

Abstract
G-protein (Gβγ)-mediated voltage-dependent inhibition of N- and P/Q-type Ca2+ channels contributes to presynaptic inhibition and short-term synaptic plasticity. The voltage dependence derives from the dissociation of Gβγ from the inhibited channels, but the underlying molecular and biophysical mechanisms remain largely unclear. In this study we investigated the role in this process of Ca2+ channel β subunit (Cavβ) and a rigid α-helical structure between the α-interacting domain (AID), the primary Cavβ docking site on the channel α1 subunit, and the pore-lining IS6 segment. Gβγ inhibition of P/Q-type channels was reconstituted in giant inside-out membrane patches from Xenopus oocytes. Large populations of channels devoid of Cavβ were produced by washing out a mutant Cavβ with a reduced affinity for the AID. These β-less channels were still inhibited by Gβγ, but without any voltage dependence, indicating that Cavβ is indispensable for voltage-dependent Gβγ inhibition. A truncated Cavβ containing only the AID-binding guanylate kinase (GK) domain could fully confer voltage dependence to Gβγ inhibition. Gβγ did not alter inactivation properties, and channels recovered from Gβγ inhibition exhibited the same activation property as un-inhibited channels, indicating that Gβγ does not dislodge Cavβ from the inhibited channel. Furthermore, voltage-dependent Gβγ inhibition was abolished when the rigid α-helix between the AID and IS6 was disrupted by insertion of multiple glycines, which also eliminated Cavβ regulation of channel gating, revealing a pivotal role of this rigid α-helix in both processes. These results suggest that depolarization-triggered movement of IS6, coupled to the subsequent conformational change of the Gβγ-binding pocket through a rigid α-helix induced partly by the Cavβ GK domain, causes the dissociation of Gβγ and is fundamental to voltage-dependent Gβγ inhibition.
Keywords: Gβγ, voltage-dependent modulation, β subunit, α helix, inhibition, patch clamp
Introduction
Voltage-gated Ca2+ channels (VGCCs) shape electrical signals in excitable cells and generate a ubiquitous and essential chemical signal, Ca2+ ion. The activity of VGCCs is subject to dynamic regulation by numerous signaling pathways and molecules (for review, see Catterall, 2000). A prevalent and versatile form of regulation is inhibition by G-protein-coupled receptors (GPCRs) of the Cav2 family of high voltage-activated (HVA) N-, P/Q- and R-type Ca2+ channels (Dunlap and Fischbach, 1978). In particular, the G-protein-mediated, membrane-delimited and voltage-dependent inhibition has received the most attention because it is likely involved in presynaptic inhibition (for review, see Hille, 1994; Dolphin, 2003a; Tedford and Zamponi, 2006). This inhibition exhibits three major biophysical characteristics (Bean, 1989; Elmslie et al., 1990): it slows down the activation kinetics of the inhibited channels; shifts the activation voltage to a more depolarized potential; and can be relieved by a strong conditioning depolarizing potential, resulting in the so-called prepulse facilitation (for review, see Dolphin, 2003a; Tedford and Zamponi, 2006). These voltage-dependent features contribute to short-term synaptic plasticity by virtue of relieving G-protein inhibition during high-frequency action potential firing (Brody et al., 1997; Williams et al., 1997; Bertram et al., 2003).
The voltage-dependent inhibition is mediated by the G-protein βγ subunits (Gβγ) (Herlitze et al., 1996; Ikeda, 1996). The Gβγ-bound channels are in a “reluctant” gating mode, requiring stronger depolarizations to open (Bean, 1989; Elmslie et al., 1990), and activation of these channels results in the dissociation of Gβγ from the channel (Boland and Bean, 1993). The voltage dependence of Gβγ inhibition thus stems mainly from the voltage-dependent unbinding of Gβγ. The molecular and biophysical mechanisms underlying this dissociation remain largely unclear.
Some functional effects of Gβγ are the opposites of those of Ca2+ channel β subunits (Cavβ), raising the possibility that Gβγ and Cavβ compete with each other (Campbell et al., 1995; Bourinet et al., 1996). However, some key issues have not been fully resolved (for review, see Dolphin, 2003a; Tedford and Zamponi, 2006). One question is whether Cavβ is required for voltage-dependent Gβγ inhibition. On the one hand, it was reported that knockdown of endogenous Cavβs in neurons enhanced receptor-induced inhibition (Campbell et al., 1995), and that coexpression of Cavβs reduced G-protein inhibition in oocytes (Bourinet et al., 1996; Qin et al., 1997). On the other hand, Cavβ was shown to be necessary for Gβγ-induced voltage-dependent inhibition in COS cells (Meir et al., 2000). Another controversial question is whether Cavβ is dislodged from Gβγ-inhibited channels. One view is that there is a competitive interaction between Gβγ and Cavβ binding to the Ca2+ channel α1 subunit (Bourinet et al., 1996; Qin et al., 1997). An opposing view is that Cavβ remains associated during G-protein inhibition (Cantí et al., 2000; Meir et al., 2000; Feng et al., 2001). Further unanswered questions are, if Cavβ is required for voltage-dependent Gβγ inhibition, which regions are needed and what is the underlying molecular mechanism for its mandatory role.
We addressed these questions by investigating Gβγ and Cavβ regulation of P/Q-type Ca2+ channels expressed in Xenopus oocytes. Purified Gβγ was applied directly to the cytoplasmic side in inside-out membrane patches. Large populations of surface channels devoid of Cavβ (β-less channels) were obtained by washing out a mutant Cavβ, allowing us to directly compare the effect of Gβγ on channels with or without Cavβ. We also investigated the importance in Gβγ inhibition of a rigid linker between the IS6 segment and the α-interacting domain (AID) of the channel α1 subunit. The AID is located in the cytoplasmic loop (I-II loop) connecting the first two homologous repeats of the α1 subunit and constitutes the high-affinity Cavβ-binding site (Pragnell et al., 1994; Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004). Our results highlight an essential role of Cavβ and a rigid coupling between IS6 and the AID in voltage-dependent Gβγ inhibition.
Materials and Methods
Purification of Gβγ from Sf9 insect cells.
As described by Kozasa (2004), Sf9 insect cells (Novagen) were coinfected with recombinant baculoviruses encoding bovine Gβ1 (NM_175777) and hexahistidine-tagged bovine Gγ2 (BC112789). Cells were cultured in suspension at 27°C for 48 h, then collected and sonicated. Membranes were isolated by ultracentrifugation and resuspension in a solution containing 1% sodium cholate. After a 10 h incubation with stirring at 4°C, the mixture was centrifuged, and the supernatant containing the membrane extract was collected. Recombinant Gβγ protein was purified from the membrane extract with BD TALON metal affinity resin (BD Biosciences). The elution from the resin was concentrated and exchanged into another solution containing (in mm) 20 HEPES, 0.5 EDTA, 2 MgCl2, 1 DTT, 11 CHAPS, and 100 NaCl, pH7.8 with NaOH, after a further purification with a Superdex 200 gel-filtration column (Pharmacia).
Protein synthesis in E. coli.
βARK_PH domain protein was obtained by expressing the cDNA encoding residues Q546-S670 of rat adrenergic receptor kinase (NM_012776), which was subcloned into pET28a vector (Novagen), in BL21(DE3) bacteria. The proteins were purified from cell lysates with BD TALON metal affinity resin. The elution was further purified with a Superdex 200 gel-filtration column.
cDNA encoding residues R369-Q413 of rabbit brain Cav2.1(X57477) was subcloned into a modified pGEX4T-1 vector and expressed in BL21(DE3) bacteria to obtain the I-II loop protein. Seven or five glycine or alanine residues were inserted between F376 and L377 to produce the mutant forms of the I-II loop.
The Cavβ core domain proteins were obtained by expressing the cDNA encoding the core region of wild-type (WT) or mutant β2a (M80545) or β1b (NP-000714) in BL21(DE3) bacteria. The core regions are from G17 to N416 for β2a and from G58 to T418 for β1b; both were subcloned into pET28a vector. The proteins were purified individually.
GST pull-down assay.
GST_I-II loop was immobilized on glutathione Sepharose 4B beads (Novagen). The Cavβ core protein bound to the AID was eluted from the beads with glutathione. The elution was detected with Coomassie Blue staining on SDS-PAGE.
Oocyte preparation and expression.
cDNAs encoding various constructs were subcloned into a modified oocyte expression vector pGEMHE. The constructs include WT rabbit brain Cav2.1 (X57477) and mutant Cav2.1 with seven or five alanines or glycines inserted between F376 and L377, skeletal muscle α2-δ, human β1b (NP-000714), the core region of rat brain β2a (M80545), and the guanylate kinase (GK) domain (S228-T412) of β1b. Xenopus oocytes were prepared and maintained as described (He et al., 2007). cRNAs were synthesized in vitro and varying amounts (0.2–5 ng) were injected into oocytes in various combinations. Recordings were performed 3–7 d after injection.
Electrophysiology.
