

Abstract
The homodimeric Escherichia coli β sliding clamp contains two hydrophobic clefts with which proteins involved in DNA replication, repair and damage tolerance interact. Deletion of the C-terminal five residues of β (βC) disrupted both clefts, severely impairing interactions of the clamp with the DnaX clamp loader, as well as the replicative DNA polymerase, Pol III. In order to determine whether both clefts were required for loading clamp onto DNA, stimulation of Pol III replication and removal of clamp from DNA after replication was complete, we developed a method for purification of heterodimeric clamp proteins comprised of one wild-type subunit (β+), and one βC subunit (β+/βC). The β+/βC heterodimer interacted normally with the DnaX clamp loader, and was loaded onto DNA slightly more efficiently than was β+. Moreover, β+/βC interacted normally with Pol III, and stimulated replication to the same extent as did β+. Finally, β+/βC was severely impaired for unloading from DNA using either DnaX or the δ subunit of DnaX. Taken together, these findings indicate that a single cleft in the β clamp is sufficient for both loading and stimulation of Pol III replication, but both clefts are required for unloading clamp from DNA after replication is completed.
INTRODUCTION
The Escherichia coli β sliding clamp, encoded by the dnaN gene (1), is the founding member of the ubiquitous β sliding clamp family of proteins (2). The β clamp was initially identified based on its ability to tether the replicative DNA polymerase (Pol) to the DNA template, effectively increasing its processivity (3). However, it is now clear that sliding clamp proteins interact with a variety of proteins in addition to their cognate replicative Pols that act on DNA, and also play important roles in managing the activities of these proteins to coordinate initiation of DNA replication with elongation, as well as DNA repair, DNA damage tolerance and cell-cycle progression (2). Although several models have been proposed to describe the role(s) by which sliding clamps coordinate these processes, the precise mechanism(s) are still largely unknown.
In general, bacterial clamps (β clamps) exist as stable homodimers, whereas eukaryotic and archaeal clamps (proliferating cell nuclear antigen, or PCNA) are homotrimers (2). Despite a lack of sequence similarity, crystallographic studies have revealed that their three-dimensional structures are nearly identical (4–8). The head-to-tail arrangement of the protomers forms a ring with two distinct surfaces. This ring has a positively charged core of α-helices that associates with DNA (5,9) and is flanked by β-sheets (5). The so-called N-side bears the N-terminus, while the C-side contains the C-terminus, which is part of a hydrophobic cleft that serves as a common docking site for most of its partner proteins. A structure for the β clamp on DNA was recently reported (9). In the crystal, the clamp makes specific contacts with DNA, consistent with computational modeling studies (10). These contacts play poorly defined roles in clamp loading in vitro (9), and are essential for viability of E. coli (11).
Sliding clamps are loaded around DNA by a conserved multi-subunit AAA+ ATPase (expanded group of ATPases Associated with various cellular Activities) referred to as the clamp loader (12). The complete form of the E. coli clamp loader, DnaX complex (also called γ complex), is comprised of two copies of the τ protein, and one copy each of γ, δ, δ′, χ, and ψ (τ2γδδ′χψ). In addition to τ2γδδ′χψ, alternative forms of DnaX differing in the stoichiometry of τ and γ (i.e. τ3δδ′χψ or γ3δδ′χψ), and/or lacking χψ (i.e. τ3δδ′ or γ3δδ′) are active in clamp loading (3,12). Both τ and γ are expressed from the same gene (dnaX) (13–15). τ represents the full-length product of dnaX, and is 643 amino acids (aa) in length, while γ, which is 431-aa long, is the result of a programmed ribosomal frame shift. The τ2, γ, δ and δ′ subunits associate with each other forming a pentameric ring with a partial opening between δ and δ′ through which DNA can pass during clamp loading/unloading (16). The ψ subunit interacts with χ, and also interacts with τ/γ (3,17). The DnaX complex associates with two Pol III core complexes, each of which consists of a single α catalytic (Pol IIIα), ε proofreading and θ subunit to form Pol III* ([τ2γδδ′χψ]1[αεθ]2). The complex consisting of Pol III* in association with two β clamps is referred to as Pol III holoenzyme (Pol III HE) and acts to replicate the chromosome (3).
Similar to clamp loaders from other organisms, DnaX adopts an arc-shaped structure that is proposed to interact with and stabilize an open form of the β clamp without dissociating the clamp into monomers (12,18). This form of the clamp, in which a single interface is broken, adopts a lock washer-like structure. The structure of the clamp loader matches the contour of both the open clamp and the DNA (12,16). A stable DnaX-β complex requires that the τ/γ subunits of DnaX first bind ATP (12). This complex then associates with nicked or primed DNA, ratcheting itself over the 3′-end of the primer like a screw cap on a bottle (19). Once properly positioned at the 3′-end, the τ/γ subunits hydrolyze bound ATP, triggering the immediate dissociation of DnaX from both the clamp and DNA, leaving β behind to close over the dsDNA (12,19).
The E. coli β clamp contains a hydrophobic cleft composed in part of C-terminal residues. This cleft interacts with a fairly well-conserved eubacterial clamp-binding motif (CBM) that is often reported to be QL[S/D]LF, but can be considerably more degenerate (20). Many binding partners make important contacts with the β clamp in addition to the CBM/cleft interaction (21). Another important paradigm that has become evident in recent years is that sliding clamps do not merely recruit partners to DNA, but can also stimulate their catalytic activity. For example, human PCNA enhances base excision repair (22). In addition, interaction of eukaryotic Pol κ with PCNA (23), as well as E. coli Pol IV with β clamp serves to markedly reduce their apparent Km for dNTPs (24). In light of these findings, sliding clamps should be viewed as dynamic coordinators of DNA replication, repair, and damage tolerance.
An important yet unanswered question pertains to how different partners exchange with each other, or switch, on a clamp encircling DNA. This is a particularly important issue in light of the fact that clamp proteins, and the processes that they manage, are so widely conserved throughout evolution. One well-conserved process, termed translesion DNA synthesis (25), involves specialized Pols capable of replicating over lesions in the DNA. Most Pols capable of catalyzing TLS display low fidelity on undamaged DNA (26–28). Mismanagement of TLS Pol function is widely believed to contribute to mutations that lead to human diseases such as cancers (25,27). One area that has received considerable attention in recent years pertains to Pol switching at a lesion in the DNA. One particularly popular yet untested model, referred to as the ‘toolbelt model’, postulates that two different Pols simultaneously bind to the same dimeric β clamp, with each Pol contacting a separate hydrophobic cleft in the dimeric clamp (29–31). In this model, the clamp acts like a toggle switch to regulate access of the different Pols to the DNA template while coordinating high fidelity replication with potentially error-prone TLS.
Despite its reported ability to interact with DNA (9), the clamp nonetheless slides along double strand (ds) DNA with a half-life on circular DNA of >100 min in vitro (32). Due to this remarkable stability, β clamps accumulate on DNA, particularly lagging strand during DNA replication (33,34). As there are only ∼300–600 clamps/cell (32,35), and a new clamp is required for replication of each of the ∼4000 Okazaki fragments (assuming an average length of 1000 bp), it is postulated that the clamp must be actively unloaded (i.e. recycled) from DNA during replication/repair synthesis (32). The DnaX complex, as well as the δ subunit of DnaX, which is present in excess over the other DnaX subunits in vivo, can unload β from DNA (32). Taken together, these findings highlight the need to carefully regulate both the loading and the unloading of clamps in order to ensure that partner proteins, such as TLS Pols, cannot gain access to the DNA unless their function is required. The goal of the work discussed in this report was determine whether a single cleft in the dimeric β clamp was sufficient for clamp loading, Pol III* replication, and unloading clamp from DNA. To help answer these questions, we generated a mutant form of the β clamp, called βC, which lacks its C-terminal five aa residues. Using gel filtration chromatography, surface plasmon resonance (SPR) and isothermal titration calorimetry (ITC), we demonstrated that a single β+ clamp binds two δ subunits of DnaX with negative cooperativity. In contrast, βC was severely impaired for interaction with δ, DnaX, and, albeit to a lesser degree, Pol IIIα. Using a method that we developed for purification of heterodimeric β clamp proteins, we purified a β+/βC clamp comprised of one β+ protomer in complex with one βC protomer. Biochemical characterization of the β+/βC heterodimer revealed that it bound a single δ, and interacted normally with DnaX and Pol IIIα in vitro. Moreover, the β+/βC heterodimer was proficient for loading onto DNA, as well as stimulation of Pol III* replication in vitro. In contrast, both clefts were required for efficient unloading of clamp from DNA by either DnaX or δ. Taken together, these findings provide insight into models for how the β clamp may act to coordinate the actions of its different partners on DNA, as well as how clamp loading and unloading, or recycling, may contribute to this coordination.
MATERIALS AND METHODS
Proteins
The γ3δδ′χψ form of the DnaX clamp loader complex was reconstituted from the individually purified subunits as described (21,36). The α subunit of Pol III (Pol IIIα) (21), and single-strand DNA-binding protein (SSB) (37) were each purified as described previously. Pol III* was purified as described (21). All protein concentrations were determined by densitometry of Coomassie-stained SDS–PAGE, and/or their extinction coefficient. βC lacks its C-terminal five residues, and was cloned using the Quickchange kit (Stratagene) to replace the codons for residues M362 and P363 in the dnaN gene with stop codons (underlined) using primers (Sigma Genosys) dnaNΔ362-6Top (5′-GGCTTATGTTGTCTAATGAATGAGACTG-3′) and dnaNΔ362-6Bottom (5′-CAGTCTCATTCATTAGACAACATAAGCC-3′).
