


Abstract
Replicative DNA polymerases possess 3′ → 5′ exonuclease activity to reduce misincorporation of incorrect nucleotides by proofreading during replication. To examine if this proofreading activity modulates DNA synthesis of damaged templates, we constructed a series of recombinant human DNA polymerase δ (Pol δ) in which one or two of the three conserved Asp residues in the exonuclease domain are mutated, and compared their properties with that of the wild-type enzyme. While all the mutant enzymes lost more than 95% exonuclease activity and severely decreased the proofreading activity than the wild-type, the bypass efficiency of damaged templates was varied: two mutant enzymes, D515V and D402A/D515A, gave higher bypass efficiencies on templates containing an abasic site, but another mutant, D316N/D515A, showed a lower bypass efficiency than the wild-type. All the enzymes including the wild-type inserted an adenine opposite the abasic site, whereas these enzymes inserted cytosine and adenine opposite an 8-oxoguanine with a ratio of 6:4. These results indicate that the exonuclease activity of human Pol δ modulates its intrinsic bypass efficiency on the damaged template, but does not affect the choice of nucleotide to be inserted.
INTRODUCTION
Cellular DNA is constantly damaged by various endogenous and exogenous agents. Many of resultant DNA lesions can block replication. The recent discovery of a number of DNA polymerases specialized for translesion synthesis (TLS) has uncovered one of the general mechanisms to tackle the replication blocking lesions (1–4). In a current model, when the replication fork stalls at a damaged site, proliferating cell nuclear antigen (PCNA) is monoubiquitinated, consequently recruiting TLS DNA polymerase(s). Thus the stalling of the replicative polymerases serves as a major trigger for PCNA ubiquitination and the subsequent lesion bypass by TLS polymerases.
DNA polymerase δ (Pol δ) is one of the three replicative polymerases in eukaryotes. Pol δ is essential for replication and is a major enzyme for lagging-strand synthesis (5). In mammals, it is composed of four subunits: p125, p55, p66 and p12 (6). Pol δ's activity is strongly stimulated by PCNA and replication factor C (RFC) which loads PCNA onto DNA. These factors may affect lesion bypass by Pol δ as suggested by Maga et al. (7). However, since PCNA tends to slide on DNA quite freely, stable loading of PCNA on the primer/template is critical for evaluation of their effects on Pol δ's function in vitro.
Like most other replicative DNA polymerases, Pol δ has an intrinsic 3′ → 5′ exonuclease activity in its catalytic subunit, p125. This activity participates in three distinct fidelity mechanisms: proofreading, mismatch repair and Okazaki fragment maturation (8). In addition to these functions on normal templates, the exonuclease activity of replicative polymerases is anticipated to influence DNA synthesis on damaged templates. Indeed, the exonuclease activities of DNA polymerases I, II and III of Escherichia coli suppress bypass of apurinic/apyrimidinic (AP) sites (9,10). In eukaryotes, Pol δ mutants deficient in the exonuclease activity were constructed in yeast (8,11,12) and in mice (13). While they were examined for replication fidelity on normal templates, their effect on damaged DNA synthesis has not been reported.
To investigate the effects of Pol δ's exonuclease activity on lesion replication, we developed a bacterial expression system for recombinant human Pol δ, which allowed the purification of Pol δ by two column chromatography steps. This expression/purification system for human Pol δ facilitated the screening of a number of mutant enzymes. Using this system, we constructed exonuclease-deficient mutants. Eukaryotic Pol δ enzymes share three conserved Asp residues in the exonuclease domain with bacteriophage DNA polymerases in which these clustered residues are essential for the exonuclease activity (14,15). Here, we introduced one or two mutations into the three conserved Asp residues of human Pol δ, prepared three mutant enzymes as well as the wild-type Pol δ, and compared their proofreading activities and translesion capabilities. Their activities were assayed on oligonucleotide primer/templates which were blocked at both ends with Lac repressor proteins and a biotin/streptavidin, respectively, to ensure stable loading of PCNA onto the DNA. Consequently PCNA and its loading factor, replication factor C (RFC), were indispensable in this assay system as for replication in vivo. Under these conditions, we examined the Pol δ's behavior not only for stall/bypass efficiency but also for error-free/error prone ratio. The nucleotide inserted opposite the lesion was quantitatively determined by pyrosequencing of the fully extended products.
We focused on two lesions: AP site and 8-oxoguanine (8-OG). The AP site and 8-OG are two of the most abundant lesions generated during normal cell growth (16), so that the replication machinery must encounter these lesions frequently. A previous study demonstrated that AP sites strongly block yeast Pol δ (17). As cited above, however, exonuclease-mutants of some bacterial DNA polymerases were able to bypass this lesion (9,10). In contrast, the 8-OG does not strongly block replication but it is highly mutagenic and introduces a CA transversion (18). Using our assay system, we found that, among the three exonuclease-mutant Pol δ enzymes we tested, two of them slightly increased the bypass efficiency at the AP site, while one mutant significantly decreased the translesion efficiency at this lesion than the wild-type enzyme. The effect of the exonuclease mutations on the bypass efficiency of the 8-OG template was in the same tendency but milder than that of the AP site template. Pyrosequencing of the bypass products revealed that the wild-type Pol δ and all the mutants inserted predominantly A opposite the AP site, and inserted C and A opposite the 8-OG with a 6:4 ratio. Thus the exonuclease mutations of Pol δ affected the efficiency of lesion bypass but not the choice of nucleotides inserted opposite lesions.
MATERIALS AND METHODS
Construction of recombinant Pol δ expression vectors
cDNAs encoding human Pol δ subunits (p125, p55, p66 and p12) were obtained by RT–PCR of mRNA from HEK293T cells with specific primers. An NdeI site and a XhoI site were introduced into the N-terminus and C-terminus, respectively, of each cDNA. The p125 cDNA was introduced into a pET32 (Novagen)-derived vector in which the Lac repressor gene was deleted. The three other cDNAs were introduced into a pCOLADuet-1 (Novagen)-derived vector, producing pCOLA-hPold234. In this plasmid, each cDNA was placed under its own T7 promoter. N-terminal 16× His and Flag tags and an N-terminal Myc tag were attached to p12 and p66, respectively (Figure 1A). Site-directed mutations shown in Figure 5A were introduced into the p125 cDNA by PCR using appropriate primers. Throughout this study, the DNAs obtained by PCR were confirmed by sequencing to be free from undesired mutations.
Figure 1.