For two-electrode voltage-clamp, the electrodes were filled with 3 mm KCl and had a resistance of 0.5–1 MΩ. The bath solution contained (in mm) 10 BaCl2, 5 KCl, 60 TEA-OH, 20 NaOH and 5 HEPES, pH 7.4 with HCl. The current was evoked every 6 s by a +20 mV pulse for 40 ms from the holding potential of −80 mV. For inside-out macropatch recording, the electrode had a diameter of 15–30 μm and a resistance of 0.2–0.5 MΩ. It was filled with a solution containing (in mm) 45 BaCl2, 80 KCl and 10 HEPES, pH 7.3 with KOH. The bath (i.e., cytoplasmic) solution contained (in mm) 125 KCl, 4 NaCl, 10 HEPES, 10 EGTA (pH 7.3 with KOH). Phosphatidylinositol-4,5-bisphosphate (PIP2; 0.3 μm) and Mg-ATP (3 mm) were added freshly to the bath solution to attenuate rundown. Control and test solutions were fed by a pressurized system through separate tubes to a manifold attached to a single outlet tube and were switched on/off individually. After obtaining the inside-out patches, the recording pipette was inserted into the perfusion tube to achieve a rapid and complete exchange of solution. To obtain the β-less channels described in Figures 2, 3, and 5, a fast perfusion speed (∼1.5 ml/min) was used. Experiments were performed at 22°C.
Figure 2.

Producing abundant β-less surface P/Q-type channels with a mutant Cavβ. A, Close-up of the AID-Cavβ interaction interface in β2a. The two labeled AID-binding residues, M245 and L249, were mutated to alanine in the β2a core, producing a mutant named β2a_Mut2. B, Coomassie Blue staining illustrating the interaction between WT β2a_core or β2a_Mut2 and a I-II loop fragment from Cav2.1. GST_I-II loop was immobilized in a GST column and was used to pull down WT β2a_core or β2a_Mut2 protein. B, Bound; U, unbound. A large excess of WT β2a_core and β2a_Mut2 proteins were present in the unbound fraction. The two bands close to the size of GST_I-II loop in the unbound fraction of β2a_Mut2 are degraded products of the β2a_Mut2 protein. C, Whole-oocyte peak Ba2+ current from oocytes expressing Cav2.1 and α2-δ alone (β−), or with WT β2a_core or β2a_Mut2. D, Representative current traces evoked by a +20 mV pulse, showing the inactivation kinetics under the indicated condition of channels containing WT β2a_core (left) or β2a_Mut2 (middle), or without exogenous Cavβ (β− channels, right). The traces were obtained from different time points as indicated in E: immediately after patch excision (0 min,  ), at the end of wash (wash, ⊗), or after the application of purified β2a_core protein (β2a_core, ·). E, Time course of changes of the inactivation kinetics of channels containing WT β2a_core (left) or β2a_Mut2 (middle), or of β− channels (right). After washing the cytoplasmic side of the membrane patch for ∼ 5 min (∼100 s for β− channels), 1 μm WT β2a_core protein was applied. Current was evoked every 20 s by a 2 s pulse to +20 mV. The half-time of inactivation (T1/2_inact), defined as the time for the current to decay from the peak to the 50% size, was normalized to that obtained immediately after patch excision (0 min). n = 5–14. F, Voltage dependence of activation under the indicated conditions for channels containing WT β2a_core (left) or β2a_Mut2 (middle), or for β− channels (right). G, H, Bar graph comparing the half-time of inactivation (T1/2_inact) and the midpoint (V1/2) and slope factor (k) of activation under the indicated conditions for channels containing WT β2a_core or β2a_Mut2, or for β− channels. n = 5–14. *p < 0.05.
), at the end of wash (wash, ⊗), or after the application of purified β2a_core protein (β2a_core, ·). E, Time course of changes of the inactivation kinetics of channels containing WT β2a_core (left) or β2a_Mut2 (middle), or of β− channels (right). After washing the cytoplasmic side of the membrane patch for ∼ 5 min (∼100 s for β− channels), 1 μm WT β2a_core protein was applied. Current was evoked every 20 s by a 2 s pulse to +20 mV. The half-time of inactivation (T1/2_inact), defined as the time for the current to decay from the peak to the 50% size, was normalized to that obtained immediately after patch excision (0 min). n = 5–14. F, Voltage dependence of activation under the indicated conditions for channels containing WT β2a_core (left) or β2a_Mut2 (middle), or for β− channels (right). G, H, Bar graph comparing the half-time of inactivation (T1/2_inact) and the midpoint (V1/2) and slope factor (k) of activation under the indicated conditions for channels containing WT β2a_core or β2a_Mut2, or for β− channels. n = 5–14. *p < 0.05.
Figure 3.

β-Less P/Q-type channels lack voltage-dependent Gβγ inhibition. Macroscopic currents were recorded in inside-out macropatches from oocytes expressing Cav2.1, α2-δ and β2a_Mut2. β-Less channels were generated by washing β2a_Mut2 off the surface channels, as determined by the change of the activation curve before and after a 5 min wash, as in Figure 2F. All data shown in this figure were collected after the 5 min wash. Voltage protocols for assessing voltage-dependent Gβγ inhibition were as described in Figure 1. A, Superposition of currents evoked by a +20 mV test pulse in the absence or presence of 20 nm Gβγ, without or with a 20 ms, +100 mV prepulse, showing Gβγ-induced inhibition and the lack of prepulse facilitation. The PFI was 0.97 ± 0.11 and 0.99 ± 0.12 (n = 6) without and with Gβγ, respectively. Con, Control; PP, prepulse. Notice that the current amplitude was still sizable after washing out β2a_Mut2. B, Time course of Gβγ inhibition. Data points represent tail currents recorded at −30 mV after a depolarization to +20 mV (n = 6). Filled circles indicate the tail current of a test pulse following the prepulse. Arrows and numbers indicate the time point at which the activation curve 4 displayed in Figure 5A is taken. Current is normalized by that obtained 5 min after patch excision, after β2a_Mut2 had been washed off (0 min data point). C, Voltage dependence of activation under the indicated conditions. The midpoint and slope factor are as follows: Con: 28.0 ± 3.3 and 14.9 ± 1.1 mV; Con+PP: 23.8 ± 4.5 and 13.8 ± 2.1 mV; Gβγ: 29.0 ± 3.3 and 15.9 ± 1.1 mV; Gβγ+PP: 27.0 ± 6.1 and 16.1 ± 2.0 mV (n = 6 for all). D, Time constant of activation (τact) of currents evoked at +20 mV under the indicated conditions.
Figure 5.

Cavβ does not dissociate from Gβγ-inhibited channels. A, Comparison of the voltage dependence of activation of channels containing β1b under the indicated conditions with that of the β-less channels produced as in Figure 2. The activation curve for β-less channels is reproduced from Figure 3C. The time points at which each activation curve is taken are indicated in Figure 1C (for 1–3) and in Figure 3B (for 4). The midpoint and slope factor are as follows: Con: 11.5 ± 2.3 and 8.8 ± 0.6 mV (n = 7); Gβγ: 22.3 ± 4.8 and 14.7 ± 2.3 mV (n = 7); βARK_PH: 12.3 ± 1.9 and 9.9 ± 0.9 mV (n = 5); β-less channels: 28.0 ± 3.3 and 14.9 ± 1.1 mV (n = 6). B, Comparison of the voltage dependence of steady-state inactivation of β1b-containing channels without or with Gβγ treatment, as well as that of β-less channels with Gβγ treatment. The midpoint and slope factor are −26.1 ± 4.0 and 7.2 ± 0.9 mV for β1b-containing channels without Gβγ, −24.6 ± 4.7 and 8.0 ± 1.3 mV for β1b-containing channels with Gβγ, −14.3 ± 1.5 and 5.8 ± 0.3 mV for β-less channels with Gβγ (n = 4–5). C, Averaged current traces (n = 4–5) evoked by a + 20 mV pulse, showing the inactivation kinetics of β-less channels with Gβγ and of β1b-containing channels without or with Gβγ.
Currents were sampled at 10 kHz and filtered at 2.5 kHz. The holding potential for all the following protocols was −80 mV. Macroscopic currents were evoked by 5.5 ms depolarizations ranging from −40 to + 100 mV in 10 mV increments at a 1.5 s interval. Tail currents were always recorded by repolarization to −30 mV, regardless of the preceding test pulse. To obtain the activation curves, tail currents were normalized by that following the depolarization to +100 mV and plotted against the test potentials. To study prepulse facilitation, a 20 ms, +100 mV prepulse was applied 2 ms before each of the 5.5 ms test pulses mentioned above. Steady-state inactivation was determined by a three-pulse protocol in which a 10 ms pulse to +20 mV (pulse A) was followed sequentially by a 20 s conditioning pulse (ranging from −80 to +40 mV) and a 10 ms test pulse to +20 mV (pulse B). The interval between each protocol was 1.5 min. Peak current evoked by pulse B was normalized by that evoked by pulse A and was plotted against the conditioning potentials to obtain the voltage dependence of inactivation.
Data analysis.
Data was analyzed with Clampfit. The voltage dependence of activation and inactivation was fitted with the Boltzmann function of the form 1/(1 + exp[−(V − V1/2)/k]), where V1/2 and k are the midpoint of activation or inactivation and the slope factor, respectively. To obtain the time constant of activation (τact), the rising phase of the current was fitted with a single exponential function. The prepulse facilitation index (PFI) was defined as the ratio of the current at the end of the 5.5 ms, +20 mV test pulse with and without the 20 ms, +100 mV prepulse. Data are represented as mean ± SD (number of observations). Significance was determined using two-tailed Student's t test.