Untagged forms of β+ and βC were purified as described (31) using plasmids pJRC210 (β+) and pJRCβΔ362-6 (βC). N-terminally his6-tagged forms of β+ and βC were expressed from pβHMK and pβΔ362-6HMK, respectively, while the N-terminally myc-tagged form of the β clamp was expressed from pMYCdnaN using strain BLR(DE3) (Novagen). The clamps expressed with a his6-tag also bear an N-terminal cAMP-dependent protein kinase motif (38). Following their induced expression for 3 h at 37°C by addition of 0.5 mM IPTG, cell pellets were resuspended in 50 mM Tris–HCl (pH 7.5) with 10% sucrose and lyzed by French press. The soluble fraction was precipitated with sequential 30 and 70% ammonium sulfate cuts, and the 70% pellet was resuspended in Buffer A [20 mM Tris–HCl (pH 7.5), 0.5 mM EDTA, 0.1 mM DTT and 10% glycerol]. The his6-tagged clamps were purified by cobalt chelating chromatography using an AKTA FPLC equipped with a 5 ml HiTrap chelating HP column (GE Healthcare) charged with 100 mM CoCl2 and equilibrated in Buffer B [50 mM NaPO4 (pH 6.8), 0.5 M NaCl, 10% glycerol and 5 mM imidizole]. Bound protein was eluted using a 100-ml gradient (20 column volumes) of 5–250 mM imidizole in Buffer B. The myc-tagged β protein was also purified using an AKTA FPLC (GE Healthcare). Buffer was exchanged using a 5-ml HiTrap desalting column equilibrated in Buffer A containing 50 mM NaCl according to manufacturer's recommendations. The protein was then loaded onto a MonoQ HR 16/10 column and eluted with a 50–500 mM NaCl gradient. It was then passed over a 5-ml HiTrap heparin column in Buffer A containing 50 mM NaCl to remove nuclease contaminants. Myc-tagged β was not retained on this column.
Circular dichroism spectroscopy
The structure of the βC mutant was compared to that of β+ in solution using circular dichroism spectroscopy. Clamp proteins were dialyzed against 5 mM sodium phosphate (pH 7.0), after which the concentration of each was adjusted to 0.2 mg/ml. The absorption spectra shown for each clamp protein (Figure 1B) were an average of four separate scans in the ultraviolet range (165–240 nm), and were obtained using a Jansco J-710 spectropolarimeter with a 1 cm cell length.
Figure 1.

Deletion of residues M362-L366 in the β clamp impairs the cleft without affecting the overall tertiary structure of the clamp protein. (A) Proximity of residues M362-L366 to the hydrophobic cleft in the β clamp is indicated. This structural figure was generated using imol and the coordinates for the wild type β clamp (1MMI) from the PDB. (B) Averaged results of circular dichroism spectroscopy are shown from four separate scans. Representative results from Superose 12 gel filtration chromatography examining (C) interactions of β+ and βC (2.5 μM as dimer) with the δ subunit of DnaX complex (5 μM), or (D) the α catalytic subunit of Pol III (Pol IIIα; 1.5 μM). Untagged forms of recombinant β+ and βC were used in ITC and gel filtration experiments.
Purification of β+/βC heterodimer
Simultaneous expression of both myc- and his6-tagged forms of the β clamp within the same bacterial strain was achieved using the pCOLADuet-1 vector (Stratagene). The gene encoding the N-terminally myc-tagged β+ was subcloned from pMYCdnaN into multiple cloning site-1 (MCS-1), and that for either the N-terminally his6-tagged β+ (pβHMK) or βC (pβΔ362-6HMK) was subcloned into MCS-2, resulting in plasmids pSKSP100 (β+/β+) and pSKSP101 (β+/βC), respectively. The differentially tagged β clamps were co-expressed in BLR(DE3) cells (Novagen) following induction by 0.5 mM IPTG, resulting in three different forms of the clamp: myc-tagged homodimer, his6-tagged homodimer and a myc- and his6-tagged heterodimer. Cell pellets were resuspended in Buffer B and lysed by French press. The three forms of the clamp were resolved from each other by cobalt chelating chromatography using a 5-ml HiTrap chelating HP column (GE Healthcare) charged with 100 mM CoCl2. Bound protein was eluted using a 100-ml gradient (20 column volumes) of 5–250 mM imidizole in Buffer B. As summarized in Figure 2A, three distinct peaks were observed: peak 1 contained myc-tagged homodimer, which eluted at ∼70 mM imidizole; peak 2 contained the myc-tagged and his6-tagged heterodimer, which eluted at ∼90 mM imidizole; and peak 3 contained the his6-tagged homodimer, which eluted at ∼120 mM imidizole. The myc-tagged β+/his6-tagged β+ (β+/β+) and myc-tagged β+/his6-tagged βC heterodimers (β+/βC) purified identically. For simplicity, we will refer to the wild-type clamp as β+ in the text that follows, regardless of whether it is his6-tagged (βhis), myc-tagged (βmyc) or myc and his6 tagged (β+/β+). The specific form of the clamp used in each experiment is noted in the legend to each figure.
Figure 2.

Purification of heterodimeric β clamp proteins. (A) FPLC chromatography trace of a representative HiTrap cobalt chelating HP column run for myc-β+/his6-β+. Respective positions of peaks 1–3, as well as the type of clamp present in that peak (homodimeric or heterodimeric) are indicated. Myc-tagged clamps are blue, and his6-tagged clamps are red. (B) Western blot analysis of peaks 1–3. Aliquots of each peak were separated by 12% SDS–PAGE, and probed with either anti-myc (top panel) or anti-Penta•His antibody (bottom panel). The load (load), cobalt column flow through (FT), and purified myc- (βmyc) and his6-tagged β+ clamp proteins (βhis) were included as controls. Representative SPR traces in which the his6-tagged subunit was captured on the chip surface using anti-Penta•His antibody (anti-Penta•His on chip), and anti-myc antibody was injected over the chip surface (C), or the myc-tagged clamp subunit was captured on the chip surface using anti-myc antibody and anti-Penta•His was injected (anti-myc on chip) (D) are shown. SPR analysis was performed as described in ‘Materials and Methods’ section. Although representative results for the β+/β+ heterodimer are shown, identical results were observed during purification of the myc-β+/his6-βC heterodimer (β+/βC).
Gel filtration chromatography
Size exclusion chromatography was used to evaluate interactions of the untagged β+ and βC proteins (2.5 μM as dimer) with Pol IIIα (1.5 μM) or the δ subunit of DnaX (5 μM). Proteins were incubated for 30 min at room temperature in 100 μl of Binding Buffer [20 mM Tris–HCl (pH 7.5), 10% glycerol, 2 mM DTT, 0.1 mM EDTA and 100 mM NaCl]. The 100 μl binding reactions were then injected onto a Superose 12 10/30 column (GE Healthcare) equilibrated in the same buffer at a flow rate of 0.25 ml/min at 4°C. Aliquots (80 μl) of the indicated fractions (200 μl) were analyzed by 12% SDS–PAGE and Coomassie staining.
SPR
All SPR experiments were carried out at 25°C using a BIAcore X biosensor instrument (GE Healthcare) with HBS-EP [10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA and 0.005% v/v Surfactant P20] as running buffer. Approximately 5000 response units (RUs) of either BSA free anti-Penta•His Antibody (Qiagen), or 9E10 monoclonal anti-myc antibody (Covance) were covalently immobilized to either the CM5 or C1 sensor chip surface (GE Healthcare) by amine coupling in both flow cells according to the manufacturer's recommendations. The antibody surface in flow cell 1 was used to capture the appropriately tagged form of the β clamp, while antibody in the flow cell 2 was left free to monitor nonspecific interactions for background subtraction. Immediately following capture of the clamp in flow cell 1, the indicated concentrations of the each β binding partner were injected over the chip for 2 min at a flow rate of 30 μl/min. To limit effects resulting from dissociation of clamp dimers tethered to the chip, the antibody surface was regenerated by an injection of 10 mM glycine-HCl (pH 1.5) for 1 min at 50 μl/min. This removed all clamps and interacting proteins from the antibody surface following each interaction cycle, allowing for capture of a fresh aliquot of β dimer for each interaction that was analyzed.
In control experiments, the relative retention of β on the antibody chip was used to determine the stability of heterodimeric clamps as a function of time. Initially, >95% of each heterodimeric clamp contained both the myc- and his6-tags, and were readily captured on the chip surface using either the anti-myc or anti-Penta•His antibody. Since clamp dimers dissociate into monomers and reassociate into a mixture of heterodimer and homodimers with time, we utilized an SPR assay to measure the level of each clamp captured by the tag-specific antibody over time. Results of these experiments indicated that the heterodimers were stable in the high salt buffer (Buffer B) in which they were purified for >180 min. In addition, with the exception of the experiments designed to characterize the purity of the β+/βC heterodimer discussed in the ‘Results’ section (Figure 3), SPR capture experiments utilized preparations of heterodimeric clamp that were supplemented with a 10-fold molar excess of the myc-tagged β+ clamp, which is not recognized by the anti-Penta•His chip (Figure 2C). The presence of a molar excess of myc-tagged β+ relative to the his6-tagged βC (or β+) clamp inhibited his6-tagged βC (or β+) homodimer formation, ensuring the purity of the heterodimer captured on the chip surface in SPR experiments.