Expression and purification of recombinant human Pol δ. (A) Schematic structures of expression plasmids for Pol δ. (B) Purification profile of Pol δ. Each fraction was electrophoresed in an SDS 4–12% polyacrylamide gel and stained with Coomassie Brilliant Blue. Lane 1, load to a Ni2+-Sepharose column; lane 2, flow-through of the Ni2+-Sepharose column; lane 3, a wash fraction with 1 M NaCl-containing buffer; lane 4, a wash fraction with 100 mM imidazole-containing buffer; lane 5, eluate from the Ni2+-Sepharose column with 300 mM imidazole-containing buffer; lane 6, flow-through of an SP-Sepharose column, lane 7, a wash fraction with 200 mM NaCl-containing buffer; lane 8, a wash fraction with 500 mM NaCl-containing buffer; lane 9, eluate from the SP column with 1 M NaCl-containing buffer. (C) Purified Pol δ wild-type (wt) and exonuclease mutant enzymes. (D) Purified recombinant human RFC visualized with Coomassie staining.
Figure 5.

Site-directed mutants of Pol δ exonuclease domains. (A) Amino-acid sequence comparison of the exonucelase domains of replicative DNA polymerases. Budding yeast and human Pol δ catalytic subunits are compared to DNA polymerases from bacterophages, RB69 and T4. Identical residues and similar residues conserved among the four enzymes are indicated in red and blue fonts, respectively. Three aspartic acids presumed to be critical for exonuclease activity are indicated with asterisks. Mutations introduced into human Pol δ in this study are indicated below. (B) DNA polymerase activity with wild-type and three mutant human Pol δ enzymes. The assay was conducted with the normal primer/49-nt G templates in the presence of PCNA, RFC and the Lac repressor as described in ‘Materials and Methods’ section. The activity was calculated as [intensity of the fully extended product]/([intensity of the fully extended product] + [intensity of the unextended primer]), and normalized with the activity of the wild-type enzyme as 100%. Average activities and standard deviations of mutant enzymes from three experiments were presented in the right panel. (C) Exonuclease assay of Pol δ. 5′-Labeled single-stranded oligonucleotide (ssDNA primer in Figure 2) was incubated with indicated Pol δ enzymes. The exonuclease activity was calculated as described in ‘Materials and Methods’ section. Average activities and standard deviations from three reactions were presented in the right panel.
Expression and purification of Pol δ
A host bacterial strain, BLR(DE3)(pLacRARE2), was obtained by transformation of BLR(DE3) with a pLacRARE2 plasmid from Rosetta2 strain (Novagen). Human Pol δ was expressed in BLR(DE3)(pLacRARE2) cotransformed with pET-hPold1 and pCOLA-hPold234 for 12 h at 16°C after induction with 1 mM IPTG. The bacterial cells were harvested by centrifugation and stored at –80°C until use. The subsequent procedures were performed at 4°C. The frozen cells were suspended in Buffer P-200 (40 mM HEPES–NaOH, pH 7.5, 200 mM NaCl, 10% glycerol, proteinase inhibitors [Complete, EDTA-free; Roche Diagnostics]) and lyzed with a French Press. The supernatant was recovered after centrifugation at 10 000 r.p.m. for 10 min in a JA20 rotor (Beckman), supplemented with imidazole (final 30 mM) and loaded onto a NiSO4-charged Chelating Sepharose FF column (GE Healthcare) which was pre-equilibrated with Buffer PI-30 (40 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10% glycerol, 30 mM imidazole, proteinase inhibitors). After sequential washing the column with Buffers PI-30, P-1000 (40 mM HEPES–NaOH, pH 7.5, 1 M NaCl, 10% glycerol, proteinase inhibitors) and PI-100 (40 mM HEPES-NaOH, pH 7.5, 100 mM NaCl, 10% glycerol, 100 mM imidazole, proteinase inhibitors), the Pol δ-containing fraction was eluted with Buffer PI-300 (40 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10% glycerol, 300 mM imidazole, proteinase inhibitors) supplemented with Triton X-100 (final 0.01%). The Ni-column eluate was loaded onto an SP Sepharose FF column (GE Healthcare) pre-equilibrated with Buffer PI-300 plus 0.01% Triton-X100. After washing the column with Buffer P-200 (+0.01% Triton X-100) and Buffer P-500 (+0.01% Triton X-100), the Pol δ-containing fraction was eluted with Buffer P-1000 (+0.01% Triton X-100). This fraction was dispensed in small aliquots and stored at –80°C. The site-directed mutant Pol δs were expressed and purified by the same procedures as the wild-type enzyme.
Authenticity of subunits of the purified Pol δ visible by Coomassie Blue staining was confirmed by either immunoblotting (p125, p66, p12) or mass spectrometry (p55) as described in ‘Results’ section. In addition, among concomitant bands, a band above p66 was identified as E. coli DnaK by mass spectrometry, while several bands between 25 and 50 kDa were detected with anti-p125 antibody, indicating that they resulted from breakdown products of p125.
Expression and purification of RFC, PCNA and Lac repressor
Recombinant human RFC was expressed in BLR(DE3)(pLacRARE2) cotransformed with pET-hRfc1 and pCOLA-hRfc2345 and purified by column chromatography with SP Sepharose FF, Heparin Sepharose, gel filtration and another SP Sepharose FF. Details of the RFC expression vectors and purification procedures will be described elsewhere (manuscript in preparation). Recombinant mouse PCNA attached with N-terminal 6xHis tag and protein kinase target site was expressed in BL21(DE3)(pLysS) (Novagen) and purified by chromatography on a NiSO4-charged Chelating Sepharose FF column. E. coli Lac repressor protein whose C-terminal 29 residues was deleted and attached with a 6× His tag was expressed in BL21(DE3)(pLysS) and purified by chromatography on a NiSO4-charged Chelating Sepharose FF column. Since the original C-terminal domain is responsible for tetramer formation (19), the Lac repressor protein used in this study is expected to form a homodimer but not a tetramer.
Oligodeoxynucleotides
All the oligonucleotides used in this study were synthesized by the DNA Synthesis Facility at Fox Chase Cancer Center. Phosphoramidite derivatives for synthetic AP site analog (dSpacer) and 8-oxodeoxyguanine were purchased from Glen Research. The oligonucleotides longer than 50 nt were prepared by ligation of two shorter oligonucleotides. All the oligonucleotides shown in Figure 2 were purified by denaturing polyacrylamide gel electrophoresis.
Figure 2.

Sequences of oligonucleotide primers and templates. 3′-Terminal mispaired nucleotides in primers are underlined. Lesions in templates, 8-OG (O) and tetrahydrofuran AP site analog (F), are also underlined. 5′-Terminal biotin is indicated as B. The lac operator sequence (lacO) is indicated by shading. Numbering of the sequence is based on the primer strand.
