

Abstract
The Escherichia coli transcription factor OxyR is activated by the formation of an intramolecular disulfide bond and subsequently is deactivated by enzymatic reduction of the disulfide bond. Here we show that OxyR can be activated by two possible pathways. In mutants defective in the cellular disulfide-reducing systems, OxyR is constitutively activated by a change in the thiol—disulfide redox status in the absence of added oxidants. In wild-type cells, OxyR is activated by hydrogen peroxide. By monitoring the presence of the OxyR disulfide bond after exposure to hydrogen peroxide in vivo and in vitro, we also show that the kinetics of OxyR oxidation by low concentrations of hydrogen peroxide is significantly faster than the kinetics of OxyR reduction, allowing for transient activation in an overall reducing environment. We propose that the activity of OxyR in vivo is determined by the balance between hydrogen peroxide levels and the cellular redox environment.
The Escherichia coli OxyR transcription factor activates the expression of antioxidant defensive activities such as hydroperoxidase I (katG), an alkylhydroperoxide reductase (ahpCF), a regulatory RNA (oxyS), glutathione reductase (gorA), and glutaredoxin 1 (grxA) in response to elevated levels of hydrogen peroxide. The OxyR protein is directly sensitive to oxidation, and only oxidized OxyR is capable of activating transcription. We have shown recently that oxidation of OxyR leads to the formation of an intramolecular disulfide bond between cysteine residues 199 and 208 and that OxyR is reduced (deactivated) by enzymatic reduction of this disulfide bond (1). Both glutaredoxin 1 and thioredoxin are able to reduce OxyR in vitro. However, glutaredoxin 1 seems to be the preferred reductant of OxyR in vivo because grxA−, gorA−, and gshA− (glutathione synthetase) mutants, but not trxA− (thioredoxin) and trxB− (thioredoxin reductase) mutants, show a prolonged OxyR response after hydrogen peroxide treatment. Because gorA and grxA are transcriptionally regulated by OxyR, the whole response is autoregulated.
Based on an in vitro titration assay, we determined the redox potential of OxyR to be −185 mV (1). Measurements of the in vivo ratios of reduced and oxidized glutathione and thioredoxin indicate that the thiol–disulfide redox potential of the cytoplasm is between −260 and −280 mV (2–4). This low redox potential suggests that OxyR should be reduced (deactivated) during normal growth, consistent with previous observations. However, the 100-mV difference in redox potential raises the question of how OxyR is activated by low concentrations of hydrogen peroxide (1). Here we address this question by examining the thermodynamics and kinetics of OxyR oxidation and reduction in vivo and in vitro.
MATERIALS AND METHODS
Strains and Plasmids.
The strains used in the study are listed in Table 1. Mutant alleles were moved into DHB4 by P1 transduction. The plasmid pGSO77 carries oxyR with cysteine residues C25, C143, C180, and C259 mutated to alanine cloned into pUC13 under its own promoter. The cells were grown in Luria broth (LB) with the appropriate antibiotics.
Table 1.
Bacterial strains
| Strain | Relevant genotype | Source or ref. |
|---|---|---|
| DHB4 | Lab collection | |
| WP570 | DHB4 ΔtrxA | W. Prinz* |
| WP840 | DHB4 gor522 miniTn10tet | 10 |
| FÅ378 | DHB4 ΔtrxA gor522miniTn10tet | This study |
| WP612 | DHB4 ΔtrxA gshA20∷Tn10kan | 10 |
| WP813 | DHB4 ΔtrxA grxA∷kan | W. Prinz |
| FÅ400 | DHB4 trxB∷kan | This study |
| FÅ369 | DHB4 katG∷Tn10tet ΔahpCF∷kan | This study |
| FÅ250 | DHB4 ΔoxyR∷kan | This study |
| WP812 | DHB4 grxA∷kan | W. Prinz |
| FÅ280 | DHB4 grxA∷kan gor522miniTn10tet | This study |
| FÅ371 | DHB4 + pGSO77 | This study |
*Harvard Medical School.
RNA Isolation and Primer Extension Analysis.
The cell pellet from 10 ml of a cell culture at OD600 = 0.4 or higher was resuspended in 1 ml of Trizol (GIBCO/BRL), and the RNA was purified according to the instructions (based on ref. 5). The RNA yield was typically 100–200 μg. The primer extension analysis to determine OxyS RNA levels was carried out as described (6).
Alkaline Phosphatase Assays.
Overnight cultures of strains carrying pAID135 (7) were subcultured in LB supplemented with 1 mM isopropyl β-d-thiogalactoside and grown to OD600 = 0.4–0.6. An aliquot (1 ml) of cells was added to 0.1 ml of 1 M iodoacetamide and incubated on ice for 20 min. Alkaline phosphatase activity subsequently was determined as described (7).
Glutathione (GSH) and Glutathione Disulfide (GSSG) Measurements.