Figure 3.

Interactions of the β clamp with the δ subunit of DnaX. (A) Representative SPR results analyzing interaction of δ (4 μM) with various β clamp proteins (∼100 RU). Clamps examined include the his6-tagged β+ homodimer (β+), the his6-tagged βC homodimer (βC), the his6-tagged β+/myc-tagged β+ heterodimer (β+/β+) and the his6-tagged β+/myc-tagged βC heterodimer (β+/βC). Theoretical Rmax values for binding of a single δ subunit (δ1), or for two δ subunits (δ2) are indicated by dashed lines. (B) Titration of δ with his6-tagged β+. Increasing concentrations of purified δ (0.1, 1, 10, 25, 50, 100, 250, 500, 750 and 1000 nM) were injected over ∼100 RU of his6-tagged β+ captured on the anti-Penta•His chip surface. (C) Comparison of sensorgrams from low (25 nM) and high (1 µM) concentrations of δ injected over ∼100 RU of his6-tagged β+ captured on the anti-Penta•His chip surface from panel (B) highlighting differences in association (ka) and dissociation rates (kd). The amplitudes (Rmax) of these SPR traces were normalized to each other to facilitate a direct comparison of the association and dissociation phases of the curves. (D) Titration of δ with β+/βC. Increasing concentrations of purified δ (10, 25, 50, 100 and 500 nM) were injected over ∼100 RU of β+/βC captured on the anti-Penta•His chip surface. (E) The double-injection experiment was performed as outlined in the cartoon (bottom). ∼100 RU of his6-tagged β+ was captured on the anti-Penta•His chip surface (injection 1). Saturating levels (10 µM) of δ were injected (injection 2), forming the β–δ2 complex (Rmax approaching 100 RU). Flow was switched to buffer, allowing for the more loosely bound δ (δ2) to dissociate, resulting in a β–δ1 complex. Increasing concentrations of δ (0, 1, 10, 25, 50, 100, 250, 500, 750 and 1000 nM) were then injected over the β–δ1 complex (injection 3) to determine the binding affinity for the second δ (δ2). (F) Summary table of kinetic constants for various clamp–δ interactions derived from SPR results summarized in (A–E). Results shown for β+–δ were determined using the his6-tagged β+ homodimer, and were indistinguishable from those observed with β+/β+. An interaction of βC with δ was not detected (nd).
SPR experiments to determine kinetic values describing interactions of β with δ or DnaX (γ3δδ′χψ) were conducted using an anti-Penta•His CM5 chip to capture ∼100 RU of his6-tagged clamp. The δ subunit, or DnaX, was injected at concentrations of 0.1–1000 nM, as noted. When analyzing β-DnaX interactions, the running buffer lacked EDTA and was supplemented with 10 mM MgCl2 and 1 mM ATP. An anti-Penta•His C1 chip was used to capture his6-tagged clamps to characterize their interactions with Pol IIIα, which was injected at concentrations of 10–1000 nM. The values for all kinetic constants were obtained by fitting the SPR curves to the 1:1 Langmuir binding model provided with the BIAevaluation Software 4.1. The Rmax was changed from a global to a local parameter in all curve fits to account for slight variations in the amount of β captured on the chip surface between regenerations. The equilibrium dissociation constants for β binding to the DnaX complex in the presence of ATP were obtained using the steady state model. Both the on (ka) and off rates (kd) were too rapid to fit the curves using the simultaneous ka and kd 1:1 Langmuir binding model. Therefore, the off rates were determined by separately fitting only the dissociation portion of the binding curves. The on rates were then calculated using the following equation:
Each fit (with the exception of DnaX) was assessed by the numerical value for the sum of the squared residuals (χ2), which represents the sum of the deviations between the experimental data and the model to which it was fit.
The stoichiometry (S) of partner interactions with the clamp was determined using the following equation:
where Rmax is the theoretical maximum response (RU) of binding to the clamp at a specific surface density of β (Rβ), MWA is the molecular weight of analyte, MWβ is the molecular weight of tagged β dimer (81.2 kDa as dimer), and S is the stoichiometry.
ITC
A VP-ITC microcalorimeter (MicroCal) was used to analyze interaction of myc-tagged β+ with δ. Both β+ and δ were dialyzed against HBS-EP Buffer, and then degassed at 37°C. δ (5.59 μM) was added to the cell pre-equilibrated to 37°C, and β+ (55.9 μM as dimer) was the titrant. The stir speed was 307 r.p.m., the reference power was 20 μcal/s, and injections were 15 μl each at a rate of 0.5 μl/s, except for the first injection, which was at a rate of 1.0 μl/s. A control in which δ was present in the cell and buffer served as titrant was used for background subtraction. Kinetic values were obtained by fitting the binding isotherm to the sequential binding sites model using the Origin software, version 7.02b (Origin Lab Corporation), provided with the instrument.
Clamp loading and unloading assays
Wild-type and mutant clamps were loaded onto a singly nicked, circular, dsDNA template as described previously (32,38). The nicked template was prepared by treatment of 50 μg of pBluescript SK− (Stratagene) with 500 U of Mung Bean nuclease (Promega) in the supplied Reaction Buffer at 2.5 × concentration for 30 min at 37°C. The DNA template was purified away from the reaction components using the QIAprep spin miniprep kit (Qiagen). Wild-type and mutant β clamp proteins were 32P-labeled at the N-terminal cAMP-dependent kinase tag adjacent to the his6 tag using the catalytic subunit of cAMP-dependent protein kinase (New England Biolabs) according to manufacturer's instructions. In control reactions, the myc-tagged β clamps were not labeled. The 32P-β was purified away from unincorporated label using a His-SpinTrap column (GE Healthcare). Clamp was eluted from the column using 200 μl Buffer B containing 250 mM imidizole and adjusted to a final concentration of 1 μM (as dimer).
The DnaX (γ3δδ′χψ) clamp loader complex (0.1 nM) was incubated for 10 min at 37°C with 250 nM of each 32P-β clamp dimer (β+, βC or β+/βC) and 10 nM nicked pBluescript DNA in 100 μl of Loading Buffer [20 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 5 mM DTT, 50 μg/ml BSA (Promega), 4% (v/v) glycerol, 125 mM NaCl and 1 mM ATP]. The loading reaction was then gel filtered through a 4 ml (packed bed volume; 0.5 cm × ∼20 cm) Sephacryl S-500 high-resolution media column (GE Healthcare) equilibrated in Loading Buffer containing 300 mM NaCl at room temperature. Fractions (250 μl) were collected manually, and fractions containing 32P-β that was either loaded onto DNA or free was detected by liquid scintillation counting and native agarose gel electrophoresis (as described below). The peak fractions containing 32P-β on DNA were quickly pooled to serve as substrate in clamp unloading assays.
Clamp unloading assays were done using the 32P-β+ and 32P-β+/βC clamps preloaded onto DNA, immediately after they were isolated by gel filtration. Reactions (20 μl) were initiated by adding either the DnaX clamp loader complex (10 nM to 5 μM) with 1 mM ATP, or the δ subunit (15 nM to 10 μM), to 10-μl aliquots of 32P-β–DNA complex, followed by incubation at 37°C for 10 min. An aliquot of each unloading reaction, as well as the 32P-clamp–DNA substrate used (prior to addition of DnaX or δ), were mixed with 4 μl 6 × Sample Buffer (15% ficoll 400, 0.25% bromophenyl blue and 0.25% xylene cyanol) and loaded onto 1.5% native agarose gels cast in Running Buffer (90 mM Tris-base and 90 mM EDTA), and electrophoresed at 200 V for 140 min at room temperature. Gels were stained with ethidium bromide to detect DNA, photographed, fixed by incubation in 20% (v/v) acetic acid, dried on DEAE paper (Whatmann) at 80°C overnight and exposed using a phosphorimager screen to visualize 32P-β.
Primer extension assay
The ability of β+, βC or β+/βC to stimulate Pol III* replication was measured as described previously (21). Briefly, reactions (20 μl) containing Replication Buffer [20 mM Tris–HCl (pH 7.5), 8.0 mM MgCl2, 0.1 mM EDTA, 5 mM DTT, 1 mM ATP, 5% glycerol and 0.8 mg/ml BSA] were supplemented with 0.133 mM [3H-dTTP]-dNTPs (68.2 CPM/pmol), 2 μM ssDNA binding protein (SSB) and 5 nM of M13 ssDNA primed with SP20 (5′-ACGCCTGTAGCATTCCACAG-3′; Sigma Genosys). Reactions were initiated by addition of 1 nM Pol III*, and were quenched by addition of 1 ml of 15% trichloroacetic acid. Acid insoluble products were collected on 2.4-cm glass microfiber filters (VWR) by vacuum suction, and nucleotide incorporation was quantitated by liquid scintillation spectroscopy.