DNA synthesis assays
For primer extension on an oligonucleotide primer/template, a primer indicated in each experiment (Figure 2) was labeled with 32P at the 5′ terminus and annealed to an indicated template. This primer/template was incubated with streptavidin-conjugated magnetic beads (Streptavidin MagneSphere Paramagnetic Particles; Promega) in the binding buffer (20 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10 mM MgCl2) for 5 min, and the unbound primer/template was removed by washing the magnetic beads with the binding buffer. A 20 μl standard reaction with 10 femtomole primer/template-bound magnetic beads was carried out at 37°C for 30 min with 200 ng PCNA, 140 ng RFC, 250 ng Pol δ, 800 ng Lac repressor in the buffer containing 20 mM HEPES-NaOH, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.1 mg/ml BSA, 0.01% Triton X-100, 2 mM ATP, 40 μM each of four dNTPs. The reaction was terminated by placing in ice/water. The magnetic beads were washed with the binding buffer and suspended in 10 μl of the loading solution (95% formamide, 0.5% SDS, 25 mM EDTA). After boiling, the supernatant was subjected to electrophoresis in an 8 M urea-containing 12% polyacrylamide gel. 32P-labeled reaction products resolved by gel electrophoresis were visualized by either autoradiography with X-ray film or phosphorimager, BAS-2500 (Fujifilm). Quantitative analyses were conducted with BAS-2500 and its ImageGauge software.
Exonuclease assay
The ssDNA primer shown in Figure 2 was labeled with 32P at the 5′ terminus, and 10 femtomole labeled oligonucleotide was incubated with 37 ng of indicated Pol δ enzymes at 37°C for 10 min in the 10 μl buffer containing 20 mM HEPES–NaOH, pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.1 mg/ml BSA, 0.01% Triton X-100. The reaction was terminated by adding 20 μl of the loading solution, and the samples were analyzed by electrophoresis in an 8 M urea containing 24% polyacrylamide gel followed by scanning with phosphorimager as above. The relative exonuclease activity was calculated by a method similar to that of Lin et al. (20). Briefly, dn, intensity of each band corresponding to the product with n nucleotides removed from the 3′ end, was quantitated by phosphorimager. The exonuclease activity was calculated as ∑(n × dn)/∑dn in which n = 0 to 18.
Replication assay with Xenopus egg extracts
Nucleoplasmic extract (NPE) of Xenopus laevis eggs was prepared following the standard procedure (21). Pol δ was depleted from NPE with a rat monoclonal antibody against the Xenopus Pol δ p50 subunit. Briefly, 20 μl Protein G Sepharose beads (Sigma) were precoated with the monoclonal antibody and incubated with 40 μl of NPE and 20 μl of egg lysis buffer (ELB: 10 mM HEPES-KOH, pH7.5, 250 mM sucrose, 2.5 mM MgCl2, 50 mM KCl, 1 mM DTT) at 4°C for 2.5 h. The incubation was repeated with a fresh batch of antibody-coated beads and the depleted extracts were saved as 5 μl aliquots at –80°C. Mock depletion was carried out with Protein G Sepharose beads only. A typical DNA synthesis reaction contained: 5 μl Polδ-depleted or mock-depleted NPE, 0.5 μl ATP cocktail (20 mM ATP/200 mM phosphocreatine/0.5 mg/ml creative kinase/50 mM DTT), 0.25 μl 32P-dATP, 0.375 μl pBS—ssDNA annealed with the ssDNA primer (Figure 2) (0.2 μg/μl), and 1.375 μl ELB or recombinant Pol δ (dialyzed in advance against ELB). After incubation at room temperature for the indicated times, the samples were treated with 0.5% SDS, 6 mM EDTA, and 1 μg/μl Proteinase K at room temperature for 2 h and analyzed by 1% TAE-agarose gel electrophoresis. The gels were dried and analyzed for 32P incorporation by phosphorimager.
Pyrosequencing of fully extended products
Fully extended products in Figure 6 were extracted from the polyacrylamide gel and recovered by phenol/chloroform extraction and ethanol precipitation. The recovered DNA fragments were subjected to two-step amplifications with Advantage cDNA Polymerase Mix (Clonetech): (i) unidirectional amplification with a single primer complementary to the extended product (25 cycles of 94°C for 5 s/50°C for 5 s/68°C for 30 s), and (ii) subsequent standard PCR amplification with the second primer in addition to the first primer (30 cycles of the same temperature condition). The amplified products prepared by this method were predominantly derived from the strand extended by Pol δ, and the contribution of the template strand was negligible. The nucleotide of interest in the amplified products was determined by pyrosequencing with a PSQ96MA system (Biotage) in the Biomarker and Genotyping Facility of Fox Chase Cancer Center. Primers used for amplifications and pyrosequencing were summarized in Figure S1 and Table S1 of the Supplementary Data.
Figure 6.

Extension of 3′-mispaired primers. Reactions were conducted with indicated primers annealed to the 49-nt G template (Figure 2) in the absence or presence of dNTPs as described in ‘Materials and Methods’ section. All the reactions contained PCNA, RFC and Lac repressor.
Conventional sequencing of 57 nt G:G extended products
Fully extended products from this primer were amplified as described above except that neither of the amplification primers was not biotinylated. The amplified fragments were inserted into pCR4Blunt-TOPO (Invitrogen) and used for transformation of TOP10 bacteria. Plasmid DNA was prepared from individual colonies, tested by restriction digestion for its insert. The clones carrying the inserted fragment were subjected to DNA sequencing with an ABI capillary genetic analyzer in the Sequencing Facility of Fox Chase Cancer Center.
RESULTS
Preparation of recombinant human Pol δ expressed in bacteria
We have developed a bacterial expression system for recombinant human Pol δ in which a tag of 16 histidines was attached at the N-terminus of p12 subunit (Figure 1A). In preliminary experiments, we observed that hexahistidine tagged proteins larger than 100 kDa were partially eluted from the Ni2+-charged resin during washing with 100 mM imidazole-containing buffer which still contained a significant quantity of concomitant bacterial proteins (data not shown). In contrast, a majority of His16-tagged Pol δ was steadily bound to the column in the same washing condition. Accordingly the expressed enzyme was purified by convenient two-step procedures with a Ni2+-charged column and an SP Sepharose column (Figure 1B). The SP eluate fraction contained four polypeptides that were detected by Coomassie staining (Figure 1B). The largest one was identified as the catalytic subunit of Pol δ by immunoblotting with anti-p125 antibody, while the second one was a tagged p66 by immunoblotting with anti-Myc antibody (data not shown). The fourth subunit, p12, was detected by immunoblotting for its attached FLAG tag. In addition, the third polypeptide was confirmed as p55 of Pol δ by mass spectrometric analysis (data not shown).
The activity of thus purified Pol δ was examined with magnetic beads-bound oligonucleotide primer/template (Figure 3A). As shown in Figure 3B, Pol δ's activity was stimulated with PCNA and RFC (lanes 4–6). Furthermore, the activity increased more than two-folds when both ends of the template were blocked, one by Lac repressor proteins and the other by streptavidin (compare lanes 3 and 6). This is consistent with the observation that PCNA tends to fall off from a free end of linear DNA (22). Together these results indicate that the bacteria-expressed Pol δ, similarly to the Pol δ purified from human cells, is stimulated by a sliding clamp, PCNA, loaded by RFC.
Figure 3.

