

Abstract
The current development of immunotherapy for Alzheimer's disease is based on the assumption that human-derived amyloid β protein (Aβ) can be targeted in a similar manner to animal cell-derived or synthetic Aβ. Because the structure of Aβ depends on its source and the presence of cofactors, it is of great interest to determine whether human-derived oligomeric Aβ species impair brain function and, if so, whether or not their disruptive effects can be prevented using antibodies. We report that untreated ex vivo human CSF that contains Aβ dimers rapidly inhibits hippocampal long-term potentiation in vivo and that acute systemic infusion of an anti-Aβ monoclonal antibody can prevent this disruption of synaptic plasticity. Aβ monomer isolated from human CSF did not affect long-term potentiation. These results strongly support a strategy of passive immunization against soluble Aβ oligomers in early Alzheimer's disease.
Keywords: synaptic plasticity, amyloid β protein, Alzheimer's disease, LTP, long-term potentiation, CSF, hippocampus
Introduction
Immunotherapy targeting Aβ offers a potential disease modifying treatment for Alzheimer's disease (AD) (Roberson and Mucke, 2006). Studies of the clinical efficacy of active immunization against preaggregated Aβ and passive immunization with anti-Aβ antibodies have commenced in humans (Gilman et al., 2005; Solomon, 2007) based primarily on studies of animal models that over-express Aβ (Schenk et al., 1999; Bard et al., 2000). Although endogenously generated and exogenously applied antibodies to Aβ can reduce cognitive and synaptic plasticity deficits in amyloid precursor protein (APP)-related transgenic mice (Janus et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002) and Aβ infusion models (Klyubin et al., 2005), it is unclear whether animal cell-generated human Aβ behaves in a manner similar to human-derived Aβ. In the biosynthesis of Aβ, many different lengths and conformations of the peptide are generated, including highly mobile soluble Aβ oligomers, which are believed to mediate the earliest stages of AD (Klein et al., 2001). Animal cell-derived Aβ oligomers are extremely potent at disrupting cognition and synaptic plasticity (Walsh et al., 2002; Cleary et al., 2005; Townsend et al., 2006). Because the biological activity of Aβ oligomers and the ability to target them selectively with immunotherapy is critically dependent on their conformation, it is of great interest to compare animal- and human-derived Aβ oligomers. Given the lability of Aβ conformation it is important to evaluate the peptide in its native state. One such source is human CSF (huCSF), which is known to contain many different Aβ species, including low-n oligomers of variable length (Walsh et al., 2000). Indeed, huCSF Aβ is being developed as a primary biological marker of preclinical and clinical AD (Shaw et al., 2007), but the question of its pathophysiological activity and the effects of immunotherapy on any such activity has not been elucidated. If selective immunotherapy is to be developed successfully, it is important to know whether the active Aβ species in the brain can be targeted with systemic treatment with antibody.
Here, we report that huCSF from both healthy older individuals and AD patients that contained clearly detectable dimers of Aβ completely disrupted synaptic plasticity in a manner similar to animal cell-derived low-n oligomers of Aβ. Moreover systemic passive immunization against Aβ fully prevented the inhibition of long-term potentiation (LTP) by both human and animal cell-derived Aβ oligomers providing impetus to targeting soluble Aβ oligomers in early AD.
Materials and Methods
huCSF, handling, and AD diagnosis.
The huCSF study was approved by the ethics committee of the University of Göteborg. CSF samples were collected by lumbar puncture through the L3/L4 or L4/L5 interspace. The first 12 ml of CSF was collected in a polypropylene tube, immediately transported to the local laboratory for centrifugation at 2000 × g at 4°C for 10 min. The supernatant was pipetted off, gently mixed to avoid possible gradient effects, and aliquoted in 2–5 ml portions that were stored at −80°C pending testing.
The samples were collected in three sets, set A (see Fig. 2A,B, CSF #1–3; supplemental Fig. 2B, CSF #1F–J, available at www.jneurosci.org as supplemental material) patients received a diagnosis of AD using the DSM-IIIR (Diagnostic and Statistical Manual of Mental Disorders, third edition, revised) (American Psychiatric Association, 1987) and National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association (McKhann et al., 1984) criteria of dementia and probable AD, respectively. The Mini-Mental State Examination (MMSE) score was used as a global measure of functioning (Folstein et al., 1975). Inclusion criteria for controls were that they should be physically and mentally healthy and not experiencing or exhibiting any cognitive impairment. All controls were thoroughly interviewed about their somatic and mental health by a research nurse before inclusion in the study and were cognitively stable over at least 2 years after the initial examination.
Figure 2.
