produce infection than the patterns associated with airborne transmission (high-frequency and low dose, Figure 9.C) .
For parameter set R, the probability of infection if the dose is inoculated with a single exposure (Figure 9.A) is 0.752. The same dose inoculated over 4 events, where each event is one fourth of the total dose (Figure 9.B), reduces the probability of infection to 0.443. In addition, if the dose is inoculated over 50 events (Figure 9.C) the probability decreases to 0.111. For parameter set S, the reduction of the infection probability is less pronounced: 0.740, 0.676 and 0.601 for 1, 4 and 50 inoculation events respectively.
In previous sections we showed that longer temporal exposure lengths decrease infectivity due to the action of the immune system. In this section, we show that not only the duration of the exposure matters, but also the way in which pathogens arrive within that interval can decrease infectivity. These results suggest that risk assessments based on current dose-response data might be over-estimating risk of infection. An important corollary is that risk of infection for a given exposure dose may depend on the route of transmission based on their differences in the pattern of exposure.
Discussion
We examined a dynamic mechanistic model where immune system effects generated dose response dependence on the timing of doses. The specific aspects of our model that generate these dose-timing effects are: 1) decreases in available immune effectors because they are being eliminated as they kill pathogens; and 2) increases in available immune effectors due to both pathogen dependent and independent recruitment. An additional mechanism resulting in decreases in available immune effectors that is not included in our model could be the time of immune effector engagement with pathogens in the killing process. The dose-timing effects we illustrate would be absent in a model where some effector like a T-cell instantaneously kills pathogens or pathogen generating cells, where no killing capacity is lost with each kill, and where effector dynamics are not otherwise altered by encounters with pathogens. Any such model, however, is highly unrealistic, and therefore we conclude that the dose-timing effects presented in our model could be important and warrant further study.
Dose-timing effects have implications for microbial risk assessment, for infection transmission system modeling, and for the evolution of emerging pathogens. Considering a microbial risk assessment example, the implications of our findings suggest that exposure routes with different dose-timing dynamics could have different risks and therefore result in different clean up protocols for contamination events such as a norovirus outbreak or a Katrina-like disaster. Dose timing could, therefore, affect decisions on which venues to close or what the total dose that workers would be permitted to accrue during a cleanup operation.
Considering modeling infection transmission, the standard approach is to define a contact and a transmission probability per contact while the physical route of transmission is ignored. Modeling the physical route of transmission is important when it is necessary to specify how much transmission is taking place in particular public venues and when specifying which control actions in these venues will reduce transmission. When different routes have different temporal exposure patterns, we demonstrate here that there is considerable potential for immune system effects to alter the ratio by which airborne transmitted and hand-fomite transmitted pathogens generate new infections. If we had data on infection risks under different dose-timing patterns, we could say more precisely how much difference in risk there might be from an airborne and a hand-fomite mediated pathogen. Unfortunately such data is lacking.
The evolution of emerging infection implications derive from the route of transmission effects just discussed. When pathogens first jump species, they are likely to encounter strong innate immune responses to which they must evolve some escape strategy. That means very high transmission doses will be required to sustain transmission and that low dose exposure over longer times such as occurs with airborne transmission will be the most unlikely to be effective in transmitting infection. But, as escape from innate immune responses evolves, the balance could begin to favor airborne transmission which might be more effective in disseminating infection.
We do not have enough dose timing data for any infection to evaluate either the microbial risk assessment implications, the infection transmission system implications, or the emerging infection evolution implications. Any data providing indications of the magnitude of dose-timing effects generated by any type of immunity to any agent would provide an important first step that would at least indicate what range of effects might be expected. Animal studies could compare the risks associated with a single instantaneously delivered dose with the same dose magnitude delivered over extended periods of time. Measurements of specific immune effector dynamics, such as interferon gamma [26] would improve our mechanistic understanding of a cumulative dose effect and indicate how to refine our models for different animal/pathogen systems.
The issue of dose-response trial design is crucial for advancing both quantitative microbial risk assessment and analysis of population infection transmission systems. Due to the absence of a prior theoretical framework, there has been no motivation to conduct dosing trials that take multiple doses and multiple dosing times into account. Now that the potential effects of dose timing have been demonstrated and the practical significance of such measurements for microbial risk assessment and transmission system analyses is more evident, we hope to see such experiments.
Acknowledgments
We thank John Coffin, Igor Rouzine, and Patrick Nelson for their valuable comments on early versions of this paper.
Footnotes
The authors have declared that no competing interests exist.