Primer extension on an end-blocked oligonucelotide by recombinant PCNA, RFC and Pol δ. (A) Schematic structure of the end-blocked primer/template. A biotin (B) is attached at the 5′ end of the template and is bound to a streptavidin (SA)-conjugated magnetic bead. (B) Primer extension reactions were conducted with indicated protein factors and the normal primer/49-nt G template as described in ‘Materials and Methods’ section.
Recombinant Pol δ is active in the Xenopus egg extract replication system
To further evaluate the activity of the recombinant Pol δ, we examined if it can support DNA replication in the Xenopus nucleoplasmic extract (NPE). A singly-primed circular pBS- ssDNA was used as the primer/template to monitor DNA synthesis and its conversion to supercoiled double-stranded forms. Pol δ is the major polymerase responsible for the replication of this type of substrate in NPE. As shown in Figure 4, DNA synthesis in extracts depleted of Pol δ was drastically reduced. This defect could largely be complemented by the addition of the recombinant Pol δ as measured by both DNA synthesis and supercoiled product formation. These data provided further evidence that the recombinant Pol δ is fully active in supporting the complete synthesis and maturation of the daughter strand.
Figure 4.

Complementation of Xenopus egg extracts with recombinant Pol δ. Single-stranded circular DNA was annealed with non-radioactive ssDNA primer (Figure 2) and used for replication synthesis in the presence of 32P-dATP. (A) DNA replication in Pol δ- or mock-depleted NPE. Samples taken at the indicated times were treated with SDS/EDTA/Proteinase K and separated by 1% TAE/agarose gel electrophoresis. (B) Rescue of the replication defect by the recombinant Pol δ.
Exonuclease-deficient mutants of Pol δ
Exonuclease domains of replicative DNA polymerases have been identified from studies of bacteriophage enzymes (14,15). These regions are well conserved even in eukaryote enzymes including human Pol δ p125 (Figure 5A). Analyses of site-directed mutations of phage and yeast enzymes indicate that three Asp residues in these domains are important for the exonuclease activity (8,14). Based on these data, we designed three mutants of human Pol δ: exo-1 (D515V), exo-23 (D402A/D515A) and exo-24 (D316N/D515A). They all target the Asp residues that are involved in divalent metal bindings (23), and conserved among DNA polymerases of organisms from bacteriophage to mammals. The exo-1 mutation was identical to 5DV, the mutant Pol δ from budding yeast that had the lowest exonuclease activity among a series of mutants tested by Jin et al. (8). D402/D515A for exo-23 and D316N/D515A for exo-24 were selected from mutations of T4 DNA polymerase by Abdus Sattar et al. (14) as the DNA polymerases with lowest exonuclease activities and highest polymerase activities. These mutant enzymes were expressed and purified by the same procedures as for the wild-type Pol δ (Figure 1C). Their polymerase activities were measured by the same method as shown in Figure 3. All three mutants retained polymerase activities comparable to the wild-type (Figure 5B). Compared to similar mutants of T4 DNA polymerases, among which double-mutants, D219A/D324A and D112A/D324A, lost 70% of the polymerase activity (14), our human Pol δ mutants retained the higher polymerase activities. Next we tested their 3′ → 5′ exonuclease activities with a 5′-labeled single-stranded oligonucleotide, and quantitated them as described by Lin et al. (20) (Figure 5C). All mutant enzymes decreased the exonuclease activity by more than 95% compared to the wild-type enzyme.
Proofreading of 3′-mispaired primers by Pol δ
To determine how these mutations in the exonuclease domain affect the proofreading activity of Pol δ, we employed a series of 3′-mismatched primers (Figure 2) and examined the degradation of the primers in the absence of dNTPs and their extensions in the presence of dNTPs by Pol δ (Figure 6). When dNTPs were omitted from the reactions, the wild-type Pol δ partially degraded the 3′ side of primers which were detected as smear products below the intact primers. Compared to the ssDNA degradation shown in Figure 5C, the 3′ → 5′ degradation products of dsDNA was significantly lesser even by the wild-type enzyme. Nevertheless, the reactions by mutant Pol δ enzymes did not generate such smear products or, if any, notably weaker signals than by the wild-type. This notable difference between the wild-type and mutant enzymes was observed with all mismatched primers tested. When the reactions were carried out in the presence of dNTPs, Pol δ extended all the primers to full-sized products. The extension efficiency was comparable among the wild-type, exo-1 and exo-23 Pol δ, whereas extension by the exo-24 enzyme was less efficient. In particular, the extension from two primers, 70 nt A:A and 71 nt C:C was inefficient.
Although the extension efficiency of 3′-mismatched primers by two Pol δ mutants was not significantly different from that of the wild-type enzyme, the proofreading activity should be directly tested by examining whether the mismatched nucleotide was replaced with a correct nucleotide before extension. Therefore we recovered extended products from polyacrylamide gels shown in Figure 6, amplified the primer-derived strand and determined what nucleotide resided at the originally mismatched position. For this purpose, we applied the pyrosequencing method which can generate quantitative sequencing data at a target position (24). Representative raw data of this analysis are presented in Figure 7A, in which the nucleotide sequencing of the extended product are shown in ‘pyrogram’. In this case, following ‘ATCT’, A and C were incorporated at the position X of the products extended by the wild-type Pol δ with a ratio of 66%:34%, whereas the products by the exo-1 enzyme had A and C with a ratio of 90%:10%. Since these products were derived from the reaction with 49 nt A:G primer/49-nt G template (Figure 2), ‘A’ corresponds to the mispaired nucleotide, while ‘C’ corresponds to the corrected nucleotide. All results of pyrosequencing of 3′-mispaired primer extended products are summarized in Figure 7B. It is noteworthy that the proofreading efficiency by the wild-type Pol δ was highly variable among different primers tested here. With the 49 nt A:G and 59 nt T:C primers, only 40% or less of the fully extended products were derived from the corrected primers by the wild-type Pol δ, while 80% or more of mispairs of three other primers (63 nt G:G, 70 nt A:A and 71 nt C:C) were corrected in their fully extended products.
Figure 7.