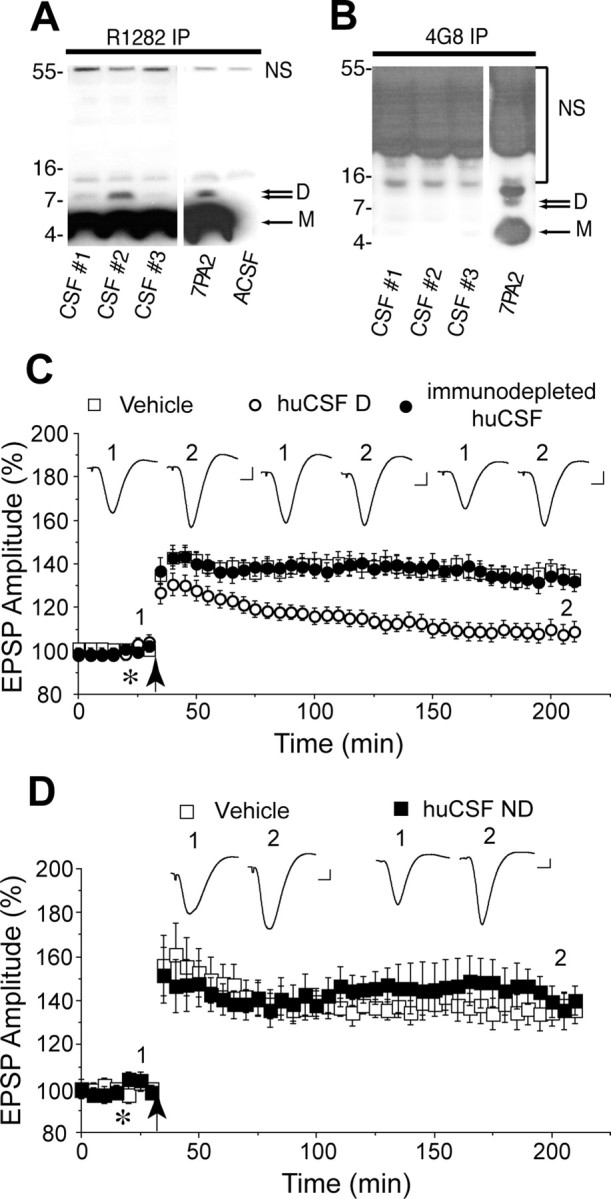
huCSF containing clearly detectable Aβ dimers disrupts synaptic plasticity in vivo. A, Immunoprecipitation of certain human CSF samples with the polyclonal anti-Aβ antibody R1282 and subsequent immunoblotting with the monoclonal anti-Aβ antibody 2G3 revealed the presence of abundant Aβ monomer (M) and a less-abundant Aβ dimer (D). These proteins migrated similarly to Aβ species detected in 7PA2 conditioned medium, but were not detected in artificial CSF (ACSF). Bands detected at ∼12 and ∼55 kDa were not specific (NS), because they were also detected in ACSF. CSF samples 1 and 2 are from a male (age 58, MMSE 13, tau 443, Aβ42 229 ng/ml) and female (age 54, MMSE 14, tau 570, Aβ42 368 ng/ml) patient with clinical AD. CSF sample 3 is from a female (age 75, MMSE 29, tau 630, Aβ42 920 ng/ml) cognitively normal healthy donor. B, A single round of immunoprecipitation of huCSF with R1282 effectively depleted samples of both Aβ monomer and dimer such that when CSF was subjected to a second round of immunoprecipitation with 4G8, no Aβ could be detected in any of the samples tested (compare A, first round of immunoprecipitation, and B, second round of immunoprecipitation). Immunoblotting, performed using 2G3, revealed a typical pattern of Aβ monomer and oligomers in a fresh sample of 7PA2 conditioned medium. C, Samples of huCSF containing Aβ dimers (huCSF D) completely inhibited LTP, and this inhibition was prevented by previous immunodepletion of Aβ. Untreated huCSF (open circles; n = 12 using samples from 6 donors, including those in A above), immunodepleted samples from the same donors (closed circles; n = 6, including those in B above), and vehicle (n = 13) were injected (10 μl, i.c.v.; asterisk) 10 min before high-frequency stimulation (arrow). D, Samples of huCSF without clearly detectable Aβ dimers but with abundant monomers (huCSF ND) had no effect on LTP. Untreated huCSF (closed squares; n = 6 using samples from 3 donors) and vehicle (open squares; n = 5) were injected (10 μl, i.c.v.; asterisk) 10 min before high-frequency stimulation (arrow). The huCSF samples were from a non-AD donor (supplemental Fig. 2A and Table 1, CSF #64, available at www.jneurosci.org as supplemental material) and two AD patients (supplemental Fig. 2B and Table 1, CSF #1G and #1J, available at www.jneurosci.org as supplemental material). Insets and calibration are as in Figure 1. Error bars indicate SEM.