Results
Reconstitution of Gβγ inhibition in inside-out membrane patch
To date, all studies on Gβγ inhibition of Ca2+ channels have been performed in whole-cell (including oocyte) or cell-attached patch recordings, and free Gβγ has been produced by GPCR activation, intracellular dialysis of GTPγS or purified Gβγ, or constitutive overexpression of Gβγ. To minimize the potential effect of and cross talk with other signaling pathways associated with these approaches, we attempted to reconstitute Gβγ inhibition in inside-out membrane patches, in which we could control the “intracellular” milieu and directly apply purified Gβγ to the cytoplasmic side. Macroscopic Ba2+ currents were recorded in giant inside-out membrane patches from Xenopus oocytes injected with the cRNA of Cav2.1 (the α1 subunit encoding the P/Q-type channel), α2-δ, and β1b. P/Q-type rather than N-type channels were chosen because their activity runs down much more slowly in inside-out macropatches (Zhen et al., 2006), and this rundown can be attenuated further by adding Mg-ATP (2–3 mm) and a low concentration (0.3 μm) of PIP2 in the cytoplasmic solution (Wu et al., 2002).
Figure 1 illustrates the basic protocol for reconstituting and detecting voltage-dependent Gβγ inhibition in our system. Macroscopic currents were evoked by a test pulse without or with a 20 ms, +100 mV prepulse (Fig. 1A), and tail currents evoked by the test pulse (Fig. 1B) were used to construct the time-course plot (Fig. 1C) and channel-activation curves (Fig. 1D). Bath application of 20 nm purified Gβγ (Gβ1 and Gγ2 dimer) caused a gradual inhibition of the current, which could be partially reversed by bath application of 5 mm purified pleckstrin homology (PH) domain of β-adrenergic receptor kinase (βARK_PH) (Fig. 1C). βARK_PH binds to Gβγ (Koch et al., 1993) and has been used to strip Gβγ from Gβγ-modulated Ca2+ channels (Meir and Dolphin, 2002). Bath application of 1 mm of a G-protein α subunit (Gα) could also partially reverse the Gβγ inhibition, but Gα itself altered some channel properties (data not shown), complicating the results. Part of the Gβγ inhibition was voltage-dependent and exhibited all the hallmarks of this form of modulation: relief by a strong depolarizing prepulse (Fig. 1A,B), depolarizing shift of the voltage dependence of activation (Fig. 1D), and slowing of the activation kinetics (Fig. 1E). The PFI, defined as the ratio of the current at the end of the 5.5 ms, +20 mV test pulse with and without the prepulse, was 0.88 ± 0.11 (n = 7) under the control condition and 1.35 ± 0.33 (n = 7) after Gβγ application (p = 0.004). Much of the Gβγ inhibition could not be relieved by the prepulse (Fig. 1B,C) and is therefore considered as voltage-independent. This study focuses mainly on the voltage-dependent Gβγ inhibition.
Figure 1.

Gβγ inhibition of P/Q-type Ca2+ channels in inside-out membrane patches. A, Typical macroscopic Ba2+ currents evoked by the indicated voltage protocols in inside-out membrane patches from oocytes expressing Cav2.1, α2-δ, and β1b, before (top row) or after application of 20 nm purified Gβγ (bottom row). Con, Control (i.e., without Gβγ). The prepulse to +100 mV is 20 ms. In this and subsequent figures showing current traces, dashed line indicates the zero current level. B, Currents evoked by the +20 mV test pulse in A are superimposed, showing Gβγ-induced inhibition and prepulse facilitation. PP, Prepulse. C, Time course of channel inhibition by 20 nm Gβγ and subsequent partial recovery produced by 5 mm βARK_PH. Data points represent tail currents recorded at −30 mV after a depolarization to +20 mV (n = 5–7). Filled circles indicate the tail current of a test pulse following the +100 mV prepulse. Arrows and numbers indicate the time points at which activation curves 1–3 displayed in Figure 5A are taken. The current is normalized by that obtained immediately after patch excision (0 min data point). D, Voltage dependence of activation under the indicated conditions. In this and subsequent figures, data points represent normalized tail currents recorded at −30 mV after depolarization to a given test potential. The midpoint (V1/2) and slope factor (k) of activation are as follows: Con: 11.5 ± 2.3 and 8.8 ± 0.6 mV; Con+PP: 9.5 ± 2.5 and 8.2 ± 0.6 mV; Gβγ: 22.3 ± 4.8 and 14.7 ± 2.3 mV; Gβγ+PP: 11.7 ± 2.0 and 9.9 ± 0.6 mV (n = 7 for all). E, Time constant of activation (τact) of currents evoked at +20 mV under the indicated conditions. *p < 0.05 (in this and subsequent figures, only statistically different pairwise comparisons within a given group are indicated).
Generating β-less surface Ca2+ channels with a mutant Cavβ
We next examined whether Cavβ was necessary for Gβγ-mediated inhibition. A major obstacle for this experiment was that in the absence of an exogenous Cavβ, HVA Ca2+ channels express poorly on the plasma membrane in oocytes. As expected, most oocytes injected with the cRNA of Cav2.1 and α2-δ alone showed little or no Ba2+ currents in macropatches (data not shown). Although WT Cavβs strongly stimulate the surface expression of HVA Ca2+ channels, they bind the AID so tightly that their dissociation rate is on the order of tens of minutes to hours (for review, see Dolphin, 2003b). Thus, conventional routes are impractical, if not impossible, to produce great numbers of surface Ca2+ channels that do not contain an associated Cavβ. Based on the recently solved crystal structure of the Cavβ/AID complex (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004), we reasoned that we might be able to create a mutant Cavβ that had the right affinity for the AID, such that it could maintain the ability to translocate channel α1 subunits to the cell surface but dissociate within minutes during perfusion in inside-out patches, leaving the surface channels β-less.
We succeeded in creating such a Cavβ, which was named β2a_Mut2 (Fig. 2). In this mutant, two residues (M245 and L249) that interact directly with the AID (Fig. 2A) were mutated to alanine in a shortened version of β2a called β2a_core, which was generated by deleting 16 N-terminal and 188 C-terminal residues of β2a. β2a_core protein expressed better than β2a protein did and was more stable in solution. This protein was used for later experiments (see below). β2a_core lacks the N-terminal palmitoylation sites, but because of its unique HOOK region, it is still able to greatly reduce the inactivation speed and shift the activation voltage in the hyperpolarized direction (He et al., 2007). This makes it much easier to detect changes in these properties if and when β2a_Mut2 dissociates from the channels. β2a_Mut2 had a reduced affinity for the AID (Fig. 2B) but was still capable of stimulating the surface expression of P/Q-type channels, nearly as strongly as WT β2a_core did, as measured by two-electrode voltage-clamp (Fig. 2C). To determine whether and how fast β2a_Mut2 dissociated from the surface channels, we compared changes in the inactivation kinetics and voltage dependence of activation during perfusion of inside-out patches isolated from oocytes expressing Cav2.1, α2-δ, and either WT β2a_core or β2a_Mut2. Furthermore, purified WT β2a_core protein was applied to the patch after extensive perfusion to see whether it could normalize the channel properties. In patches from oocytes expressing WT β2a_core, the speed of current inactivation was steady during the 5 min perfusion (Fig. 2D,E, left column; G), as was the voltage dependence of activation (Fig. 2F, left column; H). Subsequent application of purified WT β2a_core did not change these properties (Fig. 2D–F, left column; G,H). These results indicate that WT β2a_core remained associated with the channels during the 5 min perfusion. In contrast, in patches from oocytes expressing β2a_Mut2, the speed of current inactivation became faster and faster after patch excision, reaching a steady-state in ∼5 min (Fig. 2D,E, middle column; G). In the mean time, the voltage dependence of activation was shifted in the depolarized direction after the 5 min perfusion (Fig. 2F, middle column), increasing both the midpoint and slope factor of activation (Fig. 2H). Subsequent application of purified WT β2a_core fully restored all the functional parameters to the same level as before the perfusion (Fig. 2D–F, middle column; G,H). It should be noted that before the start of the perfusion (i.e., the 0 min data point) the inactivation speed and activation parameters were the same for channels from oocytes expressing β2a_Mut2 or WT β2a_core (Fig. 2G,H). Together, these results indicate that β2a_Mut2 was associated with the channels immediately after patch excision but dissociated completely in ∼5 min, producing large macroscopic currents mediated by β-less P/Q-type channels.