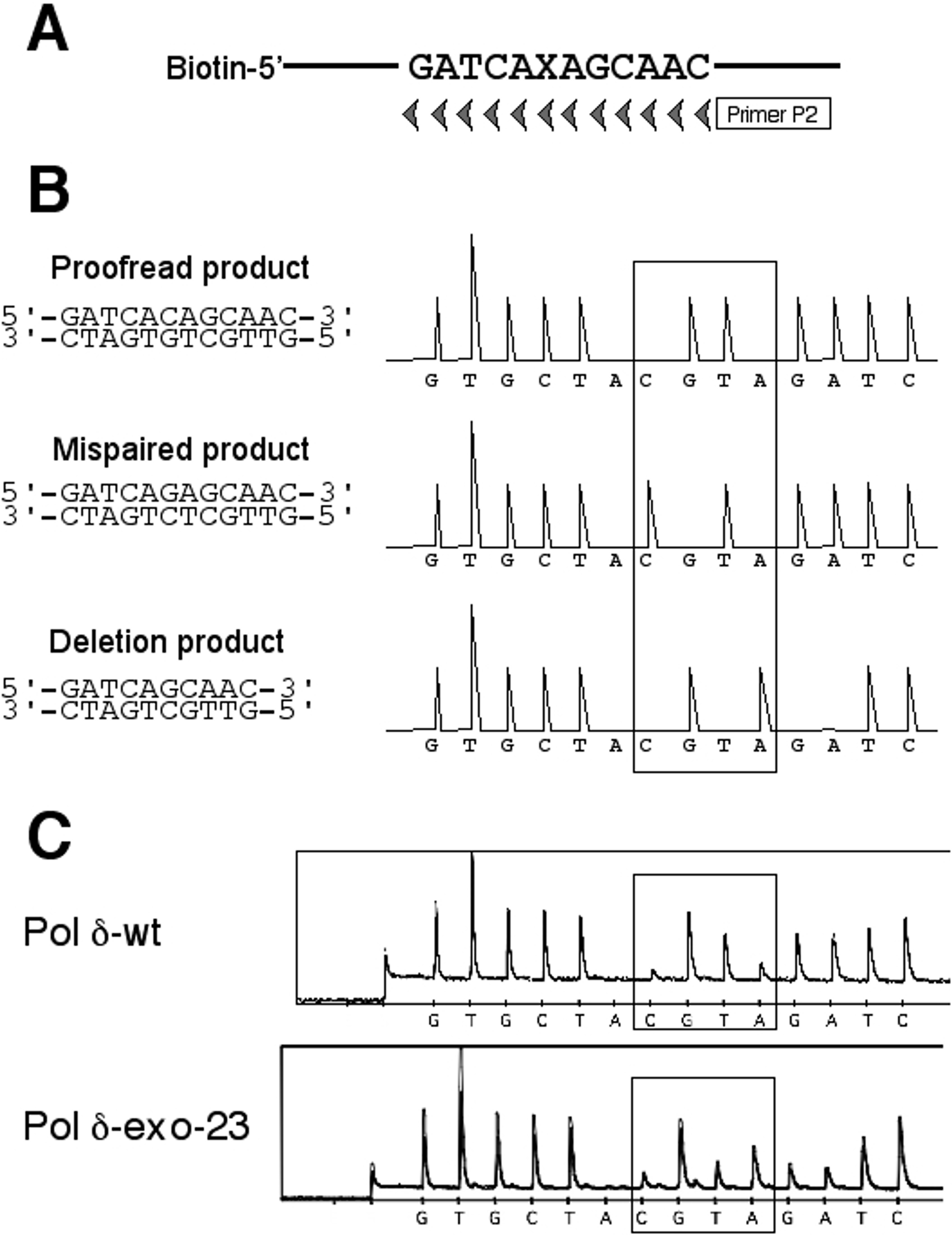
Nucleotide determination of products extended from 3′-mispaired primers. (A) Representative data of pyrosequencing. The ratio of the corrected nucleotide and the mispaired nucleotide was obtained from two peaks in the pyrogram (boxed). In this case with the 49 nt A:G primer, A is the mispaired nucleotide, while C is the corrected nucleotide opposite G. (B) Summary of pyroseqencing data. Efficiency of synthesis of fully extended products over unextended primers (quantitated from Figure 6) is presented along with the ratio of proofread products (dark area) and mispaired products (gray area). Deletion products from the 57-nt G:G primer were indicated as white area. (C) Three mechanisms for extension of the 57-nt G:G primer. The 3′-mispaired nucleotide is indicated with an asterisk.
In the case of 57 nt G:G primer, their pyrograms did not well match to those expected from a simple mixture of the proofread product and mispaired product (Figure S2 in the Supplementary Data). Re-examination of the sequence context of this primer and the template uncovered another extension mechanism in which the looping out of 2 nt in the template can allow perfect basepairing of the 3′ end of the primer (Figure 7C). Accordingly we re-analyzed the programs with three expected products: proofread product, mispaired product, and product resulting from 2-nt deletion. As shown in Figure 7B, the wild-type enzyme generated mainly the proofread product with less quantities of the deletion product and the mispaired product, whereas the major product by all three mutant enzymes was the deletion product. To confirm this interpretation of the pyrogram data, we isolated nine clones each from the wild-type Pol δ products and the exo-1 products, and determined their structures by the conventional sequencing method (Table 1). The direct sequencing data were consistent with the pyrosequencing results, although some minor products carrying additional mutations were identified. Thus the exo- mutant enzymes were deficient in proofreading. In addition, this result indicates that Pol δ, even the wild-type enzyme, can tolerate the looping-out of a few nucleotides in the template.
Table 1.
Analysis of 57 nt G:G extension products by conventional sequencing
| Pol δ | Type | Sequencea | Frequency |
|---|---|---|---|
| wt | Proofreading | ……GATCACAGCAA…… | 5/9 |
| Deletion | ……GATCAGCAA…… | 3/9 | |
| Other | ……GATCAGCGCAA…… | 1/9 | |
| exo-1 | Deletion | ……GATCAGCAA…… | 5/9 |
| Deletion + Mutation | ……GATCGGCAA…… | 1/9 | |
| Mispaired | ……GATCAGAGCAA…… | 1/9 | |
| Mispaired + Insertion | ……GATCAGCAGCAA…… | 2/9 |
aNucleotides resulting from proofreading, mispairing or additional mutation are underlined.
Lesion bypass by Pol δ
To evaluate the lesion bypass capability of Pol δ, we employed oligonucleotide templates in which either an 8-OG or synthetic AP site analog was placed at a defined position (Figure 2A). By the same in vitro system as for the proofreading analysis, the wild-type and mutant Pol δ enzymes were tested for primer extension on the templates wherein a lesion was located at either one of two different positions (Figure 8). In these templates, 3 nt before, and 4 nt after the lesion are exactly the same, but the distance from the primer end is different: 8 nt in 49-nt X templates or 22 nt in 63-nt X templates. In both types of templates, the wild-type Pol δ bypassed the 8-OG with 60–70% efficiencies (Figure 9), indicating that 8-OG caused only weak stalling. Nucleotide-level analysis of lesion-stalled products indicated that Pol δ primarily stopped just before insertion of a nucleotide opposite the lesion on the 49-nt OG template (Figure 8A, lane 7). In the case of a synthetic AP site analog, the wild-type Pol δ bypassed the lesion with efficiencies of only 32% or less (Figure 9), and its strong stalling occurred before and after a nucleotide was inserted opposite the lesion (Figure 8A, lane 12). When the bypass efficiency was compared between the two templates, Pol δ stalled at the AP site on the 63-nt X template more strongly than on the 49-nt X template. This is also the case with the 8-OG, although it is less marked. Since 7 nt surrounding the lesion are the same between two sets of templates, this difference in bypass efficiency seems to result from different distances from the primer end to the lesion, rather than the local sequence context.
Figure 8.