Set B (supplemental Fig. 2Ai,ii, available at www.jneurosci.org as supplemental material) samples were from patients who sought medical advice because of cognitive impairment. Patients were designated as normal or AD according to CSF biomarker levels using cutoffs that are >90% specific for AD (Hansson et al., 2006): total tau (T-tau) >350 ng/L and Aβ42 <530 ng/L. None of the biochemically normal subjects fulfilled these criteria. CSF T-tau and Aβ42 levels were determined using ELISA as described previously (Hulstaert et al., 1999; Vanmechelen et al., 2000). Set C (supplemental Fig. 2B, CSF #1A–E, available at www.jneurosci.org as supplemental material) samples were from de-identified non-AD cases from the clinical routine. Patients were biochemically normal according to CSF biomarker levels using the same cutoffs as set B.
APP-expressing cells.
Chinese hamster ovary (CHO) cells stably transfected with a cDNA encoding APP751 containing the Val717Phe familial AD mutation (referred to as 7PA2 cells) were cultured in DMEM with 10% fetal bovine serum as described previously (Koo and Squazzo, 1994). Nearly confluent (95–100%) 10 cm dishes of 7PA2 cells and their corresponding untransfected parental CHO cell (CHO−) line were washed with plain DMEM (5 ml, one time) and incubated in ∼5 ml of DMEM for ∼15 h. Conditioned medium (CM) was then removed and cleared of cells by centrifugation at 200 g for 10 min at 4°C and samples were stored at −20°C or used immediately.
Antibodies.
Monoclonal antibodies 6E10 and 4G8 (Covance ImmunoTechnologies, Dedham, MA) recognize epitopes within the human Aβ sequence corresponding to residues 1–16 and 17–24, respectively. R1282 is a high-titer polyclonal antiserum raised to synthetic Aβ1–40 (Walsh et al., 2000) and 5E2 is a monoclonal antibody raised against human fetal tau (Joachim et al., 1987). Monoclonal antibody 2G3 was raised to Aβ33–40 and specifically recognizes Aβ species ending at residue 40, whereas monoclonal antibody 21F12 was raised to Aβ33–42 and specifically recognizes Aβ species ending at residue 42. Both 2G3 and 21F12 were kindly provided by Drs. P. Seubert and D. Schenk (Elan Pharmaceuticals, South San Francisco, CA). 26D6 is a monoclonal antibody that specifically recognizes an epitope in the extreme N-terminal region of Aβ and was a gift from Dr. Elizabeth Chen-Dobson (Merck Research Laboratories, West Point, PA). For systemic passive transfer, preservative free antibodies were administered by intracardiac injection (in PBS, 0.6 ml) under anesthesia.
Immunoprecipitation/Western blot analysis.
A sensitive immunoprecipitation (IP)/Western blot protocol that can readily detect as little as 200 pg of naturally secreted Aβ (Walsh et al., 2000) was used for the detection of Aβ in CM and huCSF. Samples were immunoprecipitated with the high-affinity polyclonal antibody, R1282 (at a dilution of 1:75) or the monoclonal antibody, 4G8 (at 5 μg/ml). After immunoprecipitation, samples were electrophoresed on 10–20% tricine gels and transferred to 0.2 μm nitrocellulose membranes at 400 mA for 2 h. Filters were boiled for 10 min in PBS (Ida et al., 1996) and then blocked overnight at 4°C with 5% fat-free milk in 20 mm Tris-HCl, pH 7.4, containing 150 mm NaCl and 0.05% Tween 20 (TBS-T). After washing the membranes in TBS-T, they were probed with monoclonal 2G3 (1 μg/ml) alone or in combination with 21F12 (also at 1 μg/ml). Bound antibody was visualized with horseradish peroxidase-conjugated donkey anti-mouse Ig (at 1:25,000) (Jackson ImmunoResearch, West Grove, PA) and the ECL+ detection system (GE Healthcare, Little Chalfont, Buckinghamshire, UK). A single round of immunoprecipitation as described above effectively depleted CSF samples of Aβ.
Size-exclusion chromatography.
Concentrated CM (1 ml) was chromatographed on a Superdex 75 10/30 HR column attached to an AKTA FPLC system (GE Healthcare) and eluted with 50 mm ammonium acetate, pH 8.5, at a flow rate of 1 ml/min (Walsh et al., 2005). One milliliter fractions were collected and divided equally; one-half were stored frozen pending use for electrophysiology and the other half were lyophilized, resuspended in 2x SDS-tricine sample buffer (20 μl), boiled for 10 min, and then electrophoresed on 10–20% tricine gels. Proteins were transferred onto 0.2 μm nitrocellulose and used for Western blotting as described above.
Rat CSF and plasma anti-Aβ antibody ELISA.
