

Abstract
Wybutosine (yW), one of the most complicated modified nucleosides, is found in the anticodon loop of eukaryotic phenylalanine tRNA. This hypermodified nucleoside ensures correct codon recognition by stabilizing codon-anticodon pairings during the decoding process in the ribosome. TYW4 is an S-adenosylmethionine (SAM)-dependent enzyme that catalyzes the final step of yW biosynthesis, methylation and methoxycarbonylation. However, the structural basis for the catalytic mechanism by TYW4, and especially that for the methoxycarbonylation, have remained elusive. Here we report the apo and cofactor-bound crystal structures of yeast TYW4. The structures revealed that the C-terminal domain folds into a β-propeller structure, forming part of the binding pocket for the target nucleoside. A comparison of the apo, SAM-bound, and S-adenosylhomocysteine-bound structures of TYW4 revealed a drastic structural change upon cofactor binding, which may sequester solvent from the catalytic site during the reaction and facilitate product release after the reaction. In conjunction with the functional analysis, our results suggest that TYW4 catalyzes both methylation and methoxycarbonylation at a single catalytic site, and in the latter reaction, the methoxycarbonyl group is formed through the fixation of carbon dioxide.
INTRODUCTION
Non-coding RNAs (ncRNAs), including transfer RNA (tRNA), ribosomal RNA, small RNA and small nuclear RNA, are involved in various cellular functions, such as translation of the genetic code and posttranscriptional RNA processing. One of the most distinctive features of the ncRNAs is their posttranscriptional modification, which confers a greater variety of chemical properties than those of the unmodified nucleosides (i.e. A, U, G and C). More than 100 modifications have been reported in ncRNAs, and most have been found in tRNA (1). The various chemical properties of the modifications are considered to contribute to higher-order biological processes through structural stabilization and molecular recognition (2). For example, modifications in the tRNA core region are considered to be important for structural formation and stabilization (3). On the other hand, modifications found in the tRNA anticodon arm ensure the correct codon-anticodon pairing in the ribosome.
Wybutosine (yW) is a modified nucleoside that functions in the molecular recognition (Figure 1). The yW nucleotide is exclusively located at position 37 of eukaryotic phenylalanine tRNA (tRNAPhe), which is adjacent to the 3′-position of the Phe anticodon (34GAA36). It is one of the most extensively hypermodified nucleosides ever found in RNA, and it has a distinctive chemical structure: a tricyclic 1H-imidazo[1,2-α]purine core structure and a large side chain attached to the tricyclic base (4). Recent studies on the yW nucleotide have highlighted the importance of this nucleotide in the maintenance of translational fidelity. Although yW lacks a major influence on the phenylalanylation of tRNAPhe under physiological conditions (5,6), tRNAPhe lacking the yW modification was shown to enhance codon frame shifting (7), which reportedly activates the replication of human immunodeficiency virus RNA (8). Therefore, the bulky hydrophobic structure of yW may stabilize the codon-anticodon pairing in the ribosome through the base-stacking interaction, thereby maintaining the correct reading frame (9).
Figure 1.

Biosynthetic pathway of wybutosine. The chemical structures of (A) N1-methylguanosine (m1G), (B) 4-demethylwyosine (imG-14), (C) 7-(α-amino-α-carboxypropyl)wyosine (yW-72) and (D) wybutosine (yW), are shown. TRM5 transfers a methyl group to G37 to produce m1G37, utilizing SAM as a methyl donor. TYW1 catalyzes the cyclization reaction to produce a tricyclic nucleoside, imG-14. TYW2 and TYW3 attach α-amino-α-carboxypropyl and methyl groups on C7 and N4, respectively. Finally, TYW4 attaches methyl and methoxycarbonyl groups (highlighted), completing the yW formation.
Recent studies have shown that the biosynthetic pathway of yW is a multienzymatic process involving five enzymes, the S-adenosylmethionine (SAM)-dependent tRNA methylase (TRM5) and the tRNA-yW synthesizing enzymes 1–4 (TYW1–4) (10–12). First, the guanosine residue at position 37 is converted into the distinctive tricyclic structure by TRM5 (13), and the FeS-cluster-containing radical SAM enzyme, TYW1 (14,15) (Figure 1A and B). Subsequently, TYW2, 3 and 4 attach the exocyclic groups (Figure 1B, C and D) to complete yW formation. Interestingly, a recent study demonstrated that the specific intermediates in the progression of these multienzymatic steps show incremental changes in frameshift efficiency (16), underscoring the importance of completely formed yW for the correct translation of the genetic code.
TYW4 catalyzes the final step of yW biosynthesis, i.e. the modification to the α-amino-α-carboxypropyl (acp) side chain attached at the C7 position of the tricyclic ring (Figure 1C and D) (10). An in vitro biochemical analysis suggested that TYW4 catalyzes two independent reactions: methylation of the α-carboxyl group and methoxycarbonylation of the α-amino group of the acp side chain (10). Therefore, these modifications by TYW4 increase the bulkiness as well as the hydrophobicity of the side chain of the yW base, which might further stabilize the codon-anticodon pairing in the ribosome. However, the mechanism of the methoxycarbonylation, and especially the source of the methoxycarbonyl group, still remain unknown. Moreover, the mechanism by which TYW4 attaches two different chemical groups (i.e. methyl and methoxycarbonyl) to two distant sites (i.e. α-carboxyl and α-amino groups; Figure 1D) on the yW precursor also remains unknown. The N-terminal half of TYW4 shares primary structure similarity with protein phosphatase methyltransferase 1 (PPM1), which catalyzes the SAM-dependent methylation of the C-terminal α-carboxylate group of protein phosphatase 2A (PP2A). Therefore, TYW4 was formerly referred to as PPM2 (17,18). In spite of the sequence similarity, these two enzymes catalyze modification reactions on completely distinct targets, protein and tRNA. Furthermore, TYW4 catalyzes two separate chemical reactions, whereas PPM1 catalyzes only one methylation reaction. Although the tertiary structure of PPM1 was revealed (19), the structural basis for these enzymological differences between PPM1 and TYW4, as well as the tRNA modification mechanism by TYW4 itself, still remains elusive. In addition, as compared to PPM1, TYW4 has an extra C-terminal extension, which has an unknown structure and function.
In order to gain insight into the modification mechanism by TYW4, we determined the crystal structure of TYW4 from Saccharomyces cerevisiae. The apo, SAM- and S-adenosylhomocysteine (SAH)-bound structures at 2.7, 2.5 and 1.7 Å resolutions, respectively, revealed a structural transition dependent on cofactor binding, which may sequester the active site from solvent during the reaction and then expel the products after the reaction. The C-terminal domain forms a six-bladed β propeller structure, which participates in the formation of the putative binding pocket for yW at tRNA position 37. In conjunction with biochemical analyses by mass spectrometry, we showed that TYW4 catalyzes two reactions (i.e. methylation and methoxycarbonylation) with a single catalytic site. Furthermore, we showed that the latter methoxycarbonylation reaction proceeds through the fixation of CO2, which is distinctive from any other RNA modification mechanism.