Primer extension by Pol δ on lesion-containing templates. (A) Extension on the 49-nt X templates. Positions of the primer end and the lesion (X) are indicated on the right of the gel. (B) Extension on the 63-nt X templates. Both reactions were conducted in the presence of PCNA, RFC and Lac repressor as described in ‘Materials and Methods’ section.
Figure 9.

Bypass efficiency of Pol δ on damaged templates. The efficiency was calculated as [intensity of the full extended product]/([intensity of the fully extended product] + [intensity of the lesion-stalled product]) from the data shown in Figure 8. The efficiency on the normal templates was calculated with the intensity of the products stopped at the position corresponding to the lesions.
When compared to the wild-type Pol δ, two mutant enzymes, exo-1 and exo-23, increased the bypass efficiencies at the AP site analog and slightly at the 8-OG (Figure 9). In contrast, the exo-24 mutant significantly decreased the bypass efficiencies. These results imply that the lesion bypass capability of Pol δ does not simply correlate with its exonuclease activity. On the other hand, the direction of change of the bypass efficiency seems to be specific for each mutant of Pol δ but not affected by lesion type or template structure.
Nucleotide incorporated opposite lesions by Pol δ
We next determined what nucleotide was incorporated opposite the lesions by pyrosequencing of the fully extended products. In the experiments with the 8-OG templates, we observed that C and A were predominantly inserted opposite the lesion (Figure 10). This is consistent with previous studies reporting that these 2 nt are most likely to be incorporated opposite an 8-OG (18,25–28). In all cases we tested, Pol δ inserted C or A with a ratio of 6:4. Mutations in the exonuclease domain of Pol δ did not affect this ratio. This result indicates that Pol δ can incorporate C, a correct nucleotide, slightly better than A, incorrect nucleotide, opposite the 8-OG. Nonetheless this lesion is still mutagenic at a significant level.
Figure 10.

Nucleotides incorporated opposite the lesions. Pyrosequencing of the fully extended products on the 63-nt X templates (Figure 8B) was conducted as described in ‘Materials and Methods’ section. The pyrograms shown here are corresponding to the position opposite the lesions of damaged templates or the control G nucleotide of the normal template.
For incorporation opposite the synthetic AP site, two rules have been proposed: the ‘A-rule’ (29) and the ‘5′-rule’ (30). The ‘A-rule’ means that a number of DNA polymerases tend to insert dAMP opposite the AP site. On the other hand, the ‘5′-rule’ model proposes that some replicative DNA polymerases might loop out the baseless site on the template strand and use its 5′-adjacnet base to instruct nucleotide incorporation. Thus, we tested these two models with our template sequence, focusing on the insertion of A and G opposite the AP site analog. In all cases examined here, Pol δ, either the wild-type or any of the exonuclease-mutants, inserted predominantly A opposite the AP site analog, while G insertion was not more than the background level (Figure 10). It is reported that another mechanism called ‘C-rule’ works for bypassing AP sites in budding yeast in which C is mainly incorporated opposite the AP site (31). However, our pyrosequencing data clearly indicate that C was not inserted at more than the background level. Thus the nucleotide choice of Pol δ at the AP site can be explained by the A-rule.
DISCUSSION
We report the expression, purification and characterization of recombinant human Pol δ wild-type and exonuclease-mutant enzymes. Several laboratories have already reported recombinant human Pol δ prepared by various systems. Podust et al. (32) and Xie et al. (33) independently developed baculoviral vectors for human Pol δ expression. These systems provided high yields of the active enzyme that may be subject to post-translational modifications specific for eukaryotes. However, since transfer of recombinant DNAs to baculovirus is a time-consuming step, the baculoviral expression systems may not be convenient for preparation of numerous mutant enzymes. Recently, Masuda et al. (34) reported bacterial expression and purification of untagged human Pol δ. Their system also provided a high yield of active enzyme, but its purification included two column chromatography steps with linear gradient elution and one size-exclusion column step. The expression and purification system for human Pol δ described here is less laborious than the others and suitable for rapid screening of mutant constructs.
The exonuclease activity of Pol δ has been well studied in yeast systems. Jin et al. (8) reported that S. cerevisiae Pol δ has a strong exonuclease activity which can completely degrade a primer annealed to a template within 10 min in the absence of dNTPs. In contrast, the exonuclease activity of S. pombe reported by Chen et al. (35) appears to be much milder than that of S. cerevisiae. The human Pol δ described here did not extensively degrade primers annealed to templates in the absence of dNTPs (Figure 6, lanes with the wild-type enzyme). This seems to be consistent with the relationship between humans and two yeasts in which humans are biologically closer to S. pombe than S. cerevisiae. To quantitatively detect the exonuclease activity of human Pol δ, we employed a single-stranded oligonucleotide as a substrate for exonuclease assay (Figure 5C). This assay demonstrated that the wild-type human enzyme indeed has 3′ → 5′ exonucleae activity. Furthermore, the mutant enzymes in which the conserved exonuclease domain was altered decreased their exonuclease activities by more than 95% compared to the wild type. These results indicate that the ssDNA exonuclease activity is derived from the conserved exonuclease domain of human Pol δ.
In spite of the apparently weak exonuclease activity on dsDNA, the human Pol δ showed significant proofreading activities on 3′-mispaired primers (Figure 7B). In the same assay, three exonuclease-mutant enzymes showed constantly lower proofreading activities than the wild-type enzyme. Nevertheless, comparison of the mispair-extension and proofreading activities by the wild-type and mutant enzymes on a series of 3′-mispaired primer/template combinations suggest that not only mispaired nucleotides but also their surrounding sequences and/or distances from the primer's 5′ terminus may affect Pol δ's behavior when encountering a 3′-mispair. Using a more simple enzyme, Bacillus stearothermophilus DNA polymerase I large fragment, Johnson and Beese (36) analyzed all 12 possible mispairs, and accordingly categorized into four groups of mispairs (as shown as primer nucleotide:template nucleotide): (i) G:T, G:G, A:C, T:C to disrupt template strand and pre-insertion site; (ii) T:T, C:T to disrupt primer strand assembly of catalytic site; (iii) A:G, T:G to disrupt template and primer strands; (iv) A:A, G:A, C:C to be frayed at insertion site. However, our results do not indicate similarity among data with 57 nt G:G, 59 nt T:C and 63 nt G:G while data with 70 nt A:A and 71 nt C:C appear similar (Figure 7B). Johnson and Beese also proposed the effect of long-range distortions caused by a mismatch up to 6 nt away from the primer end (36). Thus further analyses with a large collection of mispaired primer/template designed by a more systematic manner will be required to decipher universal rules governing mispair extension and proofreading by DNA polymerase.
Identification of the deletion products from the 57 nt G:G primer extension directly uncover some flexibility of human Pol δ in the pocket wherein the template/primer DNA resides. It should be noticed, however, that the experimental condition we employed may have allowed formation of the loop-out before the enzyme was assembled to DNA for polymerization. Therefore it is not clear whether the 3′-mispair can be converted inside the polymerase–DNA complex to the template loop-out.