A cisternal puncture method was used for CSF sampling under anesthesia. After exposing the atlanto-occipital membrane, CSF was collected from the cisterna magna. Whole blood was collected from the anesthetized animal into Eppendorf tubes containing ∼30 μl 0.5 m EDTA and inverted several times before being spun for 10 min at 2500 rpm. Plasma and CSF were stored frozen at −80°C.
The Aβ antibodies 6E10 and 4G8 were measured in plasma and CSF using ELISA as described previously (Lemere et al., 2002). Briefly, 96-well plates were coated with human Aβ1–40 (2 μg/ml) in 50 mm carbonate buffer overnight. A standard curve was included by adding serial dilution of normal mouse IgG (Sigma, St. Louis, MO; range, 0–32 ng/ml) in duplicate in carbonate buffer. After washing in 0.05% Tween-20 PBS (TPBS) buffer, wells were blocked with 3% bovine serum albumin in 0.5% TPBS for 2 h at room temperature. Plates were washed three times in TPBS. Rat plasma and CSF samples were diluted in TBS (150 mm NaCl, 50 mm Tris) at 1:5 and 1:30 and incubated for 2 h at room temperature. After washing in TPBS, Aβ-bound antibody was detected using horseradish peroxidase-conjugated goat anti-mouse IgG (Kirkegaard and Perry Laboratories, Gaithersburg, MD) and the TMB peroxidase substrate kit (Kirkegaard and Perry Laboratories). Titers were read at an optical density of 450 nm. Nearly identical results were obtained when the matching antibody, 6E10 or 4G8, was used for the standard curve.
In vivo electrophysiology and sample injection procedure. Animal experiments were licensed by the Department of Health and Children, Ireland. Adult male Wistar rats were anesthetized with urethane (1.5 g/kg, i.p.). Single-pathway recordings of field EPSPs were made from the stratum radiatum in the CA1 area of the dorsal hippocampus in response to stimulation of the ipsilateral Schaffer collateral/commissural pathway as described previously (Walsh et al., 2002). Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity adjusted to give an EPSP amplitude of 50% of maximum. The high-frequency stimulation (HFS) protocol for inducing LTP consisted of 10 trains of 20 stimuli, with an interstimulus interval of 5 ms and intertrain interval of 2 s. The intensity was increased to give an EPSP of 75% of maximum amplitude during the HFS. LTP is expressed as the mean (±SEM) percentage of baseline field EPSP amplitude recorded over at least a 30 min baseline period. Similar results were obtained when EPSP slope rather than amplitude was measured.
To inject samples, a cannula was implanted in the lateral cerebral ventricle (coordinates: 1 mm lateral to the midline and 4 mm below the surface of the dura) just before electrode implantation (Walsh et al., 2002). Fractionated CM diluted 1:5 with milliQ water (Millipore, Billerica, MA) was normally used as the stock solution and injected (5 μl) over a 2 min period, 10 min before HFS. The huCSF samples (10 μl) were injected over a 5 min period 10 min before HFS. Control vehicle injections (5 or 10 μl of milliQ water) were interleaved between the other experiments. An interval of 1.5 h or more was left between the implantation procedure and the intracardiac injection of the antibody because pilot experiments comparing CSF concentration of antibody (6E10, 0.4–0.6 mg, i.c.) in nonimplanted (n = 4) and implanted animals (n = 4) were consistent with resealing of the blood–brain barrier at this time. Furthermore, great care was taken only to study animals in which the synaptic responses were stable over a baseline recording period of at least 1 h. Statistical comparisons used paired and unpaired Student's t tests.
Results
Systemic passive immunization against Aβ prevents the disruption of synaptic plasticity by cell-derived Aβ low-n oligomers
Because certain cultured cell lines produce Aβ monomer and low-n oligomers of Aβ similar to those found in huCSF (Walsh et al., 2000), we commenced by determining whether systemic infusion of an anti-Aβ antibody prevented the inhibition of a neurophysiological correlate of memory, LTP, by cell-derived oligomers (Klyubin et al., 2005) in vivo (Fig. 1). We used 6E10, a monoclonal antibody that recognizes Aβ residues 1–16 and which can efficiently bind to (Fig. 1A) and directly neutralize Aβ oligomers after intracerebroventricular injection in the rat brain (Klyubin et al., 2005). Aβ oligomers in the CM from a cell line expressing APP, termed 7PA2 cells, were isolated using size-exclusion chromatography (SEC) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Acute intracerebroventricular injection of Aβ oligomers 10 min before HFS completely inhibited LTP (102 ± 7%, mean (±SEM) percentage at baseline at 2 h post-HFS, n = 5; p > 0.05 compared with baseline, Student's t test; p < 0.05 compared with vehicle injected controls, 128 ± 6%, n = 6) (Fig. 1B). However, in animals that were preadministered 6E10 systemically (0.4–0.6 mg per rat; i.c.) the oligomer-enriched fraction of 7PA2 CM no longer inhibited LTP (124 ± 5%, n = 5; p < 0.05 compared with pre-HFS baseline), the level of potentiation being indistinguishable from LTP in vehicle injected control animals (p > 0.05). In contrast, previous systemic injection of an isotype control antibody, 5E2 (0.5 mg per rat, i.c.), had no significant effect on LTP inhibition by the Aβ oligomer fraction (107 ± 5%, n = 5; p > 0.05 compared with pre-HFS baseline and Oligo 7PA2-treated group, 107 ± 6%, n = 3; p < 0.05 compared with vehicle-injected controls, 146 ± 8%, n = 5) (Fig. 1C). We next quantified the ability of the peripherally injected antibody to enter the CSF and found that 6E10 was readily detected in CSF samples taken from the cisterna magna at 30 min (11.2 ± 5 ng/ml, n = 8) and 3 h (17.6 ± 9.8 ng/ml, n = 6) after intracardiac injection. The CSF concentration of 6E10 was 0.33 ± 0.18% (n = 6) of the plasma concentration at the latter time point, similar to what has been reported for endogenously generated anti-Aβ antibodies (Lemere et al., 2004). Thus, systemically administered antibody that binds to Aβ oligomers can prevent the inhibition of LTP by cell-derived Aβ oligomers, ostensibly by directly neutralizing them in the brain.