The above conclusion is further substantiated by the results from oocytes expressing only Cav2.1 and α2-δ. As mentioned above, most of these oocytes showed little or no Ba2+ currents in macropatches, but because of the existence of two isoforms of endogenous Cavβs (Tareilus et al., 1997), a small number of oocytes displayed currents ranging from 0.5–1 nA. It has been postulated that these currents are likely mediated by channels transported to the plasma membrane by the endogenous Cavβs, which then unbind from the surface channels and remain unbound at the steady state because of their low effective concentration (He et al., 2007); these channels are refereed to as β− channels. Supporting this hypothesis, the inactivation kinetics and activation parameters of β− channels did not change during the perfusion (Fig. 2D–F, right column; G,H), and they were similar to those of channels obtained from the β2a_Mut2-expressing oocytes after the 5 min perfusion (Fig. 2G,H). Subsequent application of purified WT β2a_core greatly slowed the inactivation kinetics (Fig. 2D,E, right column; G) and shifted the activation curve in the hyperpolarized direction (Fig. 2F, right column), normalizing all the measured functional parameters (Fig. 2G,H).
Gβγ does not produce voltage-dependent inhibition of β-less channels
Thus armed, we proceeded to examine the effect of Gβγ on the β-less channels. Macroscopic Ba2+ currents were recorded in inside-out macropatches obtained from oocytes expressing Cav2.1, α2-δ, and β2a_Mut2. It should be noted that β2a_Mut2 was used in this set of experiments only to serve the purpose of generating β-less surface channels. To ensure that β2a_Mut2 was removed from the surface channels in each recording, the voltage dependence of activation was continuously monitored after patch excision. Only when the activation curve had been fully shifted was Gβγ applied. Gβγ (20 nm) was still capable of inhibiting the β-less channels (Fig. 3A,B). However, all the hallmarks of voltage dependence were totally lost: there were no prepulse facilitation (Fig. 3A,B), no depolarizing shift of the activation curve (Fig. 3C), and no slowing of the activation kinetics (Fig. 3D). The PFI was the same before and after Gβγ application [0.97 ± 0.11 and 0.99 ± 0.12 (n = 6), respectively]. These results are in agreement with the observations of Meir et al. (2000) in COS cells and unequivocally show that Cavβ is required to confer voltage dependence to Gβγ-induced inhibition.
The total degree of inhibition was notably less for the β-less channels. Thus, after 3 min of application, Gβγ inhibited 70.7 ± 8.0% (n = 7) WT channels and only 30.8 ± 9.8% (n = 6) β-less channels. Because Gβγ appears to bind the same pocket to produce both voltage-dependent and voltage-independent inhibition (see later), this result suggests that either Gβγ has a reduced affinity for the β-less channels or it is less effective in causing voltage-independent inhibition of these channels.
The Cavβ GK domain is sufficient for conferring voltage dependence to Gβγ-induced inhibition
Next, we determined the region(s) of Cavβ that are necessary for voltage-dependent Gβγ inhibition. Previous functional and structural studies show that Cavβ contains five structurally and functionally modular domains: the N terminus, a Src homology 3 (SH3) domain, a HOOK region, a GK domain, and the C terminus. The GK domain binds the AID (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004) and is necessary and sufficient for trafficking HVA Ca2+ channels to the plasma membrane (He et al., 2007). Furthermore, Ca2+ channels containing the GK domain of any given Cavβ exhibit different activation and inactivation properties from those containing its full-length counterpart (He et al., 2007). Therefore, we recorded macroscopic Ba2+ currents in giant inside-out membrane patches obtained from oocytes expressing Cav2.1, α2-δ, and the β1b GK domain and examined the effect of Gβγ on these currents. Gβγ (20 nm) inhibited these currents (Fig. 4A,B). Remarkably, the characteristic features of voltage-dependent inhibition were entirely replicated, including prepulse facilitation (Fig. 4A,B), depolarizing shift of the activation curve (Fig. 4C), and slowing of the activation kinetics (Fig. 4D). These results indicate that the voltage dependence of Gβγ inhibition can be fully bestowed by the GK domain alone, in agreement with those reported recently (Dresviannikov et al., 2008). As with full-length β1b, much of the inhibition was voltage independent (Fig. 4A,B). The total inhibition was 67.3 ± 9.5% (n = 6) 3 min after Gβγ application, similar to the 70.7 ± 8.0% (n = 7) inhibition of WT channels, suggesting that the voltage-independent inhibition was also unaffected.
Figure 4.

The Cavβ GK domain is sufficient to endow P/Q-type channels with voltage-dependent Gβγ inhibition. Macroscopic currents were recorded in inside-out macropatches from oocytes expressing Cav2.1, α2-δ, and the β1b GK domain. Voltage protocols for assessing voltage-dependent Gβγ inhibition were as described in Figure 1. A, Superposition of currents evoked by a +20 mV test pulse in the absence or presence of 20 nm Gβγ, without or with a 20 ms, +100 mV prepulse, showing Gβγ-induced inhibition and prepulse facilitation. The PFI was 0.93 ± 0.06 and 1.42 ± 0.27 (n = 6) without and with Gβγ (p = 0.001), respectively. Con, Control; PP, prepulse. B, Time course of channel inhibition by Gβγ. Data points represent tail currents recorded at −30 mV after a depolarization to +20 mV (n = 6). Filled circles indicate the tail current of a test pulse following the prepulse. Current is normalized by that obtained immediately after patch excision (0 min data point). C, Voltage dependence of activation under the indicated conditions. The midpoint and slope factor are as follows: Con: 16.4 ± 1.3 and 9.7 ± 0.9 mV; Con+PP: 13.7 ± 1.6 and 9.0 ± 0.5 mV; Gβγ: 29.8 ± 4.3 and 14.9 ± 1.1 mV; Gβγ+PP: 16.2 ± 2.3 and 10.7 ± 0.43 mV (n = 6 for all). D, Time constant of activation (τact) of currents evoked at + 20 mV under the indicated conditions. *p < 0.05.
Gβγ does not displace Cavβ from the channel complex
The functional antagonism between Gβγ and Cavβ strongly suggests that there is an intimate intermolecular interplay between them. Does this antagonism imply that Gβγ displaces Cavβ from the inhibited channels? We addressed this question by comparing the activation and inactivation properties of channels with and without Cavβ, and in the absence and presence of Gβγ. Without Gβγ, the activation curve of the channels containing β1b was left-shifted compared with that of the β-less channels (Fig. 5A, compare ● and ◇ β-less), which were obtained as described in Figure 2 by washing away β2a_Mut2. Application of Gβγ did not affect the activation curve of the β-less channels, as shown in Figure 3C, but produced a depolarizing shift of the activation curve of the β1b-containing channels (Fig. 5A, compare ● Con and ▿ Gβγ), bringing it close to that of the β-less channels (Fig. 5A, compare ▿ Gβγ and ◇ β-less). This could be caused by the loss of β1b from the Gβγ-bound channels. Had this been the case, the activation curve should have remained the same as that of the β-less channels after subsequent application of βARK_PH, because the dislodged β1b would have been washed out during perfusion. βARK_PH competes Gβγ off Gβγ-bound channels and by itself did not affect channel activation (data not shown). However, the results showed that the activation curve was shifted back to resemble that of the β1b-containing channels (Fig. 5A, compare ■ βARK_PH and ● Con). These results indicate that β1b remained associated with the Gβγ-bound channels.
Comparison of the inactivation properties also gave the same conclusion. In agreement with previous studies (Bean, 1989; Meir and Dolphin, 2002), Gβγ did not alter the voltage dependence and kinetics of inactivation of β1b-containing channels (Fig. 5B,C). This result suggests that β1b was not displaced by Gβγ. It could be argued, however, that β1b was in fact dislodged from the channels, but Gβγ compensated the effects of β1b. This argument would predict that the inactivation properties of the Gβγ-bound β1b-containing channels and the Gβγ-bound β-less channels should be the same. However, the results show that the Gβγ-bound β-less channels exhibited a right-shifted inactivation curve (Fig. 5B) and inactivated more slowly (Fig. 5C), suggesting that Gβγ did not compensate the effects of β1b on inactivation.
Together, these results support the notion that Cavβ remains associated with the Cavα1 subunit during G-protein inhibition (Cantí et al., 2000; Meir et al., 2000; Feng et al., 2001; Hümmer et al., 2003).
A rigid IS6-AID linker is essential for Cavβ modulation of HVA channel gating
Why is Cavβ required for the voltage-dependent Gβγ inhibition? How does this Cavβ function relate to its modulation of HVA channel gating? To address these questions, we first investigated how Cavβ modulates the gating properties of HVA channels. An attractive hypothesis is that Cavβ modulates the mobility of IS6 (Opatowsky et al., 2004; Van Petegem et al., 2004), which contributes to form the inner pore and the activation gate of VGCCs (Xie et al., 2005; Zhen et al., 2005) and affects their activation and inactivation properties (Zhang et al., 1994; Stotz et al., 2004; Zhen et al., 2005). The AID is connected to IS6 via a 22 aa linker (Fig. 6A, first row). The 18 aa AID rolls into an α-helix in the embrace of a Cavβ (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004), and the entire IS6-AID linker also forms an α-helix, as suggested by circular dichroism (CD) spectrum measurements (Arias et al., 2005). Thus, in the presence of Cavβ, a rigid continuous α-helix extends from the C-terminal end of IS6 to the C-terminal end of the AID. When this entire region of a HVA channel is transplanted into the I-II loop of an originally Cavβ-insensitive low voltage-activated (LVA) channel, the resultant chimeric channel becomes modulated by Cavβ (Arias et al., 2005). Moreover, disrupting the α-helical structure of the IS6-AID linker by replacing a stretch of 6 aa with 6 glycines abolishes Cavβ modulation of the chimeric LVA channel (Arias et al., 2005).