RESULTS
Generation of a β clamp hydrophobic cleft mutant
Based on a crystal structure of residues 1–140 of δ in complex with a monomeric form of the β clamp βmonomer, residues V36, L177, P242, V247 and M362 of the clamp interact directly with M71, L73 and P74 of δ (39). Furthermore, a separate study reported that substitution of residues P363, M364 or L366 in β with Ala impaired interactions of the clamp with δ (33). Based on these results, we hypothesized that deletion of the C-terminal five residues (M362-L366) of the clamp would impair the ability of the cleft to interact with the CBM present in most clamp partner proteins (Figure 1A). We will refer to this β cleft mutant as βC. In silico modeling of βC using the PDB coordinates for the wild-type E. coli β clamp (1MMI) and the Insight II software suggested that deletion of residues M362-L366 not only removed residues contributing to the cleft region, but additionally caused the cleft to partially collapse in upon itself, rendering it unable to efficiently interact with the CBM, without affecting the tertiary structure of the clamp or its ability to dimerize in solution. Consistent with our in silico modeling, purified βC chromatographed as a dimer in gel filtration (Figure 1C and D), and was indistinguishable from β+ by circular dichroism spectroscopy (Figure 1B), indicating that the wild-type and βC clamps contained comparable fractions of α-helix, β-sheet and β-turn elements. Furthermore, βC failed to interact with the δ subunit of the DnaX complex (Figure 1C), indicating that the cleft was severely impaired for interacting with the CBM of δ. In contrast, β+ interacted with δ, forming what appeared to be a mixture of complexes differing in their stoichiometries of β+ and δ, or the structures of β+ and/or δ within the complex(es), resulting in an altered mobility. These findings are consistent with a previous report concluding that as many as two δ subunits could bind a single clamp in solution (40). The βC clamp was also severely impaired for interaction with Pol IIIα (Figure 1D), consistent with previous reports concluding that the CBM of Pol IIIα contacts the cleft of the clamp (20,41,42). Based on these findings, we conclude that deletion of residues M362-L366 severely impairs the cleft region of the clamp, abrogating its ability to interact with the CBM, without significantly affecting the overall tertiary structure of the clamp.
Purification of a heterodimeric β clamp bearing one wild-type protomer and one βC protomer (β+/βC)
In order to determine whether one or both clefts were required for loading clamp onto DNA, processive replication and unloading clamp off of DNA, we developed a method to purify the β+/βC heterodimeric clamp protein. Briefly, the myc-tagged β+ and his6-tagged βC clamp proteins were co-expressed in the same E. coli cell, and the β+/βC heterodimer was purified away from both myc-tagged β+ and his6-tagged βC homodimers using cobalt affinity chromatography (see ‘Materials and Methods’ section). This same approach was also used to purify a myc-tagged β+/his6-tagged β+ heterodimer for use as a control in experiments discussed below as noted. As summarized in Figure 2A, three distinct peaks were observed following cobalt chromatography. Based on western blot analysis using monoclonal antibodies specific to either the his6- or myc-tags, the first peak (peak 1) contained myc-tagged homodimeric clamp, the third peak (peak 3) contained his6-tagged homodimeric clamp, and the middle peak (peak 2) contained both the myc-tagged and his6-tagged clamps (Figure 2B), suggesting that it was heterodimeric. The presence of the heterodimer in peak 2 was directly demonstrated using an SPR assay. For this assay, we covalently attached either anti-Penta•His or anti-myc antibody to the sensor chip surface. After antibody-capture of the appropriate his6- or myc-tagged clamp protein, we asked whether the second tag was present by injecting the corresponding antibody over the chip surface. As summarized in Figure 2, we were able to capture the clamp from peak 2 with either anti-Penta•His or anti-myc antibodies. Subsequent injection of the anti-myc antibody over the anti-Penta•His captured peak 2-clamp sample indicated that the myc-tag was present (Figure 2C). Similar results were observed for the reciprocal experiment in which the peak 2-clamp sample was captured using anti-myc antibody, followed by injection of anti-Penta•His antibody (Figure 2D). As controls, we verified that the anti-Penta•His antibody was unable to capture the myc-tagged β+ clamp (Figure 2C), and the anti-myc antibody was unable to capture the his6-tagged β+ clamp (Figure 2D). Identical results were observed using the anti-Penta•His and anti-myc antibodies in a native polyacrylamide gel electrophoretic mobility shift assay (data not shown). Taken together, these findings indicate that the myc-tagged and his6-tagged clamps present in peak 2 existed as a heterodimeric clamp. Results from additional SPR experiments discussed below further verify the purity of our β+/βC heterodimer preparation.
A single dimeric β clamp simultaneously binds two δ subunits
Residues L73-F74 of δ contact the cleft on the β clamp (20,39). Since the clamp has two clefts, we hypothesized that it could simultaneously interact with two δ subunits. Results of gel filtration experiments support this hypothesis [(40); and see Figure 1C]. In contrast, since the β+/βC heterodimer has only one functional cleft, we hypothesized that it would interact with only a single δ. As a direct test of these hypotheses, we used an SPR assay similar to that discussed above to analyze interactions of δ with β+, βC and β+/βC. Of relevance to these experiments, the magnitude of the maximal SPR signal (Rmax) is directly proportional to molecular weight of the interacting partner. Thus, the stoichiometry of interacting partners within a complex can be determined.
As summarized in Figure 3A, β+ and β+/βC each interacted with δ, while βC failed to do so. For these experiments, we captured ∼100 RU of each clamp on the chip surface. The β clamp is 81.2 kDa as dimer, and δ is 38.7 kDa. Thus, if one δ interacted with the clamp, the theoretical Rmax would be 48 RU [theoretical Rmax = (100 RU) × (38.7 kDa/81.2 kDa) × 1], while binding of two δ subunits would yield an Rmax of 96 RU [theoretical Rmax = (100 RU) × (38.7 kDa/81.2 kDa)×2]. The binding response for δ saturated at ∼100 RU (Figure 3A), indicating that two δ subunits bound a single β+ clamp. Identical results were obtained regardless of whether we utilized a his6-tagged form of β+ (β+), or a form of β+ comprised of one myc-tagged subunit in complex with a his6-tagged subunit (β+/β+) (Figure 3A). Importantly, since this clamp was captured using anti-Penta•His antibody, the result with β+/β+ indicates that upon binding two δ subunits, the clamp persists as a dimer in solution. If each δ bound to a separate cleft, and binding of each δ acted to open a dimer interface, binding of two δ subunits would have broken the clamp dimer, releasing the myc-tagged protomer from the his6-tagged β subunit captured on the chip surface, reducing the RU signal.
We next determined the stoichiometry of the δ–β+/βC complex. As summarized in Figure 3A, the binding response for δ saturated at ∼50 RU when ∼100 RU of β+/βC was captured on the chip surface. This value is similar to the theoretical Rmax limit of 48 RU for 1:1 binding under these conditions, indicating that the β+/βC heterodimer bound a single δ subunit. This result demonstrates that our β+/βC preparation is homogeneous.
Two δ subunits interact cooperatively with a single β clamp
In addition to analyzing stoichiometries, we were also interested in determining kinetic constants for each clamp-δ interaction. Attempts to model the β+–δ and the β+/βC–δ interactions as simple 1:1 (Langmuir) associations revealed a KD of 43 and 15.6 nM, respectively (Figure 3B, D and F). Although the data for the β+/βC–δ interaction fit well to the Langmuir model (χ2 = 9.38), the β+–δ data did not (χ2 = 129), suggesting that it was not a simple 1:1 interaction. Thus, the 43 nM KD value based on Langmuir binding inaccurately describes the β+–δ interaction. Closer inspection of the shapes of the association and dissociation phases of the sensorgrams suggested that the β+–δ interaction involved negative cooperativity. For example, the shape of the dissociation phase of the sensorgram changed significantly as a function of the δ concentration (Figure 3C). At low δ concentrations (<50 nM), dissociation of δ was slow (2.22 × 10−3 s−1), while at high δ concentrations (>50 nM) its dissociation was 5.3-fold faster (1.17 × 10−2 s−1). Recalculation of the KD for β+–δ utilizing only the sensorgrams resulting from lower concentrations of δ (0.1–50 nM) using the Langmuir binding model, under the assumption that these results largely reflect binding of the first δ to the clamp, revealed a KD of 17.5 nM (Figure 3B and F), which is very close to the 15.6 nM KD value calculated for the β+/βC–δ heterodimer. Furthermore, this data set fit well to the Langmuir binding model (χ2 = 3.25). Taken together, these results suggest that the β+/βC heterodimer is fully proficient for binding one δ via the β+ subunit, but is unable to bind the second δ due to the mutation affecting the cleft in the βC subunit. These findings also further demonstrate the purity of our β+/βC preparation.
In order to further characterize interactions of β+ with δ, we took advantage of the fact that dissociation of δ from the clamp was biphasic (Figure 3B and C). Since only the slow dissociation phase (2.22 × 10−3 s−1) was observed when low levels of δ were injected, it must result from loss of the tightly bound δ, while the rapid phase (1.17 × 10−2 s−1) reflects dissociation of the second, more loosely bound δ. As a result of this binding mechanism, we were able to perform a ‘double-injection’ experiment (see Figure 3E), in which we first injected a saturating level of δ over captured β+ to form a β+–δ2 complex, allowed for dissociation of the more loosely bound δ (δ2), resulting in a β+–δ1 complex, and then injected increasing levels of δ over the β+–δ1 complex to characterize binding of the second δ (δ2) to the clamp. Using this approach, δ2 bound the β+–δ1 complex with a KD of 160 nM (χ2 = 1.43), or roughly 10-fold less tightly than our estimated KD for binding of the first δ (KDδ1 = 17.5 nM; see Figure 3E and F). To confirm that our double inject experiment accurately measured the affinity of the β2–δ1 complex for δ2 we also characterized this interaction using ITC. ITC isotherms fit well to the sequential binding sites model (Figure 4), yielding KD values for binding of the two δ subunits of 259 nM and 4 μM, respectively. The ∼15-fold different in KD values calculated by ITC is similar to the ∼10-fold difference observed by SPR (Figure 3F). The KD values determined by ITC are weaker than those determined by SPR because the SPR experiments were performed at 25°C, while the ITC was performed at 37°C, and KA decreases as temperature increases. Nevertheless, these findings indicate that two δ subunits simultaneously bind a single β clamp with negative cooperativity. Finally, the intracellular concentration of δ is reported to be ∼1.4 μM (32). Thus, the observed negative cooperativity is likely to be biologically relevant.