MATERIALS AND METHODS
Sample preparation
For the overproduction of yeast TYW4 with a 6× His tag at its C-terminus, the E. coli strain BL21(DE3) CodonPlus was transformed with the plasmid pET21b, carrying the TYW4 (YOL141w) gene. The cells were grown at 37°C in LB medium supplemented with 50 μg ml–1 ampicillin, to an absorbance at 600 nm of 0.6. Expression was induced at 20°C by the addition of isopropyl-β-d-thiogalactopyranoside to a final concentration of 0.5 mM. Cells were harvested by centrifugation at 8000g for 15 min, after overnight incubation. The cell pellets were resuspended in 50 mM Tris–HCl buffer (pH 9.0), containing 100 mM NaCl, 50 mM MgCl2, 5 mM 2-mercaptoethanol, 10% glycerol and 1 mM phenylmethylsulfonyl fluoride, and were gently sonicated. After centrifugation at 20 000g for 40 min, the supernatant containing TYW4 was loaded onto a Ni-NTA SuperFlow column (Qiagen), which was eluted with 200 mM imidazole in 10 mM Tris–HCl buffer (pH 9.0), containing 300 mM NaCl, 50 mM MgCl2 and 5 mM 2-mercaptoethanol. The eluted fractions were gathered, and then loaded onto a Resource Q column (GE Healthcare), which was eluted with a 20-column volume gradient of 0–300 M NaCl in 10 mM Tris–HCl buffer (pH 9.0), containing 5 mM MgCl2 and 1 mM dithiothreitol (DTT). The fractions containing TYW4 were combined, concentrated and loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) equilibrated in 10 mM Tris–HCl buffer (pH 9.0), containing 100 mM NaCl, 5 mM MgCl2 and 5 mM 2-mercaptoethanol. The purified TYW4 fractions eluted from the gel-filtration column were collected and concentrated to 10 mg ml–1 by using an Amicon Ultra-4 filter (Millipore). The protein purity was assessed by SDS–PAGE. To obtain selenomethionine-labeled proteins, the methionine-auxotroph E. coli strain B834(DE3) CodonPlus was transformed with the same plasmid. The cells were cultivated in Core medium (Wako, Japan) containing 50 μg ml–1 selenomethionine, and the protein was purified in the same manner as native TYW4.
Crystallization and data collection
The native crystals were obtained by the sitting-drop vapor diffusion method. The drops were prepared by mixing equal volumes of a 10 mg ml–1 TYW4 solution and reservoir solution, containing 200 mM ammonium citrate (pH 7.0), 10 mM HEPES (pH 7.5), 20% (w/v) PEG3,350, 20 mM sodium citrate, and 1% 2-propanol. Two types of crystal were grown at 20°C in the same drop: the plate crystal (form I) and the column crystal (form II), which grew within 2 days and 7 days, respectively. However, the X-ray datasets revealed that only the form II crystal was suitable for the structure determination. The form II crystals of selenomethionine-labeled TYW4 were obtained by macro- and micro-seeding techniques, using the native crystals as a seed. The drops were prepared by mixing equal volumes of a 10 mg ml–1 TYW4 solution and the reservoir solution containing 200 mM ammonium citrate (pH 7.0) and 20% (w/v) PEG3,350. The form II crystals of the SAM- and SAH-bound TYW4 were also obtained by the micro-seeding technique as the selenomethionine-labeled crystals, with the exception that the protein solution contained 1 mM SAM or SAH. The crystals grew to a size of 0.02 mm × 0.02 mm × 0.01 mm in 1 day. The crystals were transferred stepwise to the harvesting solution, 240 mM ammonium citrate (pH 7.0) and 24% (w/v) PEG3,350, containing 15% (v/v) ethylene glycol as a cryoprotectant, and were flash-cooled in a liquid nitrogen stream at 100 K.
Data collection, structure determination, and refinement
All diffraction data sets were collected at the station BL41XU at SPring-8 (Hyogo, Japan) and the station NW12A at KEK PF-AR (Tsukuba, Japan). Datasets were processed with the HKL2000 suite (HKL Research). Despite the structural similarity of the N-terminal half of TYW4 and PPM1, attempts to solve the TYW4 structure by molecular replacement were unsuccessful. Therefore, the structure was solved by the multiwavelength anomalous dispersion (MAD) method, using the selenomethionine-labeled crystal. The 29 selenium sites were initially located by the programs SHELXC and SHELXD (20) with the dataset collected at the peak wavelength. The heavy-atom parameters were refined and the phases were calculated by using the program SHARP. The phase improvement, including the solvent flattening and averaging, and the automatic model building were performed with the program RESOLVE (21). Especially, the phase improvement with the solvent flattening and the 2-fold NCS averaging drastically improved the initial phases, which enabled the automatic interpretation of the electron density. The resulting initial model was manually modified to fit into the electron density maps by the program O (22). Finally, the atomic models of the apo, SAM- and SAH-bound forms of TYW4 were refined against reflections up to 2.7, 2.4 and 1.7 Å resolutions, respectively, using the program PHENIX (23). Molecular graphics were illustrated with CueMol (http://www.cuemol.org/). Statistics on data collection, phasing and refinement are shown in Tables 1 and 2.
Table 1.
Data collection and phasing statistics
| SeMet | |||
|---|---|---|---|
| Data collection statistics | |||
| X-ray source | SPring-8 BL41XU | ||
| Wavelength (Å) | |||
| Peak | 0.97941 | ||
| Edge | 0.97914 | ||
| Remote | 0.96410 | ||
| Unit cell dimensions (Å,°) | a = 78.23, b = 90.16, c = 252.0, α = β = γ = 90 | ||
| Resolution (Å) | 50–2.71 (2.76–2.71) | 50–2.72 (2.77–2.72) | 50–2.67 (2.72–2.67) |
| Unique reflections | 48 596 (2406) | 47 508 (2025) | 40 354 (1747) |
| Redundancy | 9.6 (6.0) | 4.2 (2.3) | 3.8 (2.0) |
| Completeness (%) | 99.7 (99.2) | 95.5 (83.9) | 93.3 (67.5) |
| I/σ(I) | 23.3 (3.17) | 10.6 (1.32) | 8.17 (4.81) |
| Rsym | 0.127 (0.383) | 0.105 (0.384) | 0.112 (0.424) |
| Phasing statistics | |||
| No. of Se sites | 29 | ||
| Phasing power | |||
| Iso (cen./acen.) | 0.163/0.173 | – | 0.333/0.240 |
| Ano | 0.883 | 0.252 | 0.421 |
| Rcullis | |||
| Iso (cen./acen.) | 0.959/0.858 | – | 1.021/1.168 |
| Ano | 0.866 | 0.985 | 0.964 |
| Mean FOM | |||
| Cen./Acen. | 0.163/0.248 | ||
The numbers in parentheses are for the last shell.
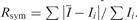 .
.
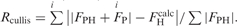 .
.
Table 2.
Data collection and refinement statistics
| Native |
|||
|---|---|---|---|
| Free | SAM | SAH | |
| Data collection statistics | |||
| X-ray source | PF-AR NW12A | SPring-8 BL41XU | SPring-8 BL41XU |
| Wavelength (Å) | 0.9789 | 1.0 | 1.0 |
| Resolution (Å) | 50–2.7 (2.75–2.7) | 50–2.5 (2.54–2.5) | 50–1.7 (1.73–1.7) |
| Unique reflections | 48 148 (1867) | 52 008 (2218) | 174 812 (8086) |
| Redundancy | 6.4 (6.1) | 4.1 (2.0) | 5.5 (2.9) |
| Completeness (%) | 96.6 (76.3) | 90.6 (76.9) | 98.7 (92.7) |
| I/σ(I) | 30.7 (4.00) | 22.6 (2.12) | 31.9 (2.37) |
| Rsym | 0.07 (0.367) | 0.07 (0.305) | 0.078 (0.344) |
| Refinement statistics | |||
| Resolution (Å) | 50–2.7 | 50–2.5 | 50–1.7 |
| No. of reflections (all/test) | 47 919/4815 | 51 990/2600 | 174 754/17319 |
| Rwork/Rfree | 0.209/0.273 | 0.193/0.245 | 0.170/0.211 |
| No. of atoms | |||
| Non-hydrogen | 10 866 | 10 810 | 12 533 |
| Water | 33 | 133 | 1543 |
| RMSD of | |||
| Bond length (Å) | 0.004 | 0.003 | 0.007 |
| Bond angle (°) | 0.826 | 0.847 | 1.142 |
| Average B factor (Å) | 73.3 | 64.2 | 29.5 |
| Ramachandran plot | |||
| Most favored (%) | 85.2 | 86.5 | 90.0 |
| Allowed region (%) | 14.7 | 13.3 | 9.9 |
| Disallowed region (%) | 0.1 | 0.2 | 0.1 |
The numbers in parentheses are for the last shell.