Figure 6.
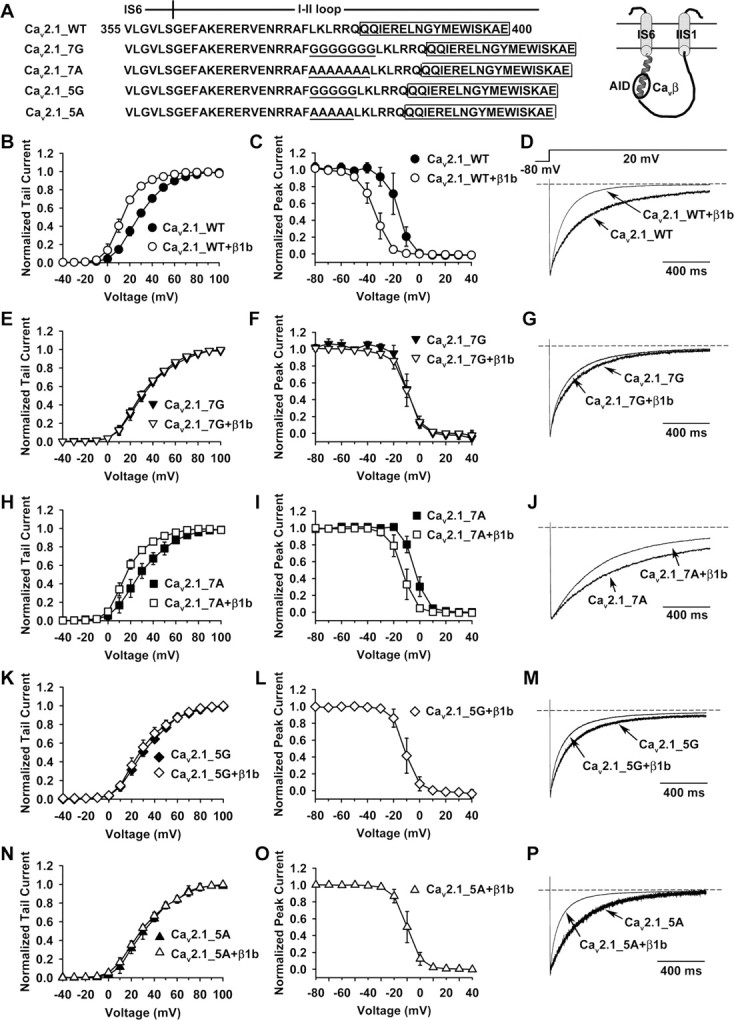
A rigid α-helical structure of the IS6-AID linker is necessary for Cavβ modulation of P/Q-type channel opening. A, The amino acid sequence of Cav2.1 from V355 to E400, which encompasses the cytoplasmic end of IS6 and the adjacent portion of the I-II loop that includes the entire AID (boxed). Seven or five glycines or alanines (underlined) were inserted in between IS6 and the AID, creating four mutant forms of Cav2.1 subunit named Cav2.1_7G, Cav2.1_7A, Cav2.1_5G, and Cav2.1_5A, respectively. Inset on the right shows a schematic of the topology of IS6, IIS1, and the connecting I-II loop. The AID is bound with a Cavβ (circle). B–P, Comparison of activation and inactivation properties of channels formed by WT Cav2.1, Cav2.1_7G, Cav2.1_7A, Cav2.1_5G, or Cav2.1_5A, without or with β1b (α2-δ was present in all cases). In this study, macroscopic currents were recorded in cell-attached patches. B, E, H, K, N, Voltage dependence of activation of the indicated channel types. The midpoint and slope factor are as follows: B, 28.0 ± 2.0 and 12.0 ± 0.7 mV for WT Cav2.1 (n = 6), 13.1 ± 2.2 and 8.3 ± 1.2 mV for WT Cav2.1 with β1b (n = 6); E, 31.2 ± 2.9 and 13.3 ± 1.2 mV for Cav2.1_7G (n = 10), 29.9 ± 1.9 and 13.3 ± 1.4 mV for Cav2.1_7G with β1b (n = 9); H, 28.3 ± 5.1 and 13.4 ± 1.8 mV for Cav2.1_7A (n = 10), 16.1 ± 2.4 and 9.6 ± 0.8 mV for Cav2.1_7A with β1b (n = 7); K, 30.4 ± 2.8 and 13.4 ± 1.3 mV for Cav2.1_5G (n = 6), 28.0 ± 3.8 and 12.6 ± 2.1 mV for Cav2.1_5G with β1b (n = 7); and N, 31.0 ± 3.2 and 13.5 ± 0.9 mV for Cav2.1_5A (n = 7), 28.9 ± 2.2 and 14.8 ± 2.4 mV for Cav2.1_5A with β1b (n = 10). C, F, I, L, O, Voltage dependence of steady-state inactivation of the indicated channel types. We could not obtain these data for Cav2.1_5G or Cav2.1_5A channels in the absence of Cavβ because the currents were miniscule. The midpoint and slope factor are: C, −16.3 ± 4.6 and 4.4 ± 1.1 mV for WT Cav2.1 (n = 4), −34.2 ± 3.7 and 5.5 ± 0.9 mV for WT Cav2.1 with β1b (n = 8); F, −9.6 ± 1.3 and 4.7 ± 1.1 mV for Cav2.1_7G (n = 4), −9.8 ± 3.9 and 4.9 ± 0.6 mV for Cav2.1_7G with β1b (n = 9); I, −3.8 ± 2.2 and 4.1 ± 0.4 mV for Cav2.1_7A (n = 5), −13.3 ± 3.7 and 4.4 ± 0.4 mV for Cav2.1_7A with β1b (n = 7); L, −11.2 ± 3.9 and 4.3 ± 0.6 mV for Cav2.1_5G with β1b (n = 7); and O, −10.0 ± 3.5 and 4.8 ± 0.7 mV for Cav2.1_5A with β1b (n = 5). D, G, J, M, P, Averaged current traces (n = 4–10) evoked by a +20 mV pulse, showing the inactivation kinetics of the indicated channel types.
Following these leads, we examined whether a rigid α-helical structure of the IS6-AID linker was necessary for Cavβ modulation of HVA P/Q-type channels. Four different poly-amino acid linkers were inserted between IS6 and the AID of Cav2.1, including either seven or five glycines, or seven or five alanines (Fig. 6A). Glycine and alanine have well established propensity to disrupt or support α-helices, respectively (Pace and Scholtz, 1998), and seven and five residues extend an α-helix by approximately two or one-and-a-half additional turns, respectively (Branden and Tooze, 1999). These mutant Cav2.1 subunits, as well as WT Cav2.1, were individually expressed in oocytes, together with α2-δ, with or without β1b, and their activation and inactivation properties were compared in cell-attached macropatches. The currents obtained from oocytes without β1b were most likely mediated by channels that were trafficked to the surface membrane by the endogenous β subunits, which became detached from the surface channels at steady-state, as described in Figure 2. β1b greatly altered the voltage dependence of activation (Fig. 6B) and steady-state inactivation (Fig. 6C) and the kinetics of inactivation (Fig. 6D) of WT channels, in the same manner as reported in previous studies (He et al., 2007). However, β1b did not change any of these properties of Cav2.1_7G channels (Fig. 6E–G). In contrast, β1b was still capable of modulating these properties of Cav2.1_7A channels (Fig. 6H–J), in the same direction as for WT channels, albeit to a lesser degree. These results suggest that the total lack of β1b modulation of Cav2.1_7G channels is not a nonspecific general effect of the amino acid insertion itself, but instead, it is because the hepta-glycine linker breaks the α-helical structure of the IS6-AID region. One could still argue, however, that the lack of β1b modulation is because β1b cannot bind the AID of Cav2.1_7G channels. This was not the case because β1b could still bind the hepta-glycine-containing I-II loop in vitro (Fig. 7A) and could strongly stimulate the expression of Cav2.1_7G subunits in oocytes (Fig. 7B). Binding of the mutated I-II loop to β1b was specific and mediated by the AID because it did not bind a mutant β1b, in which five key AID-interacting residues were mutated to alanine (β1b_Mut5) (Fig. 7A). As expected, β1b was able to bind the hepta-alanine-containing I-II loop in vitro (Fig. 7A) and to greatly enhance the surface expression of Cav2.1_7A subunits in oocytes (Fig. 7B).
Figure 7.

Cavβ is able to bind the I-II loop of Cav2.1_7G and Cav2.1_5G channels and enhance macroscopic currents. A, Coomassie Blue staining illustrating the interaction between the I-II loop fragment from the indicated WT or mutant Cav2.1 and either WT β1b_core (1) or β1b_Mut5 (2), in which five key AID-binding residues were mutated to alanine, including M248, L252, I346, V351, and L355. GST_I-II loop was immobilized in a GST column and was used to pull down WT β1b_core or β1b_Mut5 protein. B, Bound; U, unbound. B, Whole-oocyte peak Ba2+ current from oocytes expressing the indicated Cav2.1 subunit, together with α2-δ, without (β−) or with β1b. *p < 0.05.