Figure 4.

Analysis of the β+–δ interaction using isothermal titration calorimetery (ITC). ITC analysis was performed as described in ‘Materials and Methods’ section. The top panel shows heat changes associated with each injection of β+ clamp. The bottom panel shows the fit of the integrated heats to the sequential binding sites model. KD values were calculated by taking the reciprocal of K1 (3.86 × 106) and K2 (2.50 × 105) derived from the fit to the sequential binding sites model (inset).
β+/βC is proficient for interaction with DnaX and Pol IIIα
Although DnaX is thought to contact a single cleft in the dimeric clamp (12), Pol IIIα contains two CBMs (41,42), and may therefore simultaneously contact both clefts. As a direct test of these hypotheses, we asked whether the β+/βC heterodimer interacted normally with DnaX and/or Pol IIIα using the same SPR assay discussed above. As summarized in Figure 5, both β+ and β+/βC interacted with DnaX, while βC failed to do so (Figure 5A). Analysis of the RU signals under steady-state conditions revealed a KD of 27 nM for β+, and 58 nM for β+/βC (Figure 5B–D). The on rates of 7.84 × 106 M−1 s−1 for β+ and 6.92 × 106 M−1 s−1 for β+/βC were almost identical. The only significant difference in the interactions of DnaX with the two clamps was that the off rate of DnaX from β+/βC was twice as fast as that observed for β+ (Figure 5D). This 2-fold difference may be due to the fact that β+/βC has one-half the number of δ binding sites (i.e. clefts). Alternatively, other DnaX subunits in addition to δ may contact the second cleft. Nonetheless, these findings demonstrate that a single cleft is sufficient to support normal interaction of the clamp with DnaX in vitro.
Figure 5.
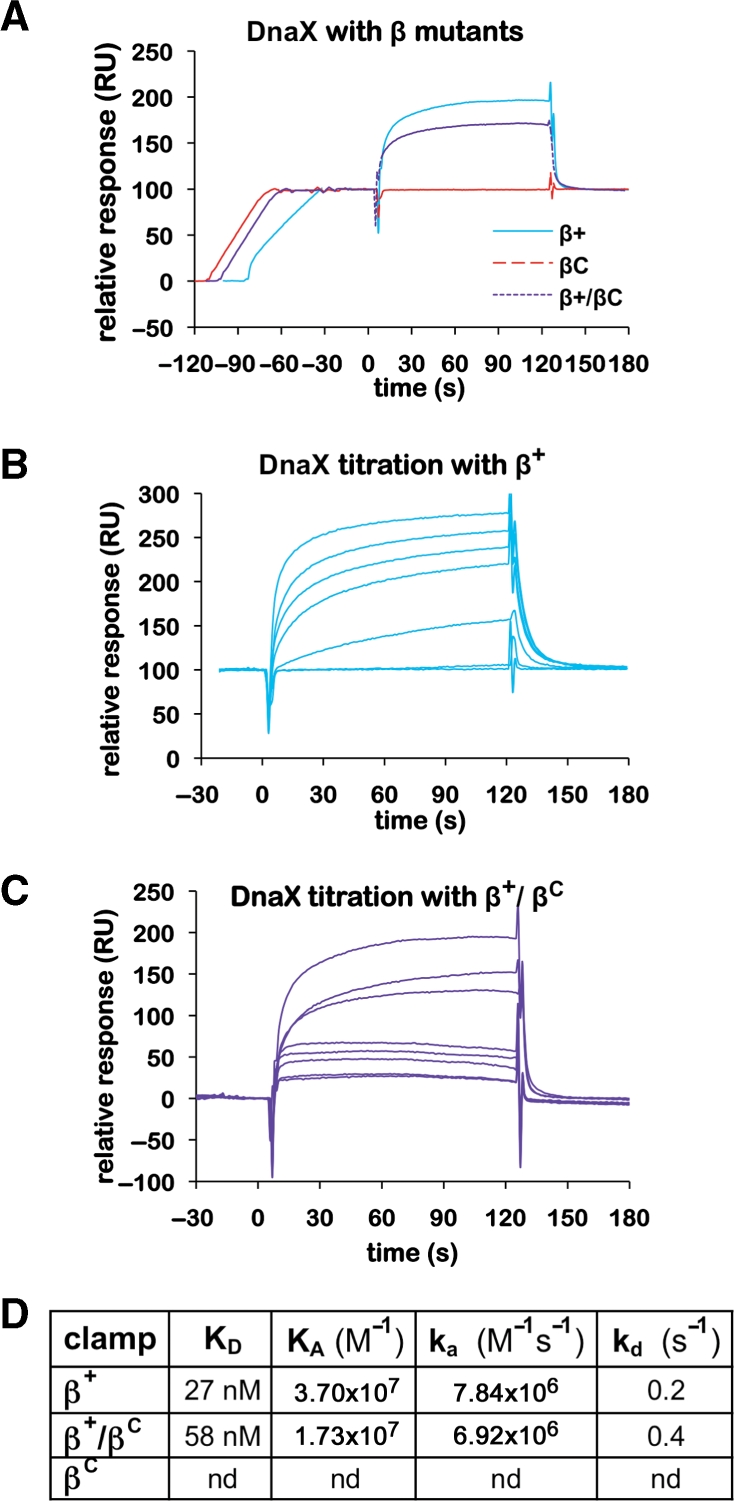
Interactions of wild-type and mutant clamps with the DnaX complex. (A) Representative SPR results analyzing interaction of DnaX (100 nM) with various β clamp proteins (∼100 RU). Clamps examined include the his6-tagged β+ homodimer (β+), the his6-tagged βC homodimer (βC) and the his6-tagged β+/myc-tagged βC heterodimer (β+/βC). (B) Titration of DnaX with β+. Increasing concentrations of the DnaX complex (0, 1, 10, 50, 100, 200 and 500 nM) were injected over ∼100 RU of his6-tagged β+ captured on the anti-Penta•His chip surface. (C) Titration of DnaX with β+/βC. Increasing concentrations of the DnaX complex [1 (in duplicate), 10, 50, 100, 500 (in duplicate) and 1000 nM] were injected over ∼100 RU of β+/βC captured on the anti-Penta•His chip surface. (D) Summary table of kinetic constants for various clamp–DnaX interactions derived from SPR results summarized in (B) and (C). The KD for the DnaX–β+ interaction was reported previously (11), and is included here for direct comparison to βC and β+/βC. An interaction of βC with DnaX was not detected (nd) by SPR.
We next examined interactions of β+, βC and β+/βC with Pol IIIα. In contrast to DnaX, all three forms of the clamp interacted with Pol IIIα, albeit with different affinities (Figure 6). With ∼100 RU of β+ captured on the chip surface, the binding response for Pol IIIα saturated at an Rmax of ∼150 RU. This is very close to the theoretical Rmax of 160 RU calculated for a 1:1 interaction, indicating that a single Pol IIIα was bound to the β+ clamp. Analysis of the SPR results indicated a KD value of 108 nM (χ2 = 4.15; Figure 6B), which is consistent with previously reported values for the β–α interaction (33,43). The KD for the βC–α interaction was 1.78 μM (χ2 = 7.28) (Figure 6B). Since the cleft is impaired in βC, this KD most likely represents contacts of Pol IIIα with noncleft surfaces of the clamp. The off rate of 4.23 × 10−3 s−1 for βC was similar to that of β+ (7.26 × 10−3 s−1), demonstrating that the contact(s) limiting dissociation of α remained intact in βC (Figure 6B). In contrast, the on rate of 2.38 × 103 M−1 s−1 for βC was 28-fold slower than that of β+ (6.75 × 104 M−1 s−1), indicating that the cleft represents the primary contact site on the clamp for Pol IIIα.
Figure 6.

Interactions of wild-type and mutant clamps with Pol IIIα. (A) Representative SPR results analyzing interaction of Pol IIIα (500 nM) with various β clamp proteins (∼100 RU). Clamps examined include the his6-tagged β+ homodimer (β+), the his6-tagged βC homodimer (βC) and the his6-tagged β+/myc-tagged βC heterodimer (β+/βC). (B) Summary table of kinetic constants for various clamp–Pol IIIα interactions derived from SPR experiments in which ∼100 RU of clamp was captured on the chip surface, and 10, 50, 100, 250, 500 and 1000 nM Pol IIIα was injected.