For analyses of the proofreading activity and also for the lesion bypass analyses, we employed pyrosequencing to determine the nucleotide inserted into the fully extended product at the position of interest. This method enabled us to obtain quantitative data in a fast and cost-effective manner compared to the conventional method which requires subcloning of the products and subsequent sequencing of at least 20 clones. The pyrosequencing technique also has its own disadvantage, such as ambiguity derived from deletion, insertion and homonucleotide regions, relatively high background up to 10%. As described for the 57 nt G:G primer extension, analysis of more than two types of products may be further complicated. Nevertheless, the result obtained from pyrosequencing was in good agreement with that from conventional sequencing (Figure 7B, Table 1).
Translesion activity of Pol δ was tested with templates carrying either an 8-OG or AP site analog. DNA synthesis by Pol δ efficiently passed through the 8-OG, whereas the AP site analog was a relatively tight blocker. Haracska et al. (17) demonstrated that AP site cannot be bypassed by S. cerevisiae Pol δ alone. In contrast, Mozzherin et al. (37) reported that PCNA increases AP site bypass by calf thymus Pol δ. In our system including PCNA and RFC, human Pol δ proceeded DNA synthesis through an AP site-containing template by 20–30% efficiency. This result suggests that while a majority of AP sites encountered by the replication machinery may be bypassed with help by TLS-type DNA polymerase(s), some part of AP site bypass can be carried out by Pol δ. Recently Liao et al. (38) characterized replication of AP site-containing DNA by Xenopus egg extract under the condition in which the AP site repair was suppressed. In this study, resultant replication products were classified into three groups by nucleotides inserted opposite the AP site: (i) a nucleotide instructed by the complementary strand (error-free products); (ii) dAMP regardless of the complementary strand sequence (error-prone, A-rule); (iii) dCMP regardless of the complementary strand sequence (error-prone, C-rule). The result in the present study implies that some, if not all, of the error-prone products carrying A opposite the AP site could result from bypass by Pol δ.
It is a striking observation that one of the exonuclease-mutant Pol δ, exo-24, had a lower lesion-bypass activity than the wild-type. This unique character of exo-24 may be related to the severe inability to extend mispaired primers (Figure 6). The higher proofreading efficiency in the products fully extended by the exo-24 enzyme compared to the other mutants (Figure 7B) can result from its stringent extension activity. One possible explanation for the exo-24's salient property is that this mutated exonuclease domain may bind to the primer end more tightly than the wild-type and other mutants, leading to inefficient accessibility of the primer end by the polymerase domain. Hogg et al. (39) reported an RB69 polymerase mutant which affects active site switching, although this mutant preferentially hold the primer DNA in the polymerase active site. Our model on exo-24 could be tested by similar structural analysis.
Nucleotide preference of Pol δ to insert opposite 8-OG is the important factor in this lesion's mutagenicity. Shibutani et al. (18) demonstrated that calf thymus Pol δ with PCNA inserted C and A with a ratio of 1:5 opposite 8-OG. In addition, Haracska et al. (28) reported that S. cerevisiae Pol δ predominantly inserted A opposite 8-OG. On the other hand, Maga et al. (7) reported that, when combined with PCNA ± RPA, human Pol δ inserted C and A by 3:1 ratio, suggesting that this lesion may be less mutagenic. Our data also indicated that human Pol δ slightly preferred C insertion to A insertion at this lesion. One possible reason for the apparent discrepancy in these observations is that replication auxiliary proteins may affect nucleotide selection as suggested by Maga et al. (7). Their data revealed that the addition of PCNA reverted Pol δ's nucleotide preference from mutagenic A to correct C. All the experimental systems discussed here except ours employed oligonucleotides with free ends as a primer/template and therefore PCNA may not be stably loaded on them. In contrast, our assay system employed end-blocked oligonucleotides, and PCNA and RFC were essential for primer extension by Pol δ in this system (Figure 3B). Another possible reason for different results on 8-OG bypass is that it may due to different lengths of extension beyond the lesion. Most previous studies examined the nucleotide inserted opposite the lesion after limited lengths of extension (0–5 nt), whereas the reaction products we analyzed here resulted from extension of at least 30 nt after the lesion. While studies of steady-state kinetics on insertion and extension (7,18) can dissect the lesion bypass reaction by DNA polymerase at a single nucleotide level, the distorted pair of the inserted nucleotide and the lesion may affect the extension at relatively distant positions as suggested with a study of mispaired primer extensions (36). Our results seem to represent products preferred for long extension and simulate the natural lesion bypass by Pol δ itself. Previously we observed that A:8-OG mispair was repaired in the replication-dependent manner in MYH-deficient mouse cells at a significant efficiency although the wild-type cells repaired better (40). Taking account of Pol δ's efficient bypassing of this lesion (Figure 9), the replication-associated repair of the A:8-OG mispair in the absence of MYH may be explained by the suboptimal C insertion in translesion synthesis by Pol δ in vivo, although partial contribution of TLS polymerases is not ruled out.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (CA06315, CA092584; core grant CA06927); appropriation from the Commonwealth of Pennsylvania. Funding for open access charge: Fox Chase Cancer Center.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Anna Marie Skalka's laboratory for generosity on our use of their French Press; Rita Michielli for pyrosequencing for Supplementary Data; Mike Schmitt and Lawrence A. Loeb of University of Washington for helpful discussion and sharing of their unpublished data; and Haruo Ohmori of Kyoto University for critical reading of the manuscript.