Likewise, the I-II loop bearing either penta-glycines or penta-alanines could bind WT β1b but not β1b_Mut5 (Fig. 7A), and β1b was able to robustly stimulate the surface expression of Cav2.1_5G and Cav2.1_5A subunits (Fig. 7B). Despite these effects, β1b failed to affect the voltage dependence of activation and the kinetics of inactivation of Cav2.1_5G channels (Fig. 6K,M). This is not surprising because penta-glycines are just as likely as hepta-glycines to introduce random coils into the IS6-AID linker. Intriguingly, however, the voltage dependence of activation of Cav2.1_5A channels was also unperturbed by β1b (Fig. 6N). The penta-alanine insertion most likely preserved the α-helical structure of the IS6-AID linker, as predicted by all the secondary structure prediction algorithms we tested (data not shown). So why was β1b ineffective in modulating activation of Cav2.1_5A channels? A likely reason is that when bound to the penta-alanine-containing I-II loop, β1b was rotated 180° with regard to the AID axis because of the one-and-a-half turn introduced by the penta-alanine insertion, thus misplacing the entire β subunit in reference to the α1 subunit. Curiously, however, β1b was still capable of speeding up inactivation of Cav2.1_5A channels (Fig. 6P), suggesting that despite the 180° rotation, some low-affinity α1-β interactions necessary for inactivation modulation are still maintained.
Together, these results indicate that a rigid IS6-AID linker is crucial for Cavβ modulation of HVA channel gating.
A rigid IS6-AID linker is essential for voltage-dependent Gβγ modulation
Having characterized Cavβ modulation of the four mutant channels, we next examined their regulation by Gβγ, using the same approach as in Figure 1. Cav2.1_7A channels (containing α2-δ and β1b subunits) showed all the characteristic features of voltage-dependent Gβγ inhibition: prepulse facilitation (Fig. 8A,B), depolarizing shift of the activation curve (Fig. 8C), and slowing of the activation kinetics (Fig. 8D). The PFI was 1.02 ± 0.07 and 1.52 ± 0.31 (n = 6) without and with Gβγ (p = 0.009), respectively. In contrast, although Gβγ strongly depressed Cav2.1_7G (Fig. 8E,F) and Cav2.1_5G channels (Fig. 8I,J), all the hallmarks of voltage-dependent inhibition were absent (Fig. 8E–L). Notably, for both channels, the 20 ms, 100 mV prepulse caused a strong suppression of the current evoked by the subsequent +20 mV test pulse (Fig. 8E,F, I,J), attributable mainly to a strong inactivation during the prepulse. In the absence and presence of Gβγ, the ratio of the current at the end and at the beginning of the prepulse was 0.48 ± 0.07 and 0.50 ± 0.10 (n = 7) for Cav2.1_7G and 0.53 ± 0.12 and 0.54 ± 0.16 (n = 6) for Cav2.1_5G. In contract, these ratios were 1.11 ± 0.05 and 1.08 ± 0.04 (n = 6) for Cav2.1_7A channels. Why the glycine insertions result in stronger inactivation awaits further elucidation. Although this inactivation could potentially overshadow the prepulse facilitation, it did not because for Cav2.1_7G the PFI was 0.57 ± 0.10 (n = 7) under the control condition and 0.59 ± 0.13 (n = 7) after Gβγ application, and for Cav2.1_5G, the PFI was 0.62 ± 0.14 (n = 6) and 0.66 ± 0.15 (n = 6), respectively. Had there been a significant prepulse facilitation, this value would have been significantly higher in the presence of Gβγ. Thus, we infer that Gβγ did not produce a significant prepulse facilitation of Cav2.1_7G and Cav2.1_5G channels. Surprisingly, Gβγ failed to produce any inhibition of Cav2.1_5A channels, either voltage-dependent or voltage-independent (Fig. 8M–P). These results indicate that a properly oriented and rigid α-helix of the IS6-AID linker is critical for endowing voltage dependence to Gβγ inhibition.
Figure 8.

A rigid α-helical structure of the IS6-AID linker is necessary for voltage-dependent Gβγ inhibition. A–P, Macroscopic currents were recorded in inside-out macropatches from oocytes expressing α2-δ, β1b, and either Cav2.1_7A (A–D), Cav2.1_7G (E–H), Cav2.1_5G (I–L), or Cav2.1_5A (M–P). Voltage protocols for assessing voltage-dependent Gβγ inhibition were as described in Figure 1. A, E, I, M, Superposition of currents evoked by a +20 mV test pulse in the absence or presence of 20 nm Gβγ, without or with a 20 ms, +100 mV prepulse. Con, Control; PP, prepulse. B, F, J, N, Time course of Gβγ inhibition. Data points represent tail currents recorded at −30 mV after a depolarization to +20 mV (n = 6–7). Filled circles indicate the tail current of a test pulse following the prepulse. Current is normalized by that obtained immediately after patch excision (0 min data point). C, G, K, O, Voltage dependence of activation under the indicated conditions. The midpoint and slope factor are as follows: C, 18.3 ± 1.0 and 10.1 ± 0.9 mV for Con, 15.9 ± 0.9 and 9.7 ± 0.8 mV for Con+PP, 27.4 ± 4.4 and 14.5 ± 1.8 mV for Gβγ, and 16.2 ± 2.2 and 11.3 ± 1.3 mV for Gβγ+PP (n = 6 for all); G, 37.0 ± 4.9 and 22.6 ± 3.0 mV for Con, 33.9 ± 5.4 and 21.8 ± 3.6 mV for Con+PP, 39.1 ± 5.8 and 21.7 ± 3.3 mV for Gβγ, and 34.9 ± 7.9 and 22.4 ± 4.7 mV for Gβγ+PP (n = 7 for all); K, 35.2 ± 3.4 and 22.1 ± 4.2 mV for Con, 31.4 ± 6.5 and 20.5 ± 2.1 mV for Con+PP, 34.8 ± 4.4 and 20.6 ± 2.3 mV for Gβγ, and 31.2 ± 5.2 and 20.6 ± 2.6 mV for Gβγ+PP (n = 6 for all); and O, 31.2 ± 2.3 and 17.2 ± 1.5 mV for Con, 29.4 ± 3.9 and 17.9 ± 1.5 mV for Con+PP, 32.9 ± 4.1 and 16.2 ± 2.2 mV for Gβγ, and 29.9 ± 3.6 and 18.4 ± 3.5 mV for Gβγ+PP (n = 6 for all). D, H, L, P, Time constant of activation (τact) of currents evoked at +20 mV under the indicated conditions. *p < 0.05.
Discussion
Advantages and limitations of reconstituting Gβγ inhibition in inside-out membrane patches
In this study, we reconstituted Gβγ-mediated inhibition of P/Q-type Ca2+ channel in inside-out macropatches. An advantage of this system is that we could directly apply purified Gβγ proteins to the intracellular side of the channel and compare various channel properties before, during, and, in cases with a high-quality giga-seal, after Gβγ modulation in the same membrane patch. This approach reduces the effect of other proteins that modulate voltage-dependent Gβγ inhibition, such as Gα (Jeong and Ikeda, 1999), protein kinase C (Zamponi et al., 1997), syntaxin (Stanley and Mirotznik, 1997), and regulators of G-protein signaling (RGS) (Diversé-Pierluissi et al., 1999; Melliti et al., 1999; Mark et al., 2000; Han et al., 2006). Furthermore, only through this approach could we wash out β2a_Mut2 and thus produce large numbers of β-less Ca2+ channels on the surface membrane.
The latter improvement is particularly useful in addressing the controversial issue of whether Cavβ is necessary for voltage-dependent Gβγ inhibition. This requires comparison of Gβγ regulation of channels with and without Cavβ. Past studies addressing this question bear two major problems. First, the currents from channels without Cavβ tend to be miniscule and are therefore difficult to analyze precisely. Second, because of the presence of endogenous Cavβ, channels may not be homogenously β-less. These problems may account, at least partially, for the discrepancies among previous studies (Roche et al., 1995; Campbell et al., 1995; Bourinet et al., 1996; Qin et al., 1997; Meir et al., 2000). Our results unambiguously show that Cavβ is required to confer voltage dependence to Gβγ-induced inhibition
Our system, however, has clear limitations, two of which are particularly obvious. First, only a small fraction of the Gβγ inhibition can be relieved by the 20 ms, +100 mV prepulse (Fig. 1B,C). It should be noted, however, that the voltage-dependent component was likely underestimated because of a number of factors. The prepulse might not be long or strong enough to produce maximum relief (longer or stronger prepulses tended to damage the giga-seal and produce more pronounced inactivation). There could be some reinhibition during the 2 ms gap between the prepulse and the test pulse (Zamponi and Snutch, 1998). The prepulse itself caused a depression of the current in the absence of Gβγ (Fig. 1A–C), presumably because of channel inactivation during the prepulse (which could explain the less-than-unity value of the PFI in the control condition). Finally, there might be a lack of proteins or factors that may facilitate the dissociation of Gβγ during the prepulse. Despite of these caveats, our results show that the main features of voltage-dependent Gβγ inhibition can be reproduced in our system.