The KD of Pol IIIα for the β+/βC heterodimer was 172 nM (χ2 = 3.03), with an on rate of 7.27 × 104 M−1 s−1. These values are similar to those observed for β+ (Figure 6B), suggesting that Pol IIIα is bound to the cleft present on the β+ subunit within the β+/βC heterodimer. The off rate was 12.5 × 10−3 s−1, which is comparable to that observed for β+. These results, taken together with those discussed above, demonstrate unambiguously that a single cleft is sufficient to support normal interaction of the clamp with both DnaX and Pol IIIα.
Only one cleft is required for clamp loading, while both clefts contribute to its unloading
There are only ∼300–600 clamps per cell (32,35), and once loaded, β remains on DNA with a half-life of >100 min (32). Since there are ∼4000 Okazaki fragments synthesized on lagging strand during each replication cycle, it is necessary that clamps be recycled ∼10 times each. This recycling presumably involves their active removal from DNA, returning them to the soluble pool that supplies clamps for lagging strand replication. DnaX and δ can each catalyze unloading of clamps from DNA (32). We therefore asked whether one or both clefts were required for clamp unloading in vitro.
In order to facilitate quantitation of the level of β clamp on and off of DNA, we took advantage of our N-terminal his6-tag, which also contains the cAMP-dependent kinase motif, to label our clamp proteins with 32P-ATP, as described previously (32). An aliquot of each loading reaction, as well as each relevant column fraction, was fractionated on a native agarose gel to separate clamp that was loaded onto nicked pBluescript plasmid DNA from free clamp. Based on densitometric analysis, ∼72% of β+/βC was loaded onto DNA by DnaX (Figure 7C), while only ∼46% of β+ was loaded (Figure 7B). In contrast, βC was not loaded (Figure 7A), consistent with our findings that this mutant protein failed to interact with DnaX (Figure 5).
Figure 7.

Ability of β+, βC and β+/βC to be loaded on DNA. Loading of βC (A), β+ (B) or β+/βC (C) onto nicked DNA by DnaX was analyzed as described in ‘Materials and Methods’ section. The myc-tagged β+/his6-tagged β+ protein (β+/β+) was used for the β+ results shown in (B). An aliquot of the clamp loading reaction prior to gel filtration chromatography (load), as well as representative gel filtration fractions we analyzed by native agarose gel electrophoresis. Positions of free clamp (free β), and clamp loaded onto nicked DNA (β on DNA) are shown. Phosphorimager analysis displaying positions of radiolabeled clamp proteins is shown at the top of each panel, while results of ethidium bromide staining highlighting positions of nicked and supercoiled DNAs are shown at the bottom of each panel. Results from one representative experiment are shown. This experiment was repeated at least three times, and the amount of clamp loaded onto DNA or free in each experiment was determined using the Molecular Analyst software (BioRad).
The ability of DnaX or δ to unload the β+ or β+/βC clamps from DNA was measured using as substrate the β+ and β+/βC clamp–DNA complexes purified in the experiments described in Figure 7. Similar levels of each purified clamp–DNA complex were incubated with either DnaX or δ, and clamp unloading was monitored using the native gel mobility shift assay. As summarized in Figure 8, titration of DnaX resulted in efficient unloading of β+ from the DNA. Clamp unloading was evidenced by both the increased abundance of free clamp observed in Figure 8A, as well as the apparent increase in the level of clamp-free nicked circular DNA in Figure 8B. Unloading was dependent on ATP (Figure 8A and B, lane 7), as previously reported (32). These same conditions promoted unloading of β+/βC, albeit with a significantly reduced efficiency (Figure 8C and D). Whereas ∼1 µM of DnaX was sufficient to unload ∼50% of β+ from DNA in 10 min, we were unable to achieve this level of unloading for β+/βC at concentrations of DnaX up to 5 μM. As with β+, unloading of the β+/βC heterodimer by DnaX was ATP-dependent (Figure 8C and D, lane 7).
Figure 8.

Ability of β+, βC and β+/βC to be unloaded from DNA by DnaX complex. The ability of DnaX to unload β+ (A, B) or β+/βC (C, D) from DNA was analyzed as described in ‘Materials and Methods’ section. The myc-tagged β+/his6-tagged β+ protein (β+/β+) was used for the β+ results shown in (A) and (B). Positions of free clamp (free β), and clamp loaded onto nicked DNA (β on DNA) are shown. Results of both phosphorimager analysis, illustrating positions of radiolabeled clamp proteins (A, C), as well as ethidium bromide staining, highlighting positions of nicked circular, linear and supercoiled DNAs (B, D) are shown. DnaX was used at the following concentrations: lane 1, 0 nM; lane 2, 10 nM; lane 3, 100 nM; lane 4, 1 μM; lane 5, 2.5 μM; lanes 6 and 7, 5 μM. We were unable to reliably quantitate the level of β+/βC clamp unloading catalyzed by DnaX, due to the poor signal-to-noise ratio.
Unloading by δ was analyzed using the same assay. Consistent with one previous report (32), δ was able to efficiently unload β+ from DNA (Figure 9A and B). Approximately 146 nM of δ was able to unload 50% of β+ on DNA in 10 min (Figure 9E). This value is remarkably similar to the KD value of 160 nM describing the binding of the second δ to the β clamp as measured by SPR (Figure 3). In contrast, δ was unable to unload the β+/βC heterodimer when added at concentrations up to 10 μM (Figure 9C–E). Disappearance of DNA in the presence of higher concentrations of δ was due to δ–DNA interactions (Figure 9B and D). Taken together, these findings indicate that both DnaX and δ require both clefts in order to efficiently unload clamp from DNA.
Figure 9.

Ability of β+, βC and β+/βC to be unloaded from DNA by the δ subunit of DnaX. The ability of δ to unload β+ (A, B) or β+/βC (C, D) from DNA was analyzed as described in ‘Materials and Methods’ section. The myc-tagged β+/his6-tagged β+ protein (β+/β+) was used for the β+ results shown in (A) and (B). Positions of free clamp (free β), and clamp loaded onto nicked DNA (β on DNA) are shown. Results of both phosphorimager analysis, illustrating positions of radiolabeled clamp proteins (A, C), as well as ethidium bromide staining, highlighting positions of nicked circular, linear and supercoiled DNAs (B, D) are shown. For β+/β+, δ was used at the following concentrations: lane 1, 0 nM; lane 2, 15 nM; lane 3, 150 nM; lane 4, 300 nM; lane 5, 450 nM; lane 6, 750 nM; lane 7, 1050 nM; lane 8, 1.2 μM; lane 9, 1.5 μM; lane 10, 2.5 μM; lane 11, 5 μM; lane 12, 10 μM. For β+/βC, δ was used at the following concentrations: lane 1, 0 nM; lane 2, 150 nM; lane 3, 750 nM; lane 4, 1.5 μM; lane 5, 5 μM; lane 6, 10 μM. (E) Quantitation of the results from (A) and (C) summarizing the ability of δ to unload β+/β+ or β+/βC from DNA. The amount of clamp loaded onto DNA or free was determined using the Molecular Analyst software (BioRad). The fraction of clamp remaining on the DNA was measured by calculating the ratio of clamp on DNA/free clamp in each lane, and dividing this ratio by the ratio observed in the absence of added δ (lane 1).
β+/βC is proficient for stimulating Pol III* replication in vitro
In light of our finding that the β+/βC heterodimer was proficient for interaction with DnaX and Pol IIIα, and was efficiently loaded onto DNA by DnaX, we asked whether it was capable of stimulating Pol III* replication in vitro using a primer extension assay. Replication activity in this assay is a reflection of both clamp loading and Pol IIIα–clamp interactions, which contribute to Pol III* processivity. As summarized in Figure 10, Pol III* was unable to synthesize significant levels of DNA in the absence of clamp. Likewise, βC was unable to stimulate Pol III* replication (Figure 10), due to the fact that it was severely impaired for interactions with both the DnaX and Pol IIIα components of Pol III*. In contrast, addition of β+ or β+/βC stimulated Pol III* replication to similar extents (Figure 10). These findings demonstrate that a single cleft in the clamp is sufficient to stimulate Pol III* replication in vitro.
Figure 10.

Ability of β+, βC, and β+/βC to stimulate Pol III* replication in vitro. Replication activity was measured as described in ‘Materials and Methods’ section. The his6-tagged β+/myc-tagged β+ form of β+ was used as the positive control (β+/β+). Results shown represent the average of three separate determinations. Error bars represent the standard deviation.
DISCUSSION
In this report, we describe a method for purification of heterodimeric E. coli β siding clamp proteins (Figure 2). Using this method, we purified a clamp (β+/βC) consisting of the β+ protomer in complex with βC, which bears a mutant cleft that we demonstrated to be severely, if not completely impaired for interaction with the eubacterial CBM (Figure 3 and 5). Our biochemical characterization of the β+/βC heterodimer illustrated that a single cleft is sufficient for efficient loading of the clamp onto DNA by DnaX (Figure 7), as well as stimulation of Pol III* replication in vitro (Figure 10). In contrast, both clefts contributed to clamp unloading, regardless of whether it was catalyzed by DnaX or δ (Figures 8 and 9). Our finding that β+/βC was loaded more efficiently than β+ (72% versus 46%; see Figure 7) may be the result of its reduced efficiency of unloading by DnaX. Regardless, these findings suggest that DnaX utilizes distinct mechanisms for loading and unloading clamps. Interactions of the clamp with the DNA template may underlie this difference. A crystal structure of the β clamp on DNA was recently described (9). In the crystal, residues R24 and Q149, among others, interacted specifically with dsDNA. In addition, the cleft of the clamp interacted with ssDNA located downstream of the 3′-end of the primer. Thus, inefficient unloading of the β+/βC clamp catalyzed by DnaX may be due to competition between ssDNA and DnaX for the single cleft. Alternatively, interaction(s) of the clamp with DNA may contribute to unloading. Further work is required in order to test these hypotheses.