Rwork=Σ|Fo–Fc|/ΣFo for reflections of work set.
Rfree=Σ|Fo–Fc|/ΣFo for reflections of test set.
Site-directed mutagenesis of TYW4 and complementation test
The S. cerevisiae wild-type strain and the deletion strain were obtained from EUROSCARF: the BY4742 (Mat α; his3Δ1; leu2Δ0; lys2Δ0; ura3Δ0) series of strains ΔTYW4 (YOL141w::kanMX4). pTYW4(pYOL141w), a BG1805-amp plasmid, was obtained from the yeast ORF E. coli strain YSC3867-9521135 (Open Biosystems). Site-directed mutagenesis of TYW4 was carried out on the plasmid pTYW4 by QuikChangeTM Site-Directed Mutagenesis (Stratagene), according to the manufacturer's instructions. Introduced mutations were confirmed by DNA sequencing. ΔTYW4 was transformed by pTYW4 or pTYW4 mutated plasmids. Each transformant was cultivated in SC-ura/glucose media (0.67% yeast nitrogen base without amino acids, 0.5% casamino acids and 2% glucose, supplemented by auxotrophic nutrients, as specified, without uracil) overnight, and then diluted into SC-ura/raffinose media (0.67% yeast nitrogen base without amino acids, 0.5% casamino acids and 2% raffinose, supplemented by auxotrophic nutrients, as specified, without uracil) at a starting OD600 ≈ 0.3. The mutants were grown to an OD600 ≈ 1.2, and then protein production was induced by culturing in YPG media (2% peptone, 1% yeast extract and 2% galactose) for 20 h. Total RNA from each mutant was extracted, and the modified nucleosides were analyzed by LC/MS, as follows.
Mass spectrometry
Total RNA (20 μg) was digested to nucleosides with nuclease P1(Yamasa) and bacterial alkaline phosphatase derived from E. coli strain C75 (BAP.C75, Takara) for 3 h at 37°C, and was analyzed by LC/MS using ion trap mass spectrometry, as described previously (10). Nucleosides were separated by an ODS reverse-phase column (Intertsil ODS3 5 μm, 2.1 × 250 mm, GL Science), using an HP1100 liquid chromatography system (Agilent). The solvent consisted of 0.1% acetonitrile in 5 mM NH4OAc (pH 5.3) (Solvent A) and 60% acetonitrile in H2O (Solvent B) in the following gradients: 1–35% B in 0–35 min, 35–99% B in 35–40 min, 99% B in 40–50 min, 99–1% B in 50–50.1 min and 1% B in 50.1–60 min. The chromatographic effluent was directly conducted to the electrospray ionization (ESI) source to ionize the separated nucleosides, which were analyzed on a LCQ DUO ion trap mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was operated with a spray voltage of 5 kV and a capillary temperature of 270°C. The sheath gas flow rate was 95 arb, and the auxiliary gas flow rate was 5 arb. Positive ions were scanned over an m/z range of 103–900.
In vitro reconstitution of yW with TYW4
Yeast tRNAPhe with the yW-72 intermediate (tRNAPhe-yW-72) was isolated from the TYW4 deletion strain, as described previously (10). Reaction mixtures (10 μl), containing 50 mM Tris–HCl (pH 8.0), 0.5 mM DTT, 10 mM MgCl2, 1 mM spermidine, 2 μg of tRNAPhe-yW-72 and 1.4 μM recombinant TYW4 protein, with or without 0.5 mM SAM, were incubated for 1 h at 30°C. For the CO2 incorporation experiment, we added 13C-labeled NaHCO3 (Isotech) or non-labeled NaHCO3 at a final concentration of 100 mM. The tRNAPhe was recovered by ISOGEN (Wako), precipitated by ethanol and then subjected to RNaseT1 and RNaseA digestions. The RNA fragment analysis by LC/MS was performed as described below.
Capillary LC nano ESI/mass spectrometry
To analyze the RNA fragments generated by the RNaseT1 and RNaseA digestions, we devised a system with a capillary LC coupled with a nanoESI-MS (24). A linear ion trap-orbitrap hybrid mass spectrometer (LTQ Orbitrap XL, Thermo Fisher Scientific) was equipped with a custom-made nanospray ion source, a Nanovolume Valve (Valco Instruments), and a splitless nano HPLC system (DiNa, KYA Technologies). The analyte mixed with TEAA, was loaded onto the nano-LC trap column (C18, Φ 0.5 × 1.0 mm), desalted, and then concentrated with 0.1 M TEAA (pH 7.0). RNA fragments were eluted from the trap column and directly injected into a C18 capillary column (HiQ Sil; 3 µm C18, 100 Å pore size; Φ 0.1 × 100 mm, KYA Technologies). The solvent system consisted of 0.4 M 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP; pH 7.0, adjusted with triethylamine; solvent A) and 0.4 M HFIP in 50% methanol (solvent B), and the samples were chromatographed at a flow rate of 300 nl/min, using a linear gradient of 10–90% solvent B over 35 min. The chromatographic eluent was sprayed from a sprayer tip attached to the capillary column. The ionization voltage was set to –1.9 kV, and ions were scanned in the negative polarity mode. The mass spectrum was acquired in the range of m/z 600–2000, with mass resolution of 30 000 (FWHM).
RESULTS
Overall structure
The crystal structure of TYW4 from S. cerevisiae was determined by the MAD method, using a selenomethionine-labeled crystal (Figure 2). The final models of the apo, SAM-bound and SAH-bound forms were refined at 2.7, 2.5 and 1.7 Å resolutions to free R-factors of 26.6%, 24.5% and 21.1%, respectively (Tables 1 and 2). The overall structure of TYW4 consists of an N-terminal domain (TYW4N, 1–350) and a C-terminal domain (TYW4C, 371–694), connected by a linker region (351–370) (Figure 2A). Both the apo and cofactor-bound form crystals contain two molecules per asymmetric unit. To determine whether TYW4 forms a dimer in solution, purified TYW4 was subjected to sedimentation velocity analytical ultracentrifugation. The result clearly showed that TYW4 exists as a monomer in solution with a molecular mass of ∼72 430 Da, which agrees with the molecular mass of 80 021.8 Da, calculated from the amino-acid sequence. This result is consistent with the observation that the molecular interface in the asymmetric unit contains many water molecules and lacks hydrophobic interactions.
Figure 2.

Overall structure of TYW4. (A) Ribbon representation of the crystal structure of TYW4 in the apo form. For TYW4N, each subdomain (i.e. the core MT domain, regions I, II, III and IV) is defined according to the PPM1 structure (19) and is shown in a different color. For TYW4C, each blade of the propeller structure is shown in a different color. The border between TYW4N and TYW4C is indicated by a dashed line. (B) Topology diagram of the N-terminal domain of TYW4, TYW4N. β sheets and α helices are indicated by triangles and circles, respectively. The same color code as in (A) is used. (C) Topology diagram of the C-terminal domain of TYW4, TYW4C. The same color code as in (A) is used.
The N-terminal domain shares a common fold with PPM1
The amino-acid sequence of TYW4N exhibits considerable homology (identity of 26%) with yeast PPM1, which catalyzes the methylation of the C-terminus of protein phosphatase 2A (PP2A) (17). PPM1 recognizes the C-terminal residues (-TPDYFL) of PP2A and transfers the methyl group from SAM to the α-carboxyl group of the Leu residue. This methylation at the C-terminus regulates the assembly of PP2A holoenzyme. The crystal structure of PPM1 revealed that it belongs to the class I SAM-dependent methyltransferases (MTases) and recognizes the cofactor with the conserved GXG motif (19).