The second major deficiency of our system is that the speed of onset and offset of Gβγ inhibition is very slow. Instead of the tens-of-milliseconds to seconds time scale seen in native cells (for review, see Hille, 1994; Dolphin, 2003a; Tedford and Zamponi, 2006), the inhibition takes minutes to develop (Fig. 1C). Part of this slowness could be caused by the time needed for the applied Gβγ to associate with the membrane before it becomes effective. The recovery is even slower and is incomplete, even in the presence of an excess amount of a Gβγ scavenger βARK_PH (Fig. 1C). Local signaling and various proteins (such as Gα and RGS) probably greatly speed up the kinetics of Gβγ inhibition in native cells. These are lacking in our system. The slowness of our system, albeit undesirable, probably does not compromise the main conclusions of this study because it centers on steady-state properties rather than the kinetics of Gβγ inhibition.
Model for voltage-dependent Gβγ inhibition
The voltage dependence of Gβγ inhibition results from the dissociation of Gβγ from the inhibited channels as they transit from the closed state to the open state (Boland and Bean, 1993). That Cavβ and a rigid IS6-AID linker are essential for voltage-dependent Gβγ inhibition suggests that they contribute to the structural elements and conformational changes that trigger this dissociation. Indeed, expression of exogenous Cavβ increases the rate of prepulse facilitation, which reflects the rate of Gβγ dissociation, during a strong depolarizing prepulse (Roche and Treistman, 1998; Cantí et al., 2000, 2001). Figure 9 depicts an allosteric model linking the movement of IS6 and the obligatory role of Cavβ to the unbinding of Gβγ. This model incorporates several known features concerning the interaction between Gβγ or Cavβ and the α1 subunit of HVA Ca2+ channels. First, several distinct regions in the channel α1 subunit play a role in voltage-dependent Gβγ inhibition, including the N terminus (Page et al., 1998; Agler et al., 2005), the I-II loop (De Waard et al., 1997; Page et al., 1997; Zamponi et al., 1997), and the C terminus (Zhang et al., 1996; Qin et al., 1997), although the role of the latter may be modulatory rather than obligatory (Hümmer et al., 2003; Li et al., 2004; Agler et al., 2005). Protein fragments from these regions are able to bind Gβγ in vitro (De Waard et al., 1997; Qin et al., 1997; Zamponi et al., 1997; Li et al., 2004; Agler et al., 2005). Although the site where Gβγ binds in the holo-channel to produce the voltage-dependent inhibition is still unknown, it may be formed collectively by these (and possibly some yet unknown) regions (Fig. 9). Second, the N terminus binds directly to the I-II loop, and together they form a Gβγ-gated inhibitory module (Agler et al., 2005). Third, the AID forms a random coil absent Cavβ, as determined by CD spectrum (Opatowsky et al., 2004), but adopts an α-helical structure when bound to Cavβ (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004). Thus, in the holo-channel, with Cavβ present, an uninterrupted α-helix is formed from IS6 to the AID (Fig. 9A).
Figure 9.

Model for the voltage dependence of Gβγ inhibition. The Gβγ-binding pocket in the holo-channel is postulated to be formed by a region of the I-II loop distal to the AID (which is buried by Cavβ) and the N and C termini of the channel α1 subunit. A, WT channel. Depolarization moves IS6, consequently altering the conformation of the Gβγ-binding pocket through a rigid α-helix and resulting in Gβγ dissociation. B, β-Less channel. The AID relaxes into a random coil in the absence of Cavβ, uncoupling IS6 from the Gβγ-binding pocket. No voltage-dependent dissociation of Gβγ. C, Channel containing Cavβ and a flexible IS6-AID linker. A random coil in the IS6-AID linker, such as that produced by a heptaglycine or pentaglycine insertion, uncouples IS6 from the Gβγ-binding pocket, abolishing voltage-dependent dissociation of Gβγ.
A distinct element of this model is that the Gβγ-binding pocket in the holo-channel is located primarily on the C-terminal end of the AID (Fig. 9A, left). Gβγ binds in vitro to two distinct regions in the I-II loop of HVA channels: one extends from the C-terminal end of IS6 to the N-terminal end of the AID and contains a signature Gβγ-interacting QXXER motif (QQIER in Cav2.1, Fig. 6A), and the other is located further downstream of the AID (De Waard et al., 1997; Zamponi et al., 1997). The first region may serve only as a secondary Gβγ-binding site in the holo-channel for three reasons. First, the QQIER motif is partially buried by Cavβ, as shown by the Cavβ-AID crystal structures (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004). Indeed, we found that Gβγ binding to a fragment of the I-II loop of Cav2.1 containing the QQIER motif was significantly weaker in the presence of Cavβ than in the absence of Cavβ (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Second, the QQIER motif is unlikely to become available because Cavβ does not vacate from the Gβγ-bound channels (Cantí et al., 2000; Meir et al., 2000; Hümmer et al., 2003) (Fig. 5). Third, Gβγ inhibition remains intact with the hepta-alanine insertion in this region (Fig. 8A–D). The second region, however, could potentially play a key role in Gβγ binding to the holo-channel.
How does this model explain our experimental observations? Under the resting condition and with Gβγ present, the channel is closed and in a “reluctant” state (Fig. 9A, left). The four S6 segments of the α1 subunit are tilted such that their cytoplasmic ends come near each other to form an activation gate (Xie et al., 2005), akin to that occurring in K+ channels (MacKinnon, 2003). On depolarization, the S6 segments spray outward, dilating the cytoplasmic orifice to >10 Å (Zhen et al., 2005). Because of the continuous rigid α-helical structure, partly induced by Cavβ, the movement of IS6 is propagated to the AID and beyond, resulting in a movement of Cavβ and the distal I-II loop and consequently a conformational change of the Gβγ-binding pocket. Such a chain of events ultimately leads to the dissociation of Gβγ from the channel and then the disassembly of the N terminus-I-II loop inhibitory module (Fig. 9A, right). Alternatively, the movement of the I-II loop, triggered by that of IS6, first causes the N terminus-I-II loop module to disassemble, which then leads to a conformational change of the Gβγ-binding pocket and the eventual dissociation of Gβγ.
In the absence of Cavβ (as in the case of the β-less channels), Gβγ can still bind to the holo-channel, as evidenced by its inhibitory effect (Fig. 3A,B), but it cannot be discharged by the depolarizing potential (Fig. 9B). This occurs because of one or both of the following reasons. First, without Cavβ, the AID unwinds into a random coil, disrupting the coupling between IS6 and the Gβγ-binding pocket. Second, the Cavβ movement itself, triggered by the voltage-dependent IS6 movement, is a prerequisite for weakening Gβγ binding. Thus, in the β-less channels, although depolarization still reorients IS6, it does not affect Gβγ binding and the N terminus–I-II loop inhibitory module (Fig. 9B, right). Similarly, disrupting the α-helical structure of the linker between IS6 and the AID with the heptaglycine or pentaglycine insertion uncouples the two regions, making Gβγ binding to the holo-channel insensitive to membrane depolarization (Fig. 9C).
The observation that the Cavβ GK domain alone was able to support voltage-dependent Gβγ inhibition adds another fine detail to the model: the other regions of Cavβ and their interactions with the channel α1 subunit are not necessary for this process. The GK domain harbors the entire AID-binding pocket (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004), thus, it can perfectly substitute full-length Cavβ in enticing the AID to form an α-helix, thereby maintaining the rigid coupling between IS6 and the Gβγ-binding pocket.
This model is applicable to the slowing of the activation kinetics and prepulse facilitation accompanying voltage-dependent Gβγ inhibition of both N- and P/Q-type channels, but not to the “reluctant openings” (i.e., the opening of Gβγ-bound channels) that have been detected in N-type but not P/Q-type channels (Colecraft et al., 2000; Lee and Elmslie, 2000). Those openings are much briefer than the normal ones, and the underlying molecular mechanism remains to be determined. This model also does not explicitly address the mechanism of voltage-independent Gβγ inhibition. However, one can envisage that Gβγ binding and the N terminus–I-II loop inhibitory module remain unaltered in some channels whose IS6 has undergone a depolarization-driven movement, especially in the absence of other proteins or factors that enhance the rate of Gβγ unbinding. The complete lack of both voltage-dependent and voltage-independent Gβγ inhibition of Cav2.1_5A channels (Fig. 8M–P) supports the notion that Gβγ binds to a single pocket to produce both forms of inhibition. The loss of these inhibitions in Cav2.1_5A channels is presumably because Gβγ can no longer bind the mutant channels, since their Gβγ binding pocket is deformed by the one-and-a-half turn introduced by the penta-alanine insertion. Further studies are needed to substantiate this possibility.
In conclusion, our study suggests that the voltage dependence of Gβγ inhibition of HVA Ca2+ channels arises from the voltage-dependent movement of IS6, and that Cavβ and a rigid IS6-AID linker play a pivotal role in translating this movement to Gβγ dissociation.
Footnotes
This work was supported by National Institutes of Health Grants NS045819, NS053494 (to J.Y.), and GM61454 (to T.K.) and the Established Investigator Award (to J.Y.) from the American Heart Association. We thank Claudia Bauer and Li Wu for their participation at the early stage of this work, Yasuo Mori for Cav2.1 cDNA, Tsutomu Tanabe for α2-δ cDNA, and Edward Perez-Reyes for Cavβ cDNAs. We also thank Zafir Buraei and Ioannis Michailidis for comments on this manuscript.