REFERENCES
- 1.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, Green CM. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair. 2007;6:891–899. doi: 10.1016/j.dnarep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 4.Ulrich HD. Conservation of DNA damage tolerance pathways from yeast to humans. Biochem. Soc. Trans. 2007;35:1334–1337. doi: 10.1042/BST0351334. [DOI] [PubMed] [Google Scholar]
- 5.Nick McElhinny SA, Gordenin DA, Stith CM, Burgers PM, Kunkel TA. Division of labor at the eukaryotic replication fork. Mol. Cell. 2008;30:137–144. doi: 10.1016/j.molcel.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacNeill SA, Baldacci G, Burgers PM, Hubscher U. A unified nomenclature for the subunits of eukaryotic DNA polymerase delta. Trends Biochem. Sci. 2001;26:16–17. doi: 10.1016/s0968-0004(00)01709-6. [DOI] [PubMed] [Google Scholar]
- 7.Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 8.Jin YH, Garg P, Stith CM, Al-Refai H, Sterling JF, Murray LJ, Kunkel TA, Resnick MA, Burgers PM, Gordenin DA. The multiple biological roles of the 3′–>5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol. Cell Biol. 2005;25:461–471. doi: 10.1128/MCB.25.1.461-471.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paz-Elizur T, Takeshita M, Goodman M, O’Donnell M, Livneh Z. Mechanism of translesion DNA synthesis by DNA polymerase II. Comparison to DNA polymerases I and III core. J. Biol. Chem. 1996;271:24662–24669. doi: 10.1074/jbc.271.40.24662. [DOI] [PubMed] [Google Scholar]
- 10.Paz-Elizur T, Takeshita M, Livneh Z. Mechanism of bypass synthesis through an abasic site analog by DNA polymerase I. Biochemistry. 1997;36:1766–1773. doi: 10.1021/bi9621324. [DOI] [PubMed] [Google Scholar]
- 11.Morrison A, Johnson AL, Johnston LH, Sugino A. Pathway correcting DNA replication errors in Saccharomyces cerevisiae. EMBO J. 1993;12:1467–1473. doi: 10.1002/j.1460-2075.1993.tb05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy K, Darmawan H, Schultz A, Fidalgo da Silva E, Reha-Krantz LJ. A method to select for mutator DNA polymerase deltas in Saccharomyces cerevisiae. Genome. 2006;49:403–410. doi: 10.1139/g05-106. [DOI] [PubMed] [Google Scholar]
- 13.Goldsby RE, Hays LE, Chen X, Olmsted EA, Slayton WB, Spangrude GJ, Preston BD. High incidence of epithelial cancers in mice deficient for DNA polymerase delta proofreading. Proc. Natl Acad. Sci. USA. 2002;99:15560–15565. doi: 10.1073/pnas.232340999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdus Sattar AK, Lin TC, Jones C, Konigsberg WH. Functional consequences and exonuclease kinetic parameters of point mutations in bacteriophage T4 DNA polymerase. Biochemistry. 1996;35:16621–16629. doi: 10.1021/bi961552q. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Sattar AK, Wang CC, Karam JD, Konigsberg WH, Steitz TA. Crystal structure of a pol alpha family replication DNA polymerase from bacteriophage RB69. Cell. 1997;89:1087–1099. doi: 10.1016/s0092-8674(00)80296-2. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000;65:127–133. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 17.Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, Prakash L. Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev. 2001;15:945–954. doi: 10.1101/gad.882301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–432. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, Matthews KS. Deletion of lactose repressor carboxyl-terminal domain affects tetramer formation. J. Biol. Chem. 1992;267:13843–13850. [PubMed] [Google Scholar]
- 20.Lin TC, Karam G, Konigsberg WH. Isolation, characterization, and kinetic properties of truncated forms of T4 DNA polymerase that exhibit 3′-5′ exonuclease activity. J. Biol. Chem. 1994;269:19286–19294. [PubMed] [Google Scholar]
- 21.Walter J, Newport JW. Regulation of replicon size in Xenopus egg extracts. Science. 1997;275:993–995. doi: 10.1126/science.275.5302.993. [DOI] [PubMed] [Google Scholar]
- 22.Podust LM, Podust VN, Floth C, Hubscher U. Assembly of DNA polymerase delta and epsilon holoenzymes depends on the geometry of the DNA template. Nucleic Acids Res. 1994;22:2970–2975. doi: 10.1093/nar/22.15.2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beese LS, Steitz TA. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J. 1991;10:25–33. doi: 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fakhrai-Rad H, Pourmand N, Ronaghi M. Pyrosequencing: an accurate detection platform for single nucleotide polymorphisms. Hum. Mutat. 2002;19:479–485. doi: 10.1002/humu.10078. [DOI] [PubMed] [Google Scholar]
- 25.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 26.Moriya M. Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted G.C–>T.A transversions in simian kidney cells. Proc. Natl Acad. Sci. USA. 1993;90:1122–1126. doi: 10.1073/pnas.90.3.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto K, Tominaga Y, Nakabeppu Y, Moriya M. Futile short-patch DNA base excision repair of adenine:8-oxoguanine mispair. Nucleic Acids Res. 2004;32:5928–5934. doi: 10.1093/nar/gkh909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haracska L, Yu SL, Johnson RE, Prakash L, Prakash S. Efficient and accurate replication in the presence of 7,8-dihydro-8-oxoguanine by DNA polymerase eta. Nat. Genet. 2000;25:458–461. doi: 10.1038/78169. [DOI] [PubMed] [Google Scholar]
- 29.Strauss BS. The ‘A rule’ of mutagen specificity: a consequence of DNA polymerase bypass of non-instructional lesions? Bioessays. 1991;13:79–84. doi: 10.1002/bies.950130206. [DOI] [PubMed] [Google Scholar]
- 30.Simonelli V, Narciso L, Dogliotti E, Fortini P. Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res. 2005;33:4404–4411. doi: 10.1093/nar/gki749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs PE, Lawrence CW. Novel mutagenic properties of abasic sites in Saccharomyces cerevisiae. J. Mol. Biol. 1995;251:229–236. doi: 10.1006/jmbi.1995.0430. [DOI] [PubMed] [Google Scholar]
- 32.Podust VN, Chang LS, Ott R, Dianov GL, Fanning E. Reconstitution of human DNA polymerase delta using recombinant baculoviruses: the p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J. Biol. Chem. 2002;277:3894–3901. doi: 10.1074/jbc.M109684200. [DOI] [PubMed] [Google Scholar]
- 33.Xie B, Mazloum N, Liu L, Rahmeh A, Li H, Lee MY. Reconstitution and characterization of the human DNA polymerase delta four-subunit holoenzyme. Biochemistry. 2002;41:13133–13142. doi: 10.1021/bi0262707. [DOI] [PubMed] [Google Scholar]
- 34.Masuda Y, Suzuki M, Piao J, Gu Y, Tsurimoto T, Kamiya K. Dynamics of human replication factors in the elongation phase of DNA replication. Nucleic Acids Res. 2007;35:6904–6916. doi: 10.1093/nar/gkm822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Zuo S, Kelman Z, O'Donnell M, Hurwitz J, Goodman MF. Fidelity of eucaryotic DNA polymerase delta holoenzyme from Schizosaccharomyces pombe. J. Biol. Chem. 2000;275:17677–17682. doi: 10.1074/jbc.M910278199. [DOI] [PubMed] [Google Scholar]
- 36.Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- 37.Mozzherin DJ, Shibutani S, Tan CK, Downey KM, Fisher PA. Proliferating cell nuclear antigen promotes DNA synthesis past template lesions by mammalian DNA polymerase delta. Proc. Natl Acad. Sci. USA. 1997;94:6126–6131. doi: 10.1073/pnas.94.12.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao S, Matsumoto Y, Yan H. Biochemical reconstitution of abasic DNA lesion replication in Xenopus extracts. Nucleic Acids Res. 2007;35:5422–5429. doi: 10.1093/nar/gkm552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hogg M, Aller P, Konigsberg W, Wallace SS, Doublie S. Structural and biochemical investigation of the role in proofreading of a beta hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J. Biol. Chem. 2007;282:1432–1444. doi: 10.1074/jbc.M605675200. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi H, Tominaga Y, Hirano S, McKenna AE, Nakabeppu Y, Matsumoto Y. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr. Biol. 2002;12:335–339. doi: 10.1016/s0960-9822(02)00686-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}