In the crystal structure of residues 1–140 of δ in complex with a mutant form of the clamp that exists as a monomer in solution (βmonomer), residues L73-F74 of δ contacted the CBM of β (39). Thus, the simplest interpretation of our results concerning interaction of δ with the β clamp is that two δ subunits bind a single dimeric clamp, with each δ contacting a separate cleft. Our finding that β+/βC bound a single δ is consistent with this conclusion (Figure 3). It has been suggested that binding of δ to the clamp destabilizes the adjacent dimer interface, effecting clamp opening/unloading (18,32). Results discussed in Figure 3 demonstrate that binding of two δ subunits to a single clamp fails to dissociate the clamp into monomers. Thus, the ability of δ to open a dimer interface in the clamp appears to require more than it simply contacting the cleft region. This result, taken together with our finding that both clefts are required for δ to unload the clamp from DNA (Figure 9), suggests that two δ subunits may catalyze clamp unloading by simultaneously binding to both clefts and the DNA. In this way, δ could utilize energy from DNA binding to induce a conformational change in β to open one, or possibly both dimer interfaces, effectively unloading the clamp from the DNA. Further work is required in order to test this model.
Our observation that δ binds the clamp with negative cooperativity may be the result of steric clashing between the two δ subunits. Based on the crystal structure, it was argued that the positioning of δ on the βmonomer would preclude a second δ from binding the unoccupied cleft due to steric constraints (39). However, it should be stressed that the actual conformation of δ on a dimeric clamp, either in solution or encircling DNA is unknown. Alternatively, the observed negative cooperativity may result form conformational changes induced in the peptide backbone of the clamp upon binding of the first δ. These conformational changes may influence the structure of the second cleft, and/or other surfaces of the clamp involved in interactions with one or more specific partner proteins. An interesting aspect of this model is that partners could impose a plastic hierarchy in clamp–partner interactions. By inducing conformational changes in β, binding of one partner to the clamp could dictate both the identity(ies) and the order in which additional partners are recruited. Clamp–DNA and partner–DNA interactions likely influence these interactions, as well, and thus contribute to the switching mechanism(s).
Our finding that δ requires both clefts in order to unload clamp from DNA suggests that that a clamp in complex with a partner that is doing work on DNA is protected from unloading, while a clamp that is not in complex with a partner is efficiently unloaded from DNA by δ. Alternatively, since δ binds the clamp with negative cooperativity, it could effect displacement of a resident partner, provided it had access to one cleft, and was able to influence affinity of the partner bound to the other cleft. Although it would be counter productive to unload a clamp that was in complex with a partner that was doing work on DNA, it would be advantageous to unload a clamp that was in complex with a partner that was for some reason precluded from functioning properly, such as Pol III* stalled at a lesion. Conformational differences between a moving Pol III* and a stalled Pol III* may influence access of δ to the cleft of a β clamp that is not in complex with Pol IIIα. Upon binding, δ may induce a conformational change in the polypeptide backbone of the β clamp, which, in turn, may destabilize interaction of the clamp with Pol IIIα, leading to dissociation of Pol III*. This would allow for recruitment of either another Pol capable of catalyzing TLS through its association with clamp persisting on the DNA, or a second δ to effect unloading of the clamp. In the latter case, DnaX could then load a new clamp at the primer/template junction abutting the lesion for subsequent recruitment of a TLS Pol. Thus, δ and/or DnaX may influence Pol switching either directly, by disassembling clamp-Pol complexes stalled at lesions, or indirectly, by loading new clamps at primer/template junctions abutting lesions.
Our finding that the β+/βC heterodimer was indistinguishable from β+ with respect to its ability to stimulate Pol III* replication (Figure 10) indicates that processive replication by Pol III* requires that Pol IIIα interact with only a single cleft in the clamp. As such, these findings provide support for the toolbelt model, in which Pol III* is bound to one cleft, and a TLS Pol is bound to the second cleft. Alternatively, the structure of the Pol IV little finger domain in complex with the β clamp, indicating that Pol IV contacts both the cleft and the rim of the clamp (44), suggests that surfaces of the clamp in addition to the cleft play roles in Pol function and/or switching. Our finding that Pol IIIα retained weak affinity for βC is consistent with this model (Figure 6). Thus, Pol switching on the clamp may involve surfaces in addition to the cleft. Our method to purify heterodimeric clamp proteins will enable a detailed biochemical dissection of the mechanisms by which the clamp mediates Pol switching in vitro.
Human PCNA remains on circular DNA with a half-life of ∼24 min (45). Since there are ∼2 × 107 Okazaki fragments synthesized on lagging strand during each replication cycle, assuming an average length of 200 bp, and there are ∼105 PCNA trimers in HeLa cells (46), it seems likely that eukaryotes recycle their clamps on average ∼100 times each during replication/repair synthesis. The human clamp loader complex, RFC, as well as the 40-kDa subunit of RFC, RFC2, are each capable of unloading PCNA from DNA (47). It will be interesting to determine whether interactions of RFC2 with PCNA involve negative cooperativity, as well as whether three RFC2 subunits are capable of binding a single PCNA clamp. Likewise, purification of differentially tagged heterotrimeric forms of PCNA would enable the determination of the number of clefts required for loading, processive replication by Pol δ and Pol ε, and unloading, as well as DNA repair and damage tolerance functions that are managed by the clamp.
In summary, findings discussed in this report indicate that interaction of the β clamp with δ, as well as possibly other partner proteins, is far more complex than previously appreciated. Further work is required in order to understand the mechanistic basis for the observed negative cooperativity between β and δ. Likewise, it will be interesting to determine whether additional partners interact with the clamp through positive or negative cooperativity, and whether cooperativity is observed in trans between different partners. Although the goal of the work discussed in this report was focused largely on determining the roles of the clefts in clamp loading, Pol III* replication and clamp unloading/recycling, our method for purification of heterodimeric clamp proteins will facilitate detailed biochemical dissection of the mechanistic roles of the clamp in DNA replication and repair, as well as in coordinating the actions of its multiple partners on the DNA during DNA replication, repair and TLS.
FUNDING
National Institutes of Health grants GM066094 (to M.D.S.); and F31GM073586 (to S.K.S.P.). Funding for open access charges: National Institutes of Health (grant GM066094 to M.D.S.).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank the members of our laboratory for many helpful discussions, particularly Robert Maul (University at Buffalo) for cloning pMYCdnaN and purification of Pol III*, Kenneth Blumenthal and Sujith Alphy (University at Buffalo) for help with CD analysis of the β+ and βC proteins, Brian Lang (GE Healthcare) for expert advice regarding kinetic analysis of our SPR data, and the expert reviewers for their insightful comments.