The 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB)-glutathione reductase recycling method (8) was used to measure GSH and GSSG levels. The pellet from 25 ml of a cell culture at OD600 = 0.4 was resuspended in 0.2 ml of stock buffer (143 mM sodium phosphate/6.3 mM Na4EDTA, pH 7.4) and 0.1 ml of 10% 5-sulfosalicylic acid. The precipitated proteins were removed by centrifugation. Total GSH levels then were measured directly in 10 μl of supernatant. To measure GSSG levels, 200 μl of supernatant was treated with 4 μl of 2-vinylpyridine (Aldrich). The pH of the solution was adjusted to 6–7 by adding ≈6 μl of triethanolamine, and 100 μl was used for the GSSG measurement.
Hydrogen Peroxide Measurements.
The cells in 1 ml of a culture at OD600 = 0.4–0.6 were collected by centrifugation (30 sec, 10,000 rpm in an Eppendorf centrifuge, room temperature) and resuspended in freshly autoclaved PBS to an OD600 = 1.0. The cells then were diluted further 100-fold into PBS incubated at 37°C. At 3, 6, 9, and 12 min, 1-ml aliquots were added to 1 ml of assay mix (80 μM p-hydroxyphenylacetic acid/5 units/ml horseradish peroxidase in PBS) or to 5 μl of 100 μM catalase. After 30 min of incubation at room temperature, 1 ml of the assay mix was added to the latter samples. The samples without and with catalase were centrifuged (10 min, 10,000 rpm), and then the fluorescence at 415 nm (excitation at 310 nm) was recorded. The hydrogen peroxide concentration was estimated by relating the peak fluorescence value of the sample (after subtracting the fluorescence of the catalase-treated controls) to fluorescence values obtained for defined hydrogen peroxide concentrations.
Determination of OxyR4C→A Redox Status.
Aliquots of the cell cultures at OD600 = 0.4 were taken with prewarmed pipettes and mixed with 1/10 vol of 100% trichloroacetic acid (TCA). Precipitated proteins were collected by centrifugation (10 min, 10,000 rpm). After complete removal of the supernatant, the pellet was dissolved in 0.1% SDS/0.67 M Tris⋅Cl, pH 8/15 mM 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS; Molecular Probes) to an OD600 = 10. Alkylations were performed at 37°C for a minimum of 2 hr. Subsequently, aliquots (10 μl) of the alkylated samples were loaded on SDS/15% PAGE minigels (Bio-Rad). After electrophoresis, the separated proteins were blotted to nitrocellulose filters and probed with polyclonal antibodies to an OxyR-β-galactosidase fusion protein (9). The bound antibodies were detected by using the enhanced chemiluminescence Western blotting reagent kit (Amersham Pharmacia).
For the in vitro assays, OxyR4C→A was resolubilized from inclusion bodies by resuspending the insoluble pellet of OxyR4C→A-overproducing cells in 6 M guanidine hydrochloride. Subsequently, the protein was dialyzed against transcription buffer (40 mM Tris⋅Cl, pH 7.9/0.1 M KCl/10 mM MgCl2/5% glycerol/0.1% NP-40). The procedure was repeated for OxyR4C→A that went out of solution after dialysis. Samples of OxyR4C→A were incubated in transcription buffer (pH 7) containing glutaredoxin 1, GSH, and GSSG at room temperature for a minimum of 24 hr in an anaerobic chamber (Coy Laboratory Products, Ann Arbor, MI). One aliquot (100 μl) was removed and acidified with 1/10 vol of 100% TCA. The remainder of the sample was treated with hydrogen peroxide, and then aliquots were precipitated with TCA at the indicated time points. The TCA-precipitated samples were removed from the anaerobic chamber and collected by centrifugation. The precipitated proteins were washed once with cold 10% TCA and collected by centrifugation. The pellet was redissolved in 20 μl of 45 mM 0.1% SDS/0.67 M Tris, pH 8/45 mM AMS and treated as described above. For samples containing low concentrations of OxyR4C→A (<0.1 μM), 100 μl of a TCA precipitate of FÅ250 (collected at OD600 = 1.0) was added as a protein carrier.
RESULTS
Constitutive OxyR Activation by a Change in the Cellular Redox Status.
The thioredoxin and glutaredoxin systems show overlapping functions in preventing disulfide bond formation in the E. coli cytoplasm. Strains carrying mutations that affect both reducing systems, such as trxA− gorA− and trxA− gshA− double mutants, allow cytoplasmic disulfide bond formation as monitored by the activity of alkaline phosphatase devoid of its signal sequence (10). Because OxyR is activated by disulfide bond formation, we examined the expression of the OxyR-regulated oxyS gene in these mutants. Whereas we detected only low levels of OxyR activity in wild-type and trxA− and gorA− single mutant strains that were not treated with hydrogen peroxide, we found that the untreated trxA− gorA− and trxA− gshA− strains