Figure 1.
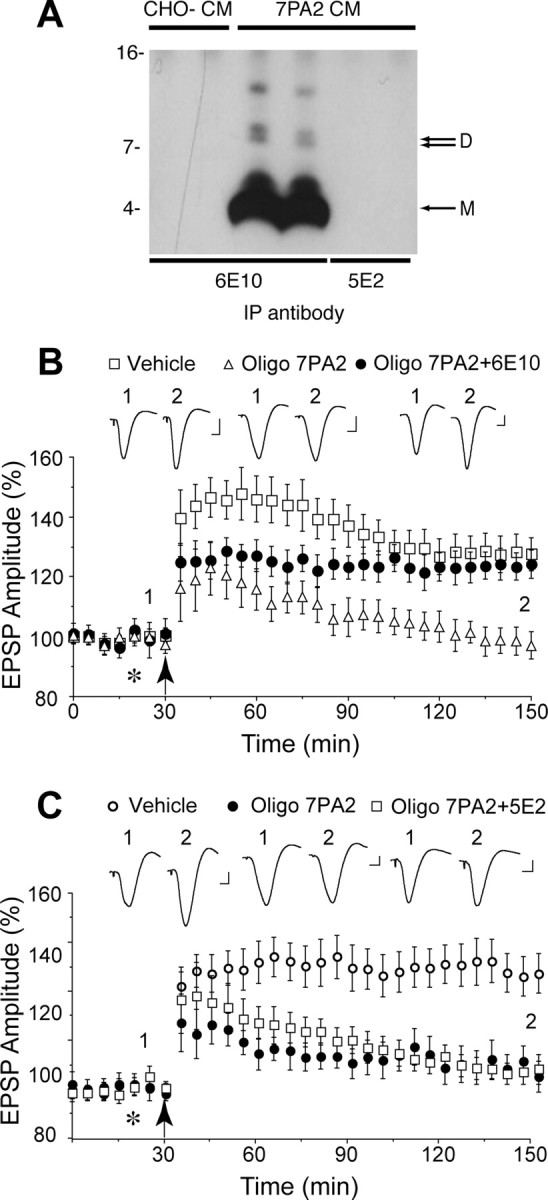
Systemic passive immunization completely prevents the disruption of synaptic plasticity by animal cell-derived human Aβ oligomers in vivo. A, The antibody 6E10 (lanes 3, 4) effectively immunoprecipitated both Aβ monomer and oligomers from the CM of CHO cells that stably express mutant human APP (7PA2 cells), whereas an isotype control antibody, 5E2 (lanes 5, 6), was not capable of binding Aβ. The specificity of the Aβ species detected by 6E10 is confirmed by the fact that no such bands were detected when CM from untransfected CHO cells (CHO−) was used (lanes 1, 2). Molecular weight markers are indicated on the left-side of the blot. The positions of Aβ monomer (M) and dimer (D) are indicated with an arrow on the right side of the blot. B, The Aβ oligomer-enriched fraction of 7PA2 CM (Oligo 7PA2) inhibited the induction of LTP induced by high-frequency stimulation (arrow) at CA3–CA1 synapses in anesthetized rats, and this inhibition was abrogated by intracardiac pretreatment with the antibody 6E10 (Oligo 7PA2 + 6E10). The Aβ oligomer treated animals received an intracerebroventricular injection of Oligo 7PA2 (5 μl) 30 min after an intracardiac injection of either vehicle (open triangles; n = 5) or the antibody 6E10 (closed circles; n = 5). Control animals (open squares; n = 6) received a peripheral injection of vehicle (0.6 ml, i.c.) followed 30 min later with an intracerebroventricular injection of vehicle (5 μl; asterisk). C, Systemic treatment with an isotype control antibody failed to affect the inhibition of LTP by cell-derived Aβ oligomers. Animals were treated with vehicle (open circles; n = 5) or Oligo 7PA2 (closed circles; n = 3) as described in A, or 5E2 (0.5 mg per rat, i.c.) followed by Oligo 7PA2 (open squares; n = 5). Values are the mean (±SEM) percentage of the baseline EPSP amplitude. Insets show representative electrophysiological traces before (1) and after (2) high-frequency stimulation. Calibration: 0.5 mV, 10 ms.