As expected from the amino-acid sequence, TYW4N has a similar tertiary structure to PPM1, and shares the core structure with the class I MTases (Figures 2A, B and 3A). The RMSD between TYW4N and PPM1 is 2.1 Å (Z score of 32.7) over 327 Cα atoms. However, there are three structural differences between the TYW4N and PPM1. First, TYW4N has a long additional α helix (14–27, helix α1′) at its N terminus, which is disordered in the apo form of PPM1 (Figure 3). Second, the helices α2 and α3 in PPM1, which pack on the helix α1, are missing in TYW4N and replaced with a short insertion, consisting of 310 helices and loops (Figure 3). Finally, the tip of the β6bc hairpin (674–684), which protrudes from TYW4C, is inserted into a gap formed between helices α5 and α8 and forms a tight interaction with them (Figure 3A). In PPM1, this gap between helices α5 and α8 was suggested to be a binding site for the C-terminal tail of PP2A (19) (Figure 3B). As a result, the helix α5, which is disordered in PPM1, is clearly observed in the density map and has a low temperature factor as low as that for the other part of the protein.
Figure 3.

Structural comparison of (A) TYW4 and (B) PPM1. The crystal structures of the apo forms of TYW4 and PPM1 (PDB ID: 1RJF) are shown in ribbon representations. The N-terminal helices α1′ and α1, the PPM1-specific helices α2 and α3, helix α5, and the β6bc hairpin of TYW4C, which have structures that differ between TYW4 and PPM1, are highlighted in red, blue, orange, and magenta, respectively. The common structures between TYW4 and PPM1 are shown in gray.
Structural change upon cofactor binding
In the apo-form structure of TYW4, the helices α1′ and α1 are arranged nearly perpendicular to each other, and helix α1′ packs against helix αB. Consequently, the SAM-binding pocket is largely exposed to the solvent (Figure 3A). The residues at the junction between helices α1′ and α1 (29–32) are partially disordered, indicating its flexibility. In some MTases, co-purified SAM bound to the catalytic site was observed, even if no SAM molecule was added during the crystallization procedure (25). However, in TYW4, we did not observe any density corresponding to co-purified SAM around the SAM-binding pocket.
To elucidate the cofactor-binding mechanism, we co-crystallized TYW4 with SAM or SAH. In both crystals, we clearly identified these cofactors’ electron densities (Supplementary Figure 1). In the SAM-complexed crystal, we observed a large conformational change of the enzyme upon SAM binding (Figure 4A); the helices α1′ and α1 straightened and formed one continuous helix. The interactions between helices α1′ and αB observed in the apo form are completely lost, and the N terminus of helix α1′ protrudes into the solvent. The SAM molecule becomes deeply buried and sequestered from the solvent by the C-terminal half of helix α1′.
Figure 4.

Catalytic site of TYW4. (A) and (B) Cofactor binding sites of TYW4 in the SAM-bound (A) and SAH-bound (B) forms. The protein main chains are shown in ribbon representations, with the same coloring scheme as in Figure 2. The bound cofactors are shown in ball-and-stick models. The protein side chains involved in the cofactor recognition are shown in stick models. (C) Stereo-view of the putative yW-72-binding pocket of TYW4 in the SAM-bound form. The protein main chain is shown in a ribbon representation, with the same coloring scheme as in Figure 2. The bound cofactors and the protein side chains involved in the cofactor recognition are shown with stick models. (D) Surface representation of the putative yW-72-binding pocket in TYW4. The solvent-accessible surface was calculated with the program MSMS (42), and is colored with the same coloring scheme as in Figure 2.
In the SAH-complexed crystal, the SAH molecule is also bound to the SAM-binding pocket in almost the same manner as the SAM molecule (Figure 4B). A similar structural change to that observed in the SAM-complexed crystal occurs in one molecule (molecule A) in the crystal asymmetric unit. However, interestingly, the structural change does not occur in the other molecule (molecule B) in the asymmetric unit, and the bound SAH molecule is largely exposed to the solvent (Figure 4B).
Catalytic site structure
As described above, there are several residues commonly conserved between TYW4N and PPM1 (Supplementary Figure 2). Among them, the residues involved in cofactor recognition, including the GXG motif, have quite similar conformations to those of PPM1. Reflecting this structural similarity, SAM or SAH is bound to TYW4N in a very similar manner to that observed in PPM1. Furthermore, there is a deep cavity in close vicinity to the bound cofactor, which is formed upon the structural change of helix α1′. In this cavity, Arg88 and Tyr229, which are conserved between TYW4N and PPM1, are located near the ε-methyl group of SAM (Figure 4C). The guanidinium and hydroxyl groups of these residues might recognize the common structure of the substrate of TYW4 and PPM1, i.e. the carboxylate moiety, to situate it correctly for the nucleophilic attack.
There are several distinctive differences between TYW4N and PPM1 in this cavity. TYW4N has many conserved Tyr and Phe residues in this deep cavity (Figure 4C), which are replaced with other amino-acid residues in PPM1. In PPM1, the gap between helices α5 and α8 forms a funnel-shaped cavity (Figure 3B), whereas in TYW4, the β6bc hairpin from TYW4C is inserted between this gap to form the small and flat cavity (Figures 3A and 4D). Thus, these TYW4-specific structures may recognize the specific structure of the yW-72 residue in tRNA, such as its planar tricyclic ring or aliphatic side chain. Hence, we hereafter refer to this deep cavity as the putative yW-72-binding pocket.
The C terminal domain of TYW4 forms a beta-propeller structure
The present crystal structure revealed that TYW4C adopts a six-bladed β-propeller structure (Figure 5A). The topology diagram of TYW4C is shown in Figure 2C. We hereafter refer to each of the blades as blade 1 to blade 6, and each β strand composing a blade as ‘βa’ to ‘βd’ (from the center to the periphery of the ring), and define the ‘top’ and ‘bottom’ sides as shown in Figures 2C and 5A. Well-known superfamilies with β-propeller structures are the WD-repeat (26,27) and Kelch-repeat (28,29), which both function as scaffold domains in macromolecular protein complexes. A structural homology search with the DALI algorithm (30) indicated that TYW4C shares high structural similarity with the Kelch-repeat proteins, especially that of the Keap1 protein (31) (Figure 5B). The RMSD between TYW4C and the Keap1 Kelch domain is 2.3 Å (Z score of 25.5) over 246 Cα atoms. Despite the lack of detectable sequence homology between TYW4C and Keap1, the structure-based sequence alignment showed that they share several features, even in their primary structures (Figure 5C). The ‘GG motif’ immediately after the βb strand, which is commonly observed in Kelch repeats, is also strictly conserved in all blades of TYW4C. An Arg residue between the βd and βa strands, which is also conserved in typical Kelch repeats, is conserved in blades 1, 2, 3 and 5 of TYW4C. However, other key signature motifs commonly observed in Kelch repeats (i.e. Tyr and Trp residues on the βc and βd strands, respectively) are not conserved. Consequently, the sequence as well as the structural similarity among the blades of TYW4C is lower than those of the Keap1 Kelch domain.
Figure 5.

The β-propeller structure of the TYW4 C-terminal domain (TYW4C). (A) and (B) Top and front views of ribbon representations of TYW4C (A) and the Keap1 Kelch domain (PDB ID: 1U6D) (31) (B). The protein main chain is colored as in Figure 2. (C) Structure-based sequence alignment of the six Kelch repeat motifs from Keap1 (upper) and TYW4 (lower). The four β strands are labeled a–b. The Arg residues involved in the interactions are boxed. Key signature residues seen in typical Kelch repeats (see text) are indicated with arrows. Structural alignments were calculated with SSM (43). (D) Interactions between TYW4N and TYW4C. The protein main chain is shown in a ribbon representation, with the same coloring scheme as in Figure 2. The protein side chains that are conserved and involved in the interaction are shown with stick models. (E) Interactions between Keap1 and the Neh2 peptide (PDB ID: 1X2R) (34). The main chain of the protein and bound peptide are shown with ribbon representations. The side chains that are conserved and involved in the interaction are shown with stick models.