This research was supported by U.S. EPA: Science to Achieve Results (STAR) Program and by U.S. Department of Homeland Security University Programs (Grant # R83236201). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Haas CN, Rose JB, Gerba CP. Quantitative Microbial Risk Assessment. New York: John Wiley & Sons, Inc., ISBN: 0-471-18397-0; 1999. [Google Scholar]
- 2.Noakes CJ, Beggs CB, Sleigh PA, Kerr KG. Modelling the transmission of airborne infection in enclosed spaces. Epidemiology and Infection. 2006;134(5):1082–91. doi: 10.1017/S0950268806005875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisenberg JNS, Lei X, Hubbard AH, Brookhart MA, Colford JM., Jr The role of disease transmission and conferred immunity in outbreaks: Analysis of the 1993 Cryptosporidium outbreak in Milwaukee. American Journal of Epidemiology. 2005:62–72. doi: 10.1093/aje/kwi005. [DOI] [PubMed] [Google Scholar]
- 4.Atkinson MP, Wein LM. Quantifying the routes of transmission for pandemic influenza. Bull Math Biol. 2008;70:820–867. doi: 10.1007/s11538-007-9281-2. [DOI] [PubMed] [Google Scholar]
- 5.Ellenberger D, et al. HIV-1 DNA/MVA vaccination reduces the per exposure probability of infection during repeated mucosal SHIV challenges. Virology. 2006;352:216–225. doi: 10.1016/j.virol.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Lerma JG, et al. Prevention of rectal SHIV transmission in macaques by daily or intermittent prophylaxis with emtricitabine and tenofovir. PLoS Med. 2008;5:e28. doi: 10.1371/journal.pmed.0050028. doi:10.1371/journal.pmed.0050028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuckwell HC, Shipman PD, Perelson AS. The probability of HIV infection in a new host and its reduction with microbicides. Math Biosci. 2008;214:81–86. doi: 10.1016/j.mbs.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 8.Van Rompay KK, Kearney BP, Sexton JJ, Colon R, Lawson JR, Blackwood EJ, Lee WA, Bischofberger N, Marthas ML. Evaluation of oral tenofovir disoproxil fumarate and topical tenofovir GS-7340 to protect infant macaques against repeated oral challenges with virulent simian immunodeficiency virus. J Acquir Immune Defic Syndr. 2006;43:6–14. doi: 10.1097/01.qai.0000224972.60339.7c. [DOI] [PubMed] [Google Scholar]
- 9.Van Rompay KK, et al. Attenuated poxvirus-based simian immunodeficiency virus (SIV) vaccines given in infancy partially protect infant and juvenile macaques against repeated oral challenge with virulent SIV. J Acquir Immune Defic Syndr. 2005;38:124–134. doi: 10.1097/00126334-200502010-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson NA, et al. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol. 2006;80:5875–5885. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brachman PS, Kaufman AF, Dalldorf FG. Industrial inhalation Anthrax. Bacteriol Rev. 1966;30:646–659. doi: 10.1128/br.30.3.646-659.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rand DA, Wilson HB. Chaotic stochasticity: a ubiquitous source of unpredictability in epidemics. Proc Royal Society B. 1991;246:179–184. doi: 10.1098/rspb.1991.0142. [DOI] [PubMed] [Google Scholar]
- 13.McKane AJ, Newman TJ. Predator-prey cycles from resonant amplification of demographic stochasticity. Physical Review Letters. 2005;94:218. doi: 10.1103/PhysRevLett.94.218102. [DOI] [PubMed] [Google Scholar]
- 14.Alonso D, McKane AJ, Pascual M. Stochastic amplification in epidemics. J R Soc Interface. 2006 doi: 10.1098/rsif.2006.0192. 10.1098/rsif.2006.0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillespie DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. Journal of Computational Physics. 1976;22:403–434. [Google Scholar]
- 16.van Kampen NG. Stochastic processes in physics and chemistry. Amsterdam: Elsevier; 1992. [Google Scholar]
- 17.Minor TE, Allen CI, Tsiatis AA, Nelson DB, d'Alesio DJ. Human infective dose determinations for oral poliovirus type 1 vaccine in infants. Journal of Clinical Microbiology. 1981;13(2):388–389. doi: 10.1128/jcm.13.2.388-389.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy workers. The New England Journal of Medicine. 1995;332(13):855–859. doi: 10.1056/NEJM199503303321304. [DOI] [PubMed] [Google Scholar]
- 19.Ward RL, Bernstein DI, Young EC, Sherwood JR, Knowlton DR, Schiff GM. Human Rotavirus studies in volunteers: determination of infectious dose and serological response to infection. Journal of Infectious Diseases. 1986;154(5):871–880. doi: 10.1093/infdis/154.5.871. [DOI] [PubMed] [Google Scholar]
- 20.Teunis PFM, van der Heijden OG, van der Giessen JWB, Havelaar AH. The dose-response relation in human volunteers for gastro-intestinal pathogens. Rijksinstituut voor Volksgezondheid en Milieu Bilthoven. 1996;284550002 [Google Scholar]
- 21.Haas CN. Estimation of risk due to low doses of microorganisms: a comparison of alternative methodologies. American Journal of Epidemiology. 1983;118(4):1097–1100. doi: 10.1093/oxfordjournals.aje.a113662. [DOI] [PubMed] [Google Scholar]
- 22.Holland JH. Adaptation in Natural and Artificial Systems. Ann Arbor: University of Michigan Press; 1975. [Google Scholar]
- 23.Koopman JS, Monto AS. The Tecumseh Study XV: Rotavirus infection and pathogenicity. American Journal of Epidemiology. 1989;130(4):750–759. doi: 10.1093/oxfordjournals.aje.a115396. [DOI] [PubMed] [Google Scholar]
- 24.Koopman JS, Monto AS, Longini IM. The Tecumseh Study XVI: Family and community sources of rotavirus infection. Am J Epidemiol. 1989;130(4):760–768. doi: 10.1093/oxfordjournals.aje.a115397. [DOI] [PubMed] [Google Scholar]
- 25.Armitage P, Spicer CC. The detection of variation in host susceptibility in dilution counting experiments. Journal of Hygiene. 1956;54:401–414. doi: 10.1017/s0022172400044661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howat TJ, Barreca C, O'Hare P, Gog JR, Grenfell TB. Modelling dynamics of the type I interferon response to in vitro viral infection. J R Soc Interface. 2006;3:699–709. doi: 10.1098/rsif.2006.0136. [DOI] [PMC free article] [PubMed] [Google Scholar]