Human CSF containing Aβ dimers disrupts synaptic plasticity
Next we determined whether huCSF that contained Aβ oligomers could inhibit LTP (Fig. 2A,B). Samples of CSF from healthy older (>50 years) donors or patients diagnosed with AD (Fig. 2A, supplemental Table 1, available at www.jneurosci.org as supplemental material) were analyzed for the presence of Aβ oligomers by immunoprecipitation with R1282 and subsequent Western blotting with 2G3. CSF from certain healthy and AD subjects contained abundant Aβ monomer together with low levels of Aβ dimers, but higher-n SDS-stable oligomers were not detected in any of the samples analyzed (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Based on these analyses, we selected six CSF samples with clearly detectable Aβ dimer and assessed their effects on LTP in vivo (Fig. 2C). Four samples were from normal subjects and the other two from patients with AD. Injection of these CSF samples (10 μl, i.c.v.) 10 min before HFS completely inhibited LTP (108 ± 5% baseline at 3 h, n = 12 animals using samples from 6 human subjects, each subject's CSF being tested in two separate experiments; p > 0.05 compared with baseline, p < 0.05 compared with vehicle-injected rats, 133 ± 4%, n = 13). When these samples of huCSF were applied in the absence of HFS they had no significant effect on baseline transmission (106 ± 6% at 3 h, n = 3 animals using samples from 3 donors, p > 0.05 compared with preinjection baseline; p > 0.05 compared with vehicle, 104 ± 4%, n = 3) (data not shown). To confirm that the inhibition of LTP was mediated by Aβ, the CSF samples were immunodepleted with the anti-Aβ antibody R1282 (Fig. 2A,B) and then tested for their ability to disrupt synaptic plasticity. Importantly, samples that had blocked LTP before immunodepeletion lost their activity after specific removal of Aβ (133 ± 5%, n = 12 animals using samples from the same six donors, p < 0.05 compared with baseline and untreated huCSF; p > 0.05 compared with vehicle) (Fig. 2C). Similarly, samples that had comparable concentrations of monomer but no detectable dimer were without effect on LTP [138 ± 8%, n = 6 animals using samples from 3 subjects (one healthy, two AD), p < 0.05 compared with baseline; p > 0.05 compared with vehicle, 137 ± 6%, n = 5] (Fig. 2D).
Because the samples with clearly detectable dimer contained variable concentrations of monomer, we directly compared the activity of two previously untested CSF samples that had equivalent monomer concentrations but clearly different dimer concentrations: CSF sample 56, which contained both monomer and dimer, and sample 60, which contained an equivalent concentration of monomer to sample 56, but no evidence of dimer (Fig. 3A, supplemental Fig. 2A and Table 1, available at www.jneurosci.org as supplemental material). Only the former sample inhibited LTP (112 ± 6%, n = 4; p > 0.05 compared with baseline; p < 0.05 compared with 145 ± 11% in animals treated with sample 60, n = 4, or 142 ± 9% in vehicle-treated animals, n = 5).
Figure 3.

Aβ dimer in huCSF is required to inhibit LTP whereas Aβ monomer is inactive. A, We directly compared the activity of a CSF sample which contained both monomer and dimer (huCSF D, open circles; n = 4) (supplemental Fig. 2A and Table 1, CSF #56, available at www.jneurosci.org as supplemental material), a sample that contained an equivalent concentration of monomer, but no evidence of dimer (huCSF ND, closed circles; n = 4) (supplemental Fig. 2A and Table 1, CSF #60, available at www.jneurosci.org as supplemental material) and vehicle (open squares; n = 5). Samples were injected (10 μl, i.c.v.; asterisk) 10 min before conditioning HFS (arrow). Only the dimer-containing sample inhibited LTP. B, An SEC fraction of huCSF enriched with Aβ monomer (for Western blot, see supplemental Fig. 3, available at www.jneurosci.org as supplemental material) (huCSF M Fr, black squares; 10 μl i.c.v., n = 4) was compared with vehicle (white squares; 10 μl i.c.v., n = 4). The monomer-enriched huCSF failed to affect LTP. Insets and calibration are as in Figure 1. Error bars indicate SEM.