Furthermore, there are several differences between TYW4C and the Keap1 Kelch domain. First, in the Keap1 Kelch domain, the propeller's ring structure is closed with the C-terminal β strand, and its N and C termini are located around the center of the ring. By contrast, in TYW4C, the ring structure is closed with the N-terminal β strand, and its N and C termini residing at the periphery of the ring (Figure 5A). Thus, the β propeller of TYW4C has a similar topology to that of the galactose oxidases (e.g. PDBID: 1GOH). Second, blade 5 of TYW4C has an extra β strand (β5e) at the periphery of the ring (Figure 5A). As a result, the N and C termini of the domain are about 21 Å apart, which is distinct from the typical β propeller structures of Kelch as well as WD repeats, in which their N and C termini are adjacent β strands in the same blade (Figure 5B). Third, there is no helix in the Keap1 Kelch domain, whereas TYW4C has several α and 310 helices between blades (Figure 5A).
In vivo complementation and in vitro reconstitution analyses by mass spectrometry
To clarify the mechanism of the methoxycarbonylation reaction, we performed in vitro reconstitution of the TYW4 reaction, using the recombinant enzyme prepared by the same method as the crystallization sample. The result clearly showed the direct conversion from yW-72 to yW by TYW4 alone (Figure 6A). We can rule out the possibility of the involvement of other proteins or organic compounds co-purified from the E. coli lysate in this catalysis, since we did not observe any electron density corresponding to them in the present crystal structure. Given that the methyl group in the methoxycarbonyl moiety originates from SAM, the compounds that can be a source of the carboxyl group are follows: SAM, Tris, DTT, and carbon dioxide (CO2) dissolved from the air atmosphere. It is unlikely that SAM acts as both a methyl and methoxycarbonyl donor, since the α-carboxyl group of the enzyme-bound SAM is distant from the catalytic pocket (Figure 4A). To further explore the methoxycarbonylation mechanism, we performed in vitro reconstitution of the TYW4 reaction with 13C-labeled sodium bicarbonate, followed mass spectrometry analysis. Intriguingly, the result showed an obvious change in the isotope distribution of the anticodon fragment (Figure 6B and C). In the presence of NaH13CO3, the mass of the most abundant fragment was shifted by +1 Da (Figure 6B, upper panel), indicating the incorporation of 13C from NaH13CO3 to yW. However, the small amount of the monoisotopic peak of the fragment containing the 12C atom (‘a’ in Figure 6B upper panel and Figure 6C left panel) was also detected. This peak may stem from 12CO2 contamination in the reaction buffer from the air atmosphere. Taken together, these results strongly suggest the incorporation of 13C from NaH13CO3 into the yW nucleotide in tRNA. Moreover, to verify the hypotheses discussed below, we constructed several mutants of TYW4 and assayed their activities by an in vivo complementation system (Table 3).
Figure 6.

In vitro reconstitution of yW synthesis using recombinant TYW4 with sodium bicarbonate. (A) LC/MS fragment analyses of RNaseT1-digested tRNAPhe with yW-72, treated by recombinant TYW4 in the absence (left panel) or the presence (right panel) of SAM. This graphs show mass chromatographs for anticodon-containing fragments containing yW-72 (m/z 681) (black line) overlaid with mass chromatographs for anticodon-containing fragments containing yW (m/z 691) (red line). (B) LC/MS fragment analyses of RNaseA-digested tRNAPhe with yW-72, treated by recombinant TYW4 in the presence of SAM and NaHCO3. The graphs on the left describe the mass chromatograms shown by the doubly charged ions of anticodon-containing fragments containing yW (GmAAyWAΨp; m/z ≈ 1119). The graphs on the right show the mass spectrum for each anticodon-containing fragment. Upper panels show the LC/MS analysis of tRNAPhe treated with TYW4 and 13C-NaHCO3. Lower panels show the LC/MS analysis of tRNAPhe treated with TYW4 and 12C-NaHCO3. In the right panels, peak ‘a’ is a monoisotopic ion, and peak ‘b’ is an isotopic ion with a mass 1 Da heavier than that of the peak ‘a’. (C) The graphs describing overlaid mass chromatograms, shown by doubly charged ions of anticodon-containing fragments containing yW (m/z 1119.60–1119.75, red line and 1119.0–1119.25, black line). The meaning of the labels ‘a’ and ‘b’ is the same as that in panel B. The theoretical masses of the doubly charged ions of peaks ‘a’ (yW-12C) and ‘b’ (yW-13C) are m/z 1119.188 and m/z 1119.692, respectively. The left panel shows the LC/MS analysis of tRNAPhe treated with TYW4 and 13C-NaHCO3. Thr right panel shows the LC/MS analysis of tRNAPhe treated with TYW4 and 12C-NaHCO3.
Table 3.
In vivo complementation analyses of the yeast TYW4 mutants
| Mutation | Complementation (%) |
|---|---|
| WT | 100.0 |
| R88A | 0.0 |
| ΔTYW4C | 46.8 |
DISCUSSION
Function and mechanism of the structural change upon cofactor binding
The present study showed that a large conformational change occurs at the N-terminal helix α1′ upon SAM binding (Figure 4A). This structural change should enable the seclusion of the catalytic site from the solvent molecules to ensure the correct reaction. Furthermore, molecule B of the present crystal structure of the SAH-bound form has an open form structure, even when SAH is bound to the SAM-binding pocket. Therefore, in this structure, the SAH molecule is exposed to the solvent and ready to be released from the SAM-binding pocket.
A detailed inspection of the present structures provides insight into the mechanism of this structural change. As already discussed, helices α1′ and α1 form a continuous helix, as shown in Figure 4A, after the SAM binding. However, the main chain conformation at the junction region deviates from that in an ideal α helix. More precisely, the main chain carbonyl oxygen of Ile27 makes hydrogen bond with the γ-OH group of the conserved Thr30 side chain (2.7 Å) and is distant from the main chain amide nitrogen of Asn31 (4.3 Å), which is a hydrogen-bonding donor in the ideal α helix. Instead, the main chain amide nitrogen of Asn31 is located near the main chain carbonyl oxygen of Gln28 (3.5 Å). Therefore, this junction region (residues 27–31) forms not an α- but a 310-helical structure, which is structurally perturbed by the interaction with the Thr30 side chain. Since a 310 helix is structurally more unstable than an α helix, it may enable the bending of the helix at this junction region.
Furthermore, we propose that the conserved Thr30 at this 310-helical junction region might function to sense the charge state of the cofactor to trigger the structural transition of the helix α1′. In the SAM-bound form, the γ-OH group of Thr30 interacts with the sulfonium group of SAM (3.8 Å, Figure 4A). The positive charge of the sulfonium group of SAM can be stabilized by the lone pair electrons of the γ-OH group of Thr30. Furthermore, the sulfonium group, the γ-OH group of Thr30 and the main chain carbonyl oxygen of Ile27 form an interaction network, involving the aforementioned 310 helical structure. Therefore, the bound SAM molecule may stabilize the closed form via this interaction network. In contrast, the thioether group of SAH is not charged and bears a lone pair of electrons. After the reaction, the interaction between the lone pair electrons of the γ-OH group of Thr30 and the sulfur atom of SAH may be destabilized. As a result, in molecule B in the SAH-complexed crystal, helix α1′ was in the open form structure, despite the cofactor binding (Figure 4B). However, it should be noted that, in molecule A in the SAH-complexed crystal, the enzyme was still in the closed form, and the γ-OH group of Thr30 was still situated near the thioether group of SAH. It is possible that some stabilization effect by the crystalline environment around molecule A might overcome the destabilization by the SAH binding discussed above.