REFERENCES
- 1.Burgers PM, Kornberg A, Sakakibara Y. The dnaN gene codes for the beta subunit of DNA polymerase III holoenzyme of escherichia coli. Proc. Natl Acad. Sci. USA. 1981;78:5391–5395. doi: 10.1073/pnas.78.9.5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 3.McHenry CS. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 2003;49:1157–1165. doi: 10.1046/j.1365-2958.2003.03645.x. [DOI] [PubMed] [Google Scholar]
- 4.Argiriadi MA, Goedken ER, Bruck I, O'Donnell M, Kuriyan J. Crystal structure of a DNA polymerase sliding clamp from a Gram-positive bacterium. BMC Struct. Biol. 2006;6:2. doi: 10.1186/1472-6807-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kong XP, Onrust R, O'Donnell M, Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: a sliding DNA clamp. Cell. 1992;69:425–437. doi: 10.1016/0092-8674(92)90445-i. [DOI] [PubMed] [Google Scholar]
- 6.Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell. 1994;79:1233–1243. doi: 10.1016/0092-8674(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 7.Moarefi I, Jeruzalmi D, Turner J, O'Donnell M, Kuriyan J. Crystal structure of the DNA polymerase processivity factor of T4 bacteriophage. J. Mol. Biol. 2000;296:1215–1223. doi: 10.1006/jmbi.1999.3511. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Jeruzalmi D, Moarefi I, Leighton L, Lasken R, Kuriyan J. Crystal structure of an archaebacterial DNA polymerase. Struct. Fold Des. 1999;7:1189–1199. doi: 10.1016/s0969-2126(00)80053-2. [DOI] [PubMed] [Google Scholar]
- 9.Georgescu RE, Kim SS, Yurieva O, Kuriyan J, Kong XP, O'Donnell M. Structure of a sliding clamp on DNA. Cell. 2008;132:43–54. doi: 10.1016/j.cell.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanov I, Chapados BR, McCammon JA, Tainer JA. Proliferating cell nuclear antigen loaded onto double-stranded DNA: dynamics, minor groove interactions and functional implications. Nucleic Acids Res. 2006;34:6023–6033. doi: 10.1093/nar/gkl744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heltzel JMH, Scouten Ponticelli SK, Sanders LH, Duzen JM, Cody V, Pace J, Snell EH, Sutton MD. Sliding clamp-DNA interactions are required for viability and contribute to DNA polymerase management in Escherichia coli. J. Mol. Biol. 2009;387:74–91. doi: 10.1016/j.jmb.2009.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeruzalmi D, O'Donnell M, Kuriyan J. Clamp loaders and sliding clamps. Curr. Opin. Struct. Biol. 2002;12:217–224. doi: 10.1016/s0959-440x(02)00313-5. [DOI] [PubMed] [Google Scholar]
- 13.Blinkova A, Hervas C, Stukenberg PT, Onrust R, O'Donnell ME, Walker JR. The Escherichia coli DNA polymerase III holoenzyme contains both products of the dnaX gene, tau and gamma, but only tau is essential. J. Bacteriol. 1993;175:6018–6027. doi: 10.1128/jb.175.18.6018-6027.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blinkowa AL, Walker JR. Programmed ribosomal frameshifting generates the Escherichia coli DNA polymerase III gamma subunit from within the tau subunit reading frame. Nucleic Acids Res. 1990;18:1725–1729. doi: 10.1093/nar/18.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flower AM, McHenry CS. The gamma subunit of DNA polymerase III holoenzyme of Escherichia coli is produced by ribosomal frameshifting. Proc. Natl Acad. Sci. USA. 1990;87:3713–3717. doi: 10.1073/pnas.87.10.3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeruzalmi D, O'Donnell M, Kuriyan J. Crystal structure of the processivity clamp loader gamma (gamma) complex of E. coli DNA polymerase III. Cell. 2001;106:429–441. doi: 10.1016/s0092-8674(01)00463-9. [DOI] [PubMed] [Google Scholar]
- 17.Xiao H, Dong Z, O'Donnell M. DNA polymerase III accessory proteins. IV. Characterization of chi and psi. J. Biol. Chem. 1993;268:11779–11784. [PubMed] [Google Scholar]
- 18.Stewart J, Hingorani MM, Kelman Z, O'Donnell M. Mechanism of beta clamp opening by the delta subunit of Escherichia coli DNA polymerase III holoenzyme. J. Biol. Chem. 2001;276:19182–19189. doi: 10.1074/jbc.M100592200. [DOI] [PubMed] [Google Scholar]
- 19.Bowman GD, Goedken ER, Kazmirski SL, O'Donnell M, Kuriyan J. DNA polymerase clamp loaders and DNA recognition. FEBS Lett. 2005;579:863–867. doi: 10.1016/j.febslet.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 20.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE, Jennings PA. A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl Acad. Sci. USA. 2001;98:11627–11632. doi: 10.1073/pnas.191384398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maul RW, Ponticelli SK, Duzen JM, Sutton MD. Differential binding of Escherichia coli DNA polymerases to the beta-sliding clamp. Mol. Microbiol. 2007;65:811–827. doi: 10.1111/j.1365-2958.2007.05828.x. [DOI] [PubMed] [Google Scholar]
- 22.Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, Aas PA, Steinsbekk K, Bakke O, Krokan HE. Post-replicative base excision repair in replication foci. EMBO J. 1999;18:3834–3844. doi: 10.1093/emboj/18.13.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haracska L, Unk I, Johnson RE, Phillips BB, Hurwitz J, Prakash L, Prakash S. Stimulation of DNA synthesis activity of human DNA polymerase kappa by PCNA. Mol. Cell. Biol. 2002;22:784–791. doi: 10.1128/MCB.22.3.784-791.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertram JG, Bloom LB, O'Donnell M, Goodman MF. Increased dNTP binding affinity reveals a nonprocessive role for Escherichia coli beta clamp with DNA polymerase IV. J. Biol. Chem. 2004;279:33047–33050. doi: 10.1074/jbc.C400265200. [DOI] [PubMed] [Google Scholar]
- 25.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2nd. Washington, D.C: ASM Press; 2006. [Google Scholar]
- 26.Friedberg EC, Feaver WJ, Gerlach VL. The many faces of DNA polymerases: strategies for mutagenesis and for mutational avoidance. Proc. Natl Acad. Sci. USA. 2000;97:5681–5683. doi: 10.1073/pnas.120152397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296:1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- 28.Sutton MD, Walker GC. Managing DNA polymerases: coordinating DNA replication, DNA repair, and DNA recombination. Proc. Natl Acad. Sci. USA. 2001;98:8342–8349. doi: 10.1073/pnas.111036998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Indiani C, McInerney P, Georgescu R, Goodman MF, O'Donnell M. A sliding-clamp toolbelt binds high- and low-fidelity DNA polymerases simultaneously. Mol. Cell. 2005;19:805–815. doi: 10.1016/j.molcel.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 30.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duzen JM, Walker GC, Sutton MD. Identification of specific amino acid residues in the E. coli beta processivity clamp involved in interactions with DNA polymerase III, UmuD and UmuD'. DNA Repair. 2004;3:301–312. doi: 10.1016/j.dnarep.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 32.Leu FP, Hingorani MM, Turner J, O'Donnell M. The delta subunit of DNA polymerase III holoenzyme serves as a sliding clamp unloader in Escherichia coli. J. Biol. Chem. 2000;275:34609–34618. doi: 10.1074/jbc.M005495200. [DOI] [PubMed] [Google Scholar]
- 33.Naktinis V, Turner J, O'Donnell M. A molecular switch in a replication machine defined by an internal competition for protein rings. Cell. 1996;84:137–145. doi: 10.1016/s0092-8674(00)81000-4. [DOI] [PubMed] [Google Scholar]
- 34.Stukenberg PT, Turner J, O'Donnell M. An explanation for lagging strand replication: polymerase hopping among DNA sliding clamps. Cell. 1994;78:877–887. doi: 10.1016/s0092-8674(94)90662-9. [DOI] [PubMed] [Google Scholar]
- 35.Sutton MD, Duzen JM, Maul RW. Mutant forms of the Escherichia coli beta sliding clamp that distinguish between its roles in replication and DNA polymerase V-dependent translesion DNA synthesis. Mol. Microbiol. 2005;55:1751–1766. doi: 10.1111/j.1365-2958.2005.04500.x. [DOI] [PubMed] [Google Scholar]
- 36.Pritchard AE, Dallmann HG, Glover BP, McHenry CS. A novel assembly mechanism for the DNA polymerase III holoenzyme DnaX complex: association of deltadelta' with DnaX(4) forms DnaX(3)deltadelta'. EMBO J. 2000;19:6536–6545. doi: 10.1093/emboj/19.23.6536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lusetti SL, Hobbs MD, Stohl EA, Chitteni-Pattu S, Inman RB, Seifert HS, Cox MM. The RecF protein antagonizes RecX function via direct interaction. Mol. Cell. 2006;21:41–50. doi: 10.1016/j.molcel.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner J, Hingorani MM, Kelman Z, O'Donnell M. The internal workings of a DNA polymerase clamp-loading machine. EMBO J. 1999;18:771–783. doi: 10.1093/emboj/18.3.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeruzalmi D, Yurieva O, Zhao Y, Young M, Stewart J, Hingorani M, O'Donnell M, Kuriyan J. Mechanism of processivity clamp opening by the delta subunit wrench of the clamp loader complex of E. coli DNA polymerase III. Cell. 2001;106:417–428. [PubMed] [Google Scholar]
- 40.Naktinis V, Onrust R, Fang L, O'Donnell M. Assembly of a chromosomal replication machine: two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particle. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 1995;270:13358–13365. [PubMed] [Google Scholar]
- 41.Dohrmann PR, McHenry CS. A bipartite polymerase-processivity factor interaction: only the internal beta binding site of the alpha subunit is required for processive replication by the DNA polymerase III holoenzyme. J. Mol. Biol. 2005;350:228–239. doi: 10.1016/j.jmb.2005.04.065. [DOI] [PubMed] [Google Scholar]
- 42.Lopez de Saro FJ, Georgescu RE, Goodman MF, O'Donnell M. Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair. EMBO J. 2003;22:6408–6418. doi: 10.1093/emboj/cdg603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DR, McHenry CS. Identification of the beta-binding domain of the alpha subunit of Escherichia coli polymerase III holoenzyme. J. Biol. Chem. 1996;271:20699–20704. doi: 10.1074/jbc.271.34.20699. [DOI] [PubMed] [Google Scholar]
- 44.Bunting KA, Roe SM, Pearl LH. Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J. 2003;22:5883–5892. doi: 10.1093/emboj/cdg568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao N, Turner J, Kelman Z, Stukenberg PT, Dean F, Shechter D, Pan ZQ, Hurwitz J, O'Donnell M. Clamp loading, unloading and intrinsic stability of the PCNA, beta and gp45 sliding clamps of human, E. coli and T4 replicases. Genes Cells. 1996;1:101–113. doi: 10.1046/j.1365-2443.1996.07007.x. [DOI] [PubMed] [Google Scholar]
- 46.Morris GF, Mathews MB. Regulation of proliferating cell nuclear antigen during the cell cycle. J. Biol. Chem. 1989;264:13856–13864. [PubMed] [Google Scholar]
- 47.Cai J, Gibbs E, Uhlmann F, Phillips B, Yao N, O'Donnell M, Hurwitz J. A complex consisting of human replication factor C p40, p37, and p36 subunits is a DNA-dependent ATPase and an intermediate in the assembly of the holoenzyme. J. Biol. Chem. 1997;272:18974–18981. doi: 10.1074/jbc.272.30.18974. [DOI] [PubMed] [Google Scholar]