In a separate set of experiments, we used SEC to isolate the Aβ monomer from huCSF (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Injection of a monomer fraction (Fig. 3B) at a concentration similar to that found in untreated samples of CSF failed to inhibit LTP (149 ± 18%, n = 4, p < 0.05 compared with baseline; p > 0.05 compared with 142 ± 8% in vehicle-injected animals, n = 4). Because of the difficulty in obtaining sufficiently large volumes of dimer-containing huCSF we have, as yet, been unable to isolate the Aβ dimer fraction from huCSF.
Systemic passive immunization prevents the disruption of synaptic plasticity by huCSF Aβ dimers
Finally, we assessed whether systemic infusion of 4G8, a monoclonal antibody directed to the midregion of Aβ could prevent the inhibition of LTP by huCSF containing Aβ dimers (Fig. 4). We chose 4G8 because we found previously that intracerebroventricular injection of 4G8 can abrogate the synaptic plasticity disruption by cell-derived Aβ oligomers (Klyubin et al., 2005). In these experiments, 4G8 was peripherally injected (0.15–0.2 mg/rat, i.c.) 30 min before intracerebroventricular injection of dimer-containing huCSF samples found to inhibit LTP (Figs. 2C, 3). In 4G8 infused animals, the huCSF failed to block HFS-induced LTP, the magnitude of which (129 ± 11%, n = 5 animals with samples from 5 subjects) was indistinguishable from that elicited in vehicle-treated controls (135 ± 8%, n = 4). Importantly, LTP was not affected in animals pretreated systemically with 4G8 and subsequently injected intracerebroventricularly with huCSF that had previously been immunodepleted of Aβ (134 ± 12%, n = 5 animals with samples from 5 subjects). We found that peripherally injected 4G8 entered the CSF, reaching a concentration of 60.0 ± 21.6 ng/ml at 4 h after intracardiac injection (n = 7). The CSF concentration was 0.39 ± 0.09% (n = 7) of the plasma concentration at this time.
Figure 4.

Systemic passive immunization prevents the disruption of synaptic plasticity by Aβ dimer-containing huCSF in vivo. Animals received an intracardiac injection of the monoclonal anti-Aβ antibody 4G8 (0.15–0.2 mg/rat), 30 min before an intracerebroventricular injection of either untreated (n = 5) or immunodepleted (n = 5) huCSF (10 μl; asterisk) and 40 min before high-frequency stimulation (arrow). Insets and calibration are as in Figure 1. Error bars indicate SEM.
Discussion
Our discovery that human CSF containing Aβ dimers inhibits LTP in vivo shows for the first time that human-derived soluble Aβ has a pathophysiological action in the brain, rapidly and potently disrupting synaptic plasticity mechanisms that are believed to underlie memory in the hippocampal network (Morris et al., 2003). Evidence that the dimers, and not the monomers, are the active Aβ species was provided by the finding that CSF samples with similar concentrations of monomer, as estimated from the Western blots and ELISA, only inhibited LTP if dimer was clearly detected. Moreover, a monomer containing fraction, isolated using size exclusion chromatography, failed to inhibit LTP, thus confirming that monomeric huCSF Aβ does not impair synaptic plasticity. Although it is possible that Aβ dimers in human CSF may form larger assemblies such as Aβ*56 (molecular weight, ∼56 kDa), which has been found in the brains of APP transgenic mice (Lesné et al., 2006), we failed to detect such species in the human CSF samples by IP/Western blot or SEC/Western blot. It will be important to determine the relationship between these different Aβ species in human CSF and brain samples in future studies.
Because Aβ conformation is critically dependent on source and the presence of cofactors, it was unclear whether synthetic and cell-derived Aβ are truly representative of human-derived Aβ. The potent inhibition of synaptic LTP by human-derived ex vivo Aβ in its native state is consistent with the assumption that animal- and human-derived Aβ exert similar synaptic actions. The requirement for human CSF to contain dimer to disrupt synaptic plasticity lends strong support to the proposal that low-n oligomers of Aβ are a key synaptotoxic species in early AD.
Although the present study was not designed to determine diagnostic potential, the presence of synaptic plasticity-disrupting Aβ dimers in CSF samples taken from people who were categorized as cognitively normal and the absence of detectable levels of dimers in CSF from patients diagnosed with AD indicates that CSF dimer concentration is unlikely to be useful on its own as a marker of clinical AD. Levels of different Aβ species in CSF do not necessarily directly match the concentration in brain extracellular fluid. Thus, for example, there is a reduced CSF concentration of Aβ42 in AD, most likely because Aβ42 is retained in the brain (Fagan et al., 2006). Moreover, the levels of ELISA-detectable Aβ in CSF from normal individuals fluctuate dramatically over the course of 36 h (Bateman et al., 2007). How CSF Aβ dimer levels change over time or what their relationship is to Aβ assemblies in the brain is currently unknown. Why Aβ dimer-containing CSF samples from elderly controls who had high CSF Aβ42 and low tau levels, supporting a normal diagnosis (Hansson et al., 2006; Fagan et al., 2007), inhibited LTP is unclear at this time.