In the previous structural analysis of yeast PPM1, Leulliot and coworkers reported that no conformational change was observed between the free- and cofactor-bound forms in PPM1 (19). However, a reinvestigation of these structures suggests that a structural change similar to that observed in TYW4 may also occur in PPM1. At first, in the free form structure of PPM1 (PDB ID: 1RJF), eight residues at its N terminus are disordered, which may corresponds to the open form. By contrast, in the SAM- and SAH-bound structures (PDB ID: 1RJD and 1RJE), seven residues (Glu2–Thr8) at its N terminus are ordered, forming a small α helix that corresponds to helix α1′ of TYW4N. The main-chain structure of the junction region between this small α1′ and α1 in PPM1 also deviates from the ideal α-helical geometry, and forms a 310-helical structure, as observed in TYW4N. Moreover, Thr30, at the junction of α1′ and α1 in TYW4N, is also conserved as Thr8 in PPM1. Therefore, this small helix α1′ and the conserved Thr residue of PPM1 may have roles similar to those proposed for TYW4.
Role of the C-terminal beta propeller domain
The present study revealed that TYW4C adopts a β-propeller structure and belongs to the Kelch repeat superfamily, which was observed in several proteins related to cellular signaling pathways, such as actin association, cell morphology and organization, and gene expression regulation (28,29). In these cellular events, these proteins act as adaptor by capturing their partners by the Kelch-repeat domain. The structural comparison revealed that TYW4C has a quite similar structure to the C-terminal Kelch repeat domain of Keap1. The C-terminal Kelch domain of Keap1 binds the Neh2 domain of the downstream transcription factor Nrf2, and the N-terminal domain of Keap1 recruits Nrf2 to the ubiquitin ligase complex for eventual degradation (32,33). In the Keap1 Kelch domain, the interaction sites for Nrf2 exist at the additional β hairpins between the βb and βc strands and at the linker between the blades, which are located at the bottom side of the ring. The complex structure of Keap1 Kelch domain and Neh2 peptide showed that their complex formation is based on a complementary electrostatic interaction, in which the basic residues at the bottom of Keap1 recognize the acidic motif conserved in the Neh2 domain (34) (Figure 5E).
In TYW4, TYW4C interacts with TYW4N on its bottom side, to insert its β6bc hairpin into the gap near the catalytic site (Figure 3). In this inter-subdomain interaction, the conserved basic residues (Arg460 and Arg492) of TYW4C provide electrostatic interactions for the conserved acidic residues (Asp331 and Glu334) in Region IV of TYW4N (Figure 5D). Although there is no distinct similarity between the TYW4C and Keap1 sequences, these basic residues involved in the interaction are conserved between TYW4C and Keap1 (Figure 5C, D and E). Consequently, the interaction manner between TYW4C and TYW4N is strikingly similar to that between Keap1 and Neh2 (34) (Figure 5E). Mutational analyses of ΔTYW4C showed that TYW4C is not required for the catalysis, but considerably enhances the catalytic efficiency (Table 3). These mutational results suggest the importance of the electrostatic interactions between TYW4C and TYW4N discussed above, for correctly inserting the β6bc hairpin into the gap between helices α5 and α8. The correct insertion of the β6bc hairpin will place Tyr678 and Phe680, which are at the tip of this hairpin (Figure 4C and D), in the gap between helices α5 and α8 to generate the flattened, hydrophobic yW-72-binding pocket
In plants, an ORF encoding a continuous TYW2, 3 and TYW4C-like sequence was found, suggesting that the TYW2, 3, and TYW4C domains are expressed as one polypeptide, TYW3-4C-2 (10). Furthermore, the species expressing TYW3-4C-2 lack the ORF corresponding to TYW4N. These species only have a single ORF belonging to the PPM1 family (hereafter referred to as PPM1/TYW4N), which strongly suggests that the PPM1/TYW4N protein has dual functions to methylate both the PP2A polypeptide and tRNA (10). The amino-acid sequence of the plant TYW4C ORF shares high homology with yeast TYW4C, which suggests that the plant TYW4C also has a similar β propeller structure to the yeast TYW4C. Moreover, the amino-acid sequence of the β6bc hairpin is also well conserved in the TYW4C domain of TYW3-4C-2 (Supplementary Figure 3). Therefore, it is likely that the TYW4C-like domain of TYW3-4C-2 may interact with PPM1/TYW4N by an electrostatic interaction and insert its β6bc hairpin into the PP2A-binding pocket. These inter-molecular interactions may switch the substrate specificity of PPM1/TYW4N from the PP2A polypeptide to yW-72 at tRNA position 37, by altering the shape of the substrate-binding cavity.
Methoxycarbonylation mechanism by TYW4
A previous in vitro biochemical analysis suggested that TYW4 may catalyze two independent reactions (10). Hereafter, we refer to the methylation of the α-carboxyl group as reaction I and to the methoxycarbonylation of the α-amino groups as reaction II (Figure 1). As discussed above, the present crystal structure strongly suggests that reaction I may have a similar mechanism to that of PPM1 (Figure 7A).
Figure 7.

Proposed catalytic mechanism of reaction I (A) and reaction II (B) by TYW4.
The present biochemical results clearly showed the incorporation of 13C from 13C-labeled bicarbonate into the yW nucleotide in tRNA (Figure 6C). Based on our results, we propose that reaction II occurs by the following two steps: i) carboxylation of the α-amino group of the α-amino-α-carboxypropyl (acp) side chain by CO2 and ii) methylation of the resultant carbamate group by SAM (Figure 7B). The formation of a carbamate group at a specific amino group by the addition of carbon dioxide has been observed in several enzymes, such as haemoglobin (35), ribulose bisphosphate carboxylase-oxygenase (Rubisco) (36) and dethiobiotin synthetase (37). In these enzymes, a carbamate group is formed by the nucleophilic attack of an uncharged protein amino group upon CO2. Therefore, we hypothesize that reaction II proceeds by the addition of not a bicarbonate ion, but a CO2 molecule. Moreover, for this reaction, a general base catalyst deprotonates the protein amino group, while a general acid catalyst, such as Arg, Lys, or a metal ion, interacts with CO2 to increase its electrophilicity (35). In TYW4, the α-amino group of the acp side chain may be deprotonated by a general base catalyst and attack the CO2 molecule bound to the enzyme. In addition, the N-C bond of the carbamate is labile, and thus the carbamate formation is highly reversible (35). We hypothesize that the carbamate group thus formed may subsequently be methylated by SAM. This methylation may form a stable methoxycarbonyl group, resulting in CO2 fixation (Figure 7B).
The catalytic site of TYW4 for two chemical reactions
The present biochemical analysis showed that TYW4 can catalyze both reactions I and II. Our crystal structure revealed that TYW4 possesses only one SAM-binding pocket in the TYW4N domain. It is unlikely that the TYW4C domain has another catalytic site for reaction II, since the ΔTYW4C mutant can complement the activity for complete yW formation (Table 3). Therefore, we propose that both reactions I and II may occur at the same catalytic site. The SAM molecule utilized in reaction II (Figure 7B) may bind the same SAM-binding pocket as that for reaction I (Figure 7A). The methylation of the carbamate by SAM in reaction II may occur via the same mechanism as the methylation in reaction I.