References
- Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (Cav2.2) Ca2+ channels. Neuron. 2005;46:891–904. doi: 10.1016/j.neuron.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Arias JM, Murbartián J, Vitko I, Lee JH, Perez-Reyes E. Transfer of β subunit regulation from high to low voltage-gated Ca2+ channels. FEBS Lett. 2005;579:3907–3912. doi: 10.1016/j.febslet.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Bertram R, Swanson J, Yousef M, Feng ZP, Zamponi GW. A minimal model for G protein-mediated synaptic facilitation and depression. J Neurophysiol. 2003;90:1643–1653. doi: 10.1152/jn.00190.2003. [DOI] [PubMed] [Google Scholar]
- Boland LM, Bean BP. Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage dependence. J Neurosci. 1993;13:516–533. doi: 10.1523/JNEUROSCI.13-02-00516.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci U S A. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branden C, Tooze J. Introduction to protein structure. New York: Garland; 1999. [Google Scholar]
- Brody DL, Patil PG, Mulle JG, Snutch TP, Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J Physiol. 1997;499:637–644. doi: 10.1113/jphysiol.1997.sp021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell V, Berrow NS, Fitzgerald EM, Brickley K, Dolphin AC. Inhibition of the interaction of G protein Go with calcium channels by the calcium channel β-subunit in rat neurones. J Physiol. 1995;485:365–372. doi: 10.1113/jphysiol.1995.sp020735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C, Bogdanov Y, Dolphin AC. Interaction between G proteins and accessory subunits in the regulation of α1B calcium channels in Xenopus oocytes. J Physiol. 2000;527:419–432. doi: 10.1111/j.1469-7793.2000.t01-1-00419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C, Davies A, Berrow NS, Butcher AJ, Page KM, Dolphin AC. Evidence for two concentration-dependent processes for β-subunit effects on α1B calcium channels. Biophys J. 2001;81:1439–1451. doi: 10.1016/S0006-3495(01)75799-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- Colecraft HM, Patil PG, Yue DT. Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J Gen Physiol. 2000;115:175–192. doi: 10.1085/jgp.115.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Diversé-Pierluissi MA, Fischer T, Jordan JD, Schiff M, Ortiz DF, Farquhar MG, De Vries L. Regulators of G protein signaling proteins as determinants of the rate of desensitization of presynaptic calcium channels. J Biol Chem. 1999;274:14490–14494. doi: 10.1074/jbc.274.20.14490. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003a;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. β subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003b;35:599–620. doi: 10.1023/b:jobb.0000008026.37790.5a. [DOI] [PubMed] [Google Scholar]
- Dresviannikov AV, Page KM, Leroy J, Pratt WS, Dolphin AC. Determinants of the voltage dependence of G protein modulation within calcium channel β subunits. Pflugers Arch. 2008 doi: 10.1007/s00424-008-0549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Fischbach GD. Neurotransmitters decrease the calcium ocmponent of sensory neurone action potentials. Nature. 1978;276:837–839. doi: 10.1038/276837a0. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron. 1990;5:75–80. doi: 10.1016/0896-6273(90)90035-e. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Arnot MI, Doering CJ, Zamponi GW. Calcium channel β subunits differentially regulate the inhibition of N-type channels by individual Gβ isoforms. J Biol Chem. 2001;276:45051–45058. doi: 10.1074/jbc.M107784200. [DOI] [PubMed] [Google Scholar]
- Han J, Mark MD, Li X, Xie M, Waka S, Rettig J, Herlitze S. RGS2 determines short-term synaptic plasticity in hippocampal neurons by regulating Gi/o-mediated inhibition of presynaptic Ca2+ channels. Neuron. 2006;51:575–586. doi: 10.1016/j.neuron.2006.07.012. [DOI] [PubMed] [Google Scholar]
- He LL, Zhang Y, Chen YH, Yamada Y, Yang J. Functional modularity of the β-subunit of voltage-gated Ca2+ channels. Biophys J. 2007;93:834–845. doi: 10.1529/biophysj.106.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hümmer A, Delzeith O, Gomez SR, Moreno RL, Mark MD, Herlitze S. Competitive and synergistic interactions of G protein β2 and Ca2+ channel β1b subunits with Cav2.1 channels, revealed by mammalian two-hybrid and fluorescence resonance energy transfer measurements. J Biol Chem. 2003;278:49386–49400. doi: 10.1074/jbc.M306645200. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Ikeda SR. Sequestration of G-protein βγ subunits by different G-protein α subunits blocks voltage-dependent modulation of Ca2+ channels in rat sympathetic neurons. J Neurosci. 1999;19:4755–4761. doi: 10.1523/JNEUROSCI.19-12-04755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Inglese J, Stone WC, Lefkowitz RJ. The binding site for the βγ subunits of heterotrimeric G proteins on the β-adrenergic receptor kinase. J Biol Chem. 1993;268:8256–8260. [PubMed] [Google Scholar]
- Kozasa T. Purification of G protein subunits from Sf9 insect cells using hexahistidine-tagged α and βγ subunits. Methods Mol Biol. 2004;237:21–38. doi: 10.1385/1-59259-430-1:21. [DOI] [PubMed] [Google Scholar]
- Lee HK, Elmslie KS. Reluctant gating of single N-type calcium channels during neurotransmitter-induced inhibition in bullfrog sympathetic neurons. J Neurosci. 2000;20:3115–3128. doi: 10.1523/JNEUROSCI.20-09-03115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhong H, Scheuer T, Catterall WA. Functional role of a C-terminal Gβγ-binding domain of Cav2.2 channels. Mol Pharmacol. 2004;66:761–769. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Potassium channels. FEBS Lett. 2003;555:62–65. doi: 10.1016/s0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- Mark MD, Wittemann S, Herlitze S. G protein modulation of recombinant P/Q-type calcium channels by regulators of G protein signalling proteins. J Physiol. 2000;528:65–77. doi: 10.1111/j.1469-7793.2000.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir A, Dolphin AC. Kinetics and Gβγ modulation of Cav2.2 channels with different auxiliary β subunits. Pflugers Arch. 2002;444:263–275. doi: 10.1007/s00424-002-0803-3. [DOI] [PubMed] [Google Scholar]
- Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel β subunit promotes voltage-dependent modulation of α1B by Gβγ. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melliti K, Meza U, Fisher R, Adams B. Regulators of G protein signaling attenuate the G protein-mediated inhibition of N-type Ca channels. J Gen Physiol. 1999;113:97–110. doi: 10.1085/jgp.113.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- Pace CN, Scholtz JM. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J. 1998;75:422–427. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type calcium channel confers aspects of G-protein sensitivity to the E-type calcium channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM, Cantí C, Stephens GJ, Berrow NS, Dolphin AC. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α1B and α1E as an essential determinant of G-protein modulation. J Neurosci. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proc Natl Acad Sci U S A. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JP, Treistman SN. Ca2+ channel β3 subunit enhances voltage-dependent relief of G-protein inhibition induced by muscarinic receptor activation and Gβγ. J Neurosci. 1998;18:4883–4890. doi: 10.1523/JNEUROSCI.18-13-04883.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche JP, Anantharam V, Treistman SN. Abolition of G protein inhibition of α1A and α1B calcium channels by co-expression of the β3 subunit. FEBS Lett. 1995;371:43–46. doi: 10.1016/0014-5793(95)00860-c. [DOI] [PubMed] [Google Scholar]
- Stanley EF, Mirotznik RR. Cleavage of syntaxin prevents G-protein regulation of presynaptic calcium channels. Nature. 1997;385:340–343. doi: 10.1038/385340a0. [DOI] [PubMed] [Google Scholar]
- Stotz SC, Jarvis SE, Zamponi GW. Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J Physiol. 2004;554:263–273. doi: 10.1113/jphysiol.2003.047068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E, Birnbaumer L. A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proc Natl Acad Sci U S A. 1997;94:1703–1708. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S, Serafin M, Mühlethaler M, Bernheim L. Facilitation of N-type calcium current is dependent on the frequency of action potential-like depolarizations in dissociated cholinergic basal forebrain neurons of the guinea pig. J Neurosci. 1997;17:1625–1632. doi: 10.1523/JNEUROSCI.17-05-01625.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. 2002;419:947–952. doi: 10.1038/nature01118. [DOI] [PubMed] [Google Scholar]
- Xie C, Zhen XG, Yang J. Localization of the activation gate of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:205–212. doi: 10.1085/jgp.200509293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gβγ subunit. Proc Natl Acad Sci U S A. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]
- Zhen XG, Xie C, Fitzmaurice A, Schoonover CE, Orenstein ET, Yang J. Functional architecture of the inner pore of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:193–204. doi: 10.1085/jgp.200509292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen XG, Xie C, Yamada Y, Zhang Y, Doyle C, Yang J. A single amino acid mutation attenuates rundown of voltage-gated calcium channels. FEBS Lett. 2006;580:5733–5738. doi: 10.1016/j.febslet.2006.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]