The complete prevention of the inhibitory effect of Aβ dimer-containing huCSF and animal cell-derived Aβ oligomers by systemic treatment with the monoclonal antibodies 4G8 and 6E10, respectively, contrasts with the situation where only partial prevention was achieved when endogenous antibodies that bound Aβ oligomers were generated by systemic active immunization with Aβ (Klyubin et al., 2005). Because a CSF antibody titer in the region of 100 pm was achieved and subnanomolar concentrations of oligomers of Aβ are required to inhibit LTP (Wang et al., 2004; Townsend et al., 2006; present study), it is likely that sufficient antibody entered the brain interstitial fluid/CSF to neutralize Aβ oligomers directly. The ability of systemic passive immunization to fully protect against Aβ oligomer-induced block of LTP in vivo augurs well for a passive immunization approach that includes targeting Aβ oligomers that disrupt synaptic plasticity in early AD.
Footnotes
This work was supported by Science Foundation Ireland, the Irish Health Research Board, the Irish Higher Education Authority (PRTLI), and the European Union (M.J.R., R.A.), National Institutes of Health Grant AG20245 (T.W.), and Wellcome Trust Grant 067660 (D.M.W.). A.W. was a recipient of an EU FP6 Marie Curie Early Stage Training Grant. D.J.S. is a consultant to Elan. We thank Dr. K. Kosik for providing 5E2 hybridoma and Dr. L. Jiang for help with antibody ELISA. 2G3 and 21F12 were kindly provided by Drs. P. Seubert and D. Schenk, and 26D6 by Dr. E. Chen-Dobson.
References
- American Psychiatric Association. Ed 3. Arlington, VA: American Psychiatric Association; 1987. Diagnostic and statistical manual of mental disorders. [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta- peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68:666–669. doi: 10.1212/01.wnl.0000256043.50901.e3. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain A beta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006;5:228–234. doi: 10.1016/S1474-4422(06)70355-6. [DOI] [PubMed] [Google Scholar]
- Hulstaert F, Blennow K, Ivanoiu A, Schoonderwaldt HC, Riemenschneider M, De Deyn PP, Bancher C, Cras P, Wiltfang J, Mehta PD, Iqbal K, Pottel H, Vanmechelen E, Vanderstichele H. Improved discrimination of AD patients using beta-amyloid(1–42) and tau levels in CSF. Neurology. 1999;52:1555–1562. doi: 10.1212/wnl.52.8.1555. [DOI] [PubMed] [Google Scholar]
- Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K. Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem. 1996;271:22908–22914. doi: 10.1074/jbc.271.37.22908. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, St., Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Joachim CL, Morris JH, Selkoe DJ, Kosik KS. Tau epitopes are incorporated into a range of lesions in Alzheimer's disease. J Neuropathol Exp Neurol. 1987;46:611–622. doi: 10.1097/00005072-198711000-00001. [DOI] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang LY, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes A beta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD. Intranasal immunotherapy for the treatment of Alzheimer's disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002;23:991–1000. doi: 10.1016/s0197-4580(02)00127-6. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Beierschmitt A, Iglesias M, Spooner ET, Bloom JK, Leverone JF, Zheng JB, Seabrook TJ, Louard D, Li D, Selkoe DJ, Palmour RM, Ervin FR. Alzheimer's disease Aβ vaccine reduces central nervous system Aβ levels in a non-human primate, the Caribbean vervet. Am J Pathol. 2004;165:283–297. doi: 10.1016/s0002-9440(10)63296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Morris RGM, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O'Carroll C. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Phil Trans Roy Soc Lond B Biol Sci. 2003;358:773–786. doi: 10.1098/rstb.2002.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer disease- like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Korecka M, Clark CM, Lee VM, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nat Rev Drug Discov. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- Solomon B. Intravenous immunoglobulin and Alzheimer's disease immunotherapy. Curr Opin Mol Ther. 2007;9:79–85. [PubMed] [Google Scholar]
- Townsend M, Shankar G, Mehta T, Walsh D, Selkoe D. Effects of secreted oligomers of amyloid {beta}-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol (Lond) 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjogren M, Andreasen N, Blennow K. Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000;285:49–52. doi: 10.1016/s0304-3940(00)01036-3. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic amyloid beta-peptide (A beta) fibrillogenesis block oligomerization of natural A beta and thereby rescue long-term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QW, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370–3378. doi: 10.1523/JNEUROSCI.1633-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]