As described above, the putative yW-72-binding pocket is formed by helix α1′, α1 of region I, α5 of region IV, and the β6bc hairpin (Figures 3A and 4D). This pocket has a flat, deep shape and may snugly fit the bulky structure of the tricyclic ring of the yW-72 base. This pocket is also deep enough to accommodate the apc side chain of the yW-72 base. The methyl group of SAM and the side chains of Arg88 and Tyr229 are situated at the bottom of this pocket. Therefore, the pocket has a shape complementary to the molecular surface of yW-72. These findings allowed us to construct a docking model of yW-72, as shown in Figure 8. In this model, the acp side chain can be accommodated in the pocket in two different modes for reactions I and II described above (Figure 8B and C). The α-carboxyl and α-amino groups are directed to the SAM side for their methylation and methoxycarbonylation, respectively. The flexible aliphatic moiety of the acp side chain may allow it to bind in two different conformations. In reaction I, the side chains of Arg88 and Tyr229 may recognize the α-carboxyl group to fix it in the vicinity of the ε-methyl group of SAM. In contrast, in reaction II, the phenolic hydroxyl group of Tyr229 can act a general base catalyst to deprotonate the α-amino group of yW-72 (Figures 7B and 8C). Arg88 may interact with CO2 to activate it for this nucleophilic attack. Actually, the R88A mutant was completely unable to complement the ΔTYW4 yeast strain, highlighting the importance of Arg88 in both of the reactions (Table 3). To verify the details of the present hypothetical reaction mechanism, the structural analysis of the complex of TYW4, yW-72 containing tRNA, and CO2 might be required.
Figure 8.

Docking model of TYW4 and the yW nucleoside precursors. (A) Close-up view of the solvent-accessible surface of the yW-72-binding pocket and the modeled yW nucleoside precursor. The surface is colored with the same scheme as in Figure 2. (B) and (C) Stereo views of the yW-72-binding pocket and the modeled yW nucleoside precursors. In (B) and (C), the yW nucleoside precursors are shown in two different conformations for reactions I and II, respectively.
tRNA recognition by TYW4
TYW4 is considered to recognize the tRNA anticodon stem loop and correctly locate nucleoside 37 at its catalytic site. The present structure, combined with a sequence alignment from other species, provides insights into the tRNA recognition mechanism. The loop encompassing residues 70–79 in Region I is conserved in TYW4N, but is replaced with helices α2 and α3 in PPM1 (Figure 3). This region contains conserved basic residues (Arg77 and Lys76) and forms a positively charged patch near the entrance of the putative yW-72-binding pocket (Figure 9A). Especially, this loop and the basic residues are also conserved in plant PPM1, which is proposed to be a dual-function enzyme (i.e. tRNA and PP2A modifications), as described above. Therefore, it is possible that this basic region recognizes the phosphate groups adjacent to the target residue, position 37, in tRNAPhe. Furthermore, another conserved basic patch is formed on the TYW4C surface near the catalytic site (Figure 9B). This region consists of the loops between β1b and β1c, and β6d and β1a, at the bottom side of the ring structure. Especially, Lys389 and Arg410 are conserved. The present biochemical analysis revealed that TYW4C is not required for the catalysis, but considerably enhances the complementation activity. We speculate that TYW4C not only generates the yW-72-specific pocket with its β6bc hairpin, but also provides a tRNA-binding site on its basic surface, thereby facilitating the efficient catalysis by TYW4N. Finer details of the tRNA recognition mechanism of TYW4 will await the structural determination of the TYW4-tRNAPhe complex.
Figure 9.

Surface representation of TYW4. The electrostatic potential in (A) is colored on the surface of the protein from red (negative, −10 kT/e) to blue (positive, +10 kT/e). The electrostatic potential was calculated by GRASP (44). The surface in (B) is colored in increasing shades of blue reflecting increasing residue conservation. In (A) and (B), the putative yW-72-binding pocket is indicated by an arrow, and the conserved basic patches are encircled.
CONCLUSIONS
The present crystal structures provide the structural basis for the creation of new substrate specificity by the combination of domains derived from different superfamilies. In TYW4, the N-terminal domain shares structural similarity with PPM1 whose substrate is a protein, whereas the C-terminal domain belongs to the Kelch repeat superfamily, which is observed in adaptor proteins involved in cellular signaling pathways. Our results revealed that the β6bc hairpin of the C-terminal domain is inserted into the gap near the catalytic site of the N-terminal domain, thereby changing the substrate specificity from a protein peptide to a hypermodified nucleoside in tRNA.
Moreover, our biochemical analysis suggested the novel catalytic mechanism of TYW4. The methoxycarbonylation reaction may proceed through carbamate formation with CO2 and methylation with SAM, which results in CO2 fixation. Since CO2 is the most oxidized state of carbon, its fixation into solid organic compounds requires a large energy input (38). In this reaction, the highly reactive sulfonium group in SAM may supply this energy required for CO2 fixation. SAM is a multifunctional cofactor that acts as a source of various chemical groups, such as methyl, ribosyl, and aminoalkyl groups, as well as a source of radicals (39). The present results would define a new role of SAM, as a high energy source in CO2 fixation.
Finally, the present crystal structures suggested the discrimination mechanism of SAM and its near-cognate ligand, SAH, by TYW4. In some MTases and a SAM-responsive riboswitch, a comparison of the SAM- and SAH-bound crystal structures revealed that the cofactors were bound in different conformations, which are considered to enable the discrimination between SAM and SAH (40,41). The discrimination mechanism by TYW4 is unique in that the structural transition of the enzyme, but not the cofactor, enables the discrimination between SAM and SAH.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Project on Protein Structural and Functional Analyses from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (to O.N. and T.S.); grants from MEXT (to R.I. and O.N.); Mitsubishi Foundation grants (to O.N.); grant from the New Energy and Industrial Technology Development Organization (NEDO) (to T.S.), JSPS Fellowship for Japanese Junior Scientists (to A.N.). Funding for open access charge: University of Tokyo.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Y. Sakaguchi (The University of Tokyo, Japan) for mass spectrometry experiments; F. Arisaka (Tokyo Institute of Technology, Japan) for the ultracentrifugation analysis; H. Nishimasu (The University of Tokyo, Japan) for helpful comments on the manuscript; and the beam-line staffs at BL41XU of SPring-8 and NW12A of KEK PF-AR for assistance in data collection.
Footnotes
† Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes: 2ZW9, 2ZWA, 2ZZK).
REFERENCES
- 1.Rozenski J, Crain PF, McCloskey JA. The RNA modification database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishitani R, Yokoyama S, Nureki O. Structure, dynamics, and function of RNA modification enzymes. Curr. Opin. Struct. Biol. 2008;18:330–339. doi: 10.1016/j.sbi.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Ishitani R, Nureki O, Nameki N, Okada N, Nishimura S, Yokoyama S. Alternative tertiary structure of tRNA for recognition by a posttranscriptional modification enzyme. Cell. 2003;113:383–394. doi: 10.1016/s0092-8674(03)00280-0. [DOI] [PubMed] [Google Scholar]
- 4.Blobstein S, Grunberg D, Weinstein I, Nakanishi K. Isolation and structure determination of the fluorescent base from bovine liver phenylalanine transfer ribonucleic acid. Biochemistry. 1973;12:188–193. doi: 10.1021/bi00726a002. [DOI] [PubMed] [Google Scholar]
- 5.Thiebe R, Zachau HG. A specific modification next to the anticodon of phenylalanine transfer ribonucleic acid. Eur. J. Biochem. 1968;5:546–555. doi: 10.1111/j.1432-1033.1968.tb00404.x. [DOI] [PubMed] [Google Scholar]
- 6.Bruce AG, Uhlenbeck OC. Enzymatic Replacement of the Anticodon of Yeast Phenylalanine Transfer Ribonucleic Acid. Biochemistry. 1982;21:855–861. doi: 10.1021/bi00534a007. [DOI] [PubMed] [Google Scholar]
- 7.Carlson BA, Kwon SY, Chamorro M, Oroszlan S, Hatfield DL, Lee BJ. Transfer RNA modification status influences retroviral ribosomal frameshifting. Virology. 1999;255:2–8. doi: 10.1006/viro.1998.9569. [DOI] [PubMed] [Google Scholar]
- 8.Hatfield D, Feng YX, Lee BJ, Rein A, Levin JG, Oroszlan S. Chromatographic analysis of the aminoacyl-tRNAs which are required for translation of codons at and around the ribosomal frameshift sites of HIV, HTLV-1, and BLV. Virology. 1989;173:736–742. doi: 10.1016/0042-6822(89)90589-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konevega AL, Soboleva NG, Makhno VI, Semenkov YP, Wintermeyer W, Rodina MV, Katunin VI. Purine bases at position 37 of tRNA stabilize codon-anticodon interaction in the ribosomal A site by stacking and Mg2+-dependent interactions. RNA. 2004;10:90–101. doi: 10.1261/rna.5142404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noma A, Kirino Y, Ikeuchi Y, Suzuki T. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J. 2006;25:2142–2154. doi: 10.1038/sj.emboj.7601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waas WF, de Crecy-Lagard V, Schimmel P. Discovery of a gene family critical to wyosine base formation in a subset of phenylalanine-specific transfer RNAs. J. Biol. Chem. 2005;280:37616–37622. doi: 10.1074/jbc.M506939200. [DOI] [PubMed] [Google Scholar]
- 12.Kalhor HR, Penjwini M, Clarke S. A novel methyltransferase required for the formation of the hypermodified nucleoside wybutosine in eucaryotic tRNA. Biochem. Biophys. Res. Commun. 2005;334:433–440. doi: 10.1016/j.bbrc.2005.06.111. [DOI] [PubMed] [Google Scholar]
- 13.Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S. Crystal structure of archaeal tRNA(m1G37)methyltransferase aTrm5. Proteins. 2008;72:1274–1289. doi: 10.1002/prot.22019. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki Y, Noma A, Suzuki T, Senda M, Senda T, Ishitani R, Nureki O. Crystal structure of the radical SAM enzyme catalyzing tricyclic modified base formation in tRNA. J. Mol. Biol. 2007;372:1204–1214. doi: 10.1016/j.jmb.2007.07.024. [DOI] [PubMed] [Google Scholar]
- 15.Goto-Ito S, Ishii R, Ito T, Shibata R, Fusatomi E, Sekine S, Bessho Y, Yokoyama S. Structure of an archaeal TYW1, the enzyme catalyzing the second step of wye-base biosynthesis. Acta Crystallogr. 2007;D63:1059–1068. doi: 10.1107/S0907444907040668. [DOI] [PubMed] [Google Scholar]
- 16.Waas WF, Druzina Z, Hanan M, Schimmel P. Role of a tRNA base modification and its precursors in frameshifting in eukaryotes. J. Biol. Chem. 2007;282:26026–26034. doi: 10.1074/jbc.M703391200. [DOI] [PubMed] [Google Scholar]
- 17.De Baere I, Derua R, Janssens V, Van Hoof C, Waelkens E, Merlevede W, Goris J. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry. 1999;38:16539–16547. doi: 10.1021/bi991646a. [DOI] [PubMed] [Google Scholar]
- 18.Kalhor HR, Luk K, Ramos A, Zobel-Thropp P, Clarke S. Protein phosphatase methyltransferase 1 (Ppm1p) is the sole activity responsible for modification of the major forms of protein phosphatase 2A in yeast. Arch. Biochem. Biophys. 2001;395:239–245. doi: 10.1006/abbi.2001.2558. [DOI] [PubMed] [Google Scholar]
- 19.Leulliot N, Quevillon-Cheruel S, Sorel I, de La Sierra-Gallay IL, Collinet B, Graille M, Blondeau K, Bettache N, Poupon A, Janin JL, et al. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 2004;279:8351–8358. doi: 10.1074/jbc.M311484200. [DOI] [PubMed] [Google Scholar]
- 20.Schneider TR, Sheldrick GM. Substructure solution with SHELXD. Acta Crystallogr. 2002;D58:1772–1779. doi: 10.1107/s0907444902011678. [DOI] [PubMed] [Google Scholar]
- 21.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr. 2000;D56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 23.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. 2002;D58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki T, Ikeuchi Y, Noma A, Suzuki T, Sakaguchi Y. Mass spectrometric identification and characterization of RNA-modifying enzymes. Methods Enzymol. 2007;425:211–229. doi: 10.1016/S0076-6879(07)25009-8. [DOI] [PubMed] [Google Scholar]
- 25.Cheng XD, Kumar S, Posfai J, Pflugrath JW, Roberts RJ. Crystal structure of the Hhal DNA methyltransferase complexed with S-Adenosyl-L-Methionine. Cell. 1993;74:299–307. doi: 10.1016/0092-8674(93)90421-l. [DOI] [PubMed] [Google Scholar]
- 26.Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith TF, Gaitatzes C, Saxena K, Neer EJ. The WD repeat: a common architecture for diverse functions. Trends Biochem.Sci. 1999;24:181–185. doi: 10.1016/s0968-0004(99)01384-5. [DOI] [PubMed] [Google Scholar]
- 28.Adams J, Kelso R, Cooley L. The kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol. 2000;10:17–24. doi: 10.1016/s0962-8924(99)01673-6. [DOI] [PubMed] [Google Scholar]
- 29.Prag S, Adams JC. Molecular phylogeny of the kelch-repeat superfamily reveals an expansion of BTB/kelch proteins in animals. BMC Bioinformatics. 2003;4 doi: 10.1186/1471-2105-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm L, Sander C. Dali – a network tool for protein-structure comparison. Trends Biochem.Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 31.Li XC, Zhang D, Hannink M, Beamer LJ. Crystal structure of the Kelch domain of human Keap1. J. Biol. Chem. 2004;279:54750–54758. doi: 10.1074/jbc.M410073200. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate for proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004;24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 2004;24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Padmanabhan B, Tong KI, Ohta T, Nakamura Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S, Yamamoto M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell. 2006;21:689–700. doi: 10.1016/j.molcel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Lorimer GH. Carbon dioxide and carbamate formation: the makings of a biochemical control system. Trends Biochem.Sci. 1983;8:65–68. [Google Scholar]
- 36.Cleland WW, Andrews TJ, Gutteridge S, Hartman FC, Lorimer GH. Mechanism of Rubisco: The carbamate as general base. Chem. Rev. 1998;98:549–561. doi: 10.1021/cr970010r. [DOI] [PubMed] [Google Scholar]
- 37.Schneider G, Lindqvist Y. Structural enzymology of biotin biosynthesis. FEBS Lett. 2001;495:7–11. doi: 10.1016/s0014-5793(01)02325-0. [DOI] [PubMed] [Google Scholar]
- 38.Sakakura T, Choi JC, Yasuda H. Transformation of carbon dioxide. Chem. Rev. 2007;107:2365–2387. doi: 10.1021/cr068357u. [DOI] [PubMed] [Google Scholar]
- 39.Fontecave M, Atta M, Mulliez E. S-adenosylmethionine: nothing goes to waste. Trends Biochem.Sci. 2004;29:243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 41.Lu C, Smith AM, Fuchs RT, Ding F, Rajashankar K, Henkin TM, Ke A. Crystal structures of the SAM-III/S-MK riboswitch reveal the SAM-dependent translation inhibition mechanism. Nat. Struct. Mol. Biol. 2008;15:1076–1083. doi: 10.1038/nsmb.1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanner MF, Olson AJ, Spehner JC. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–320. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 43.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. 2004;D60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 44.Nicholls A, Sharp KA, Honig B. Protein Folding and Association: Insights From the Interfacial and Thermodynamic Properties of Hydrocarbons. Prot. Struct. Func. Genet. 1991;11:281–296. doi: 10.1002/prot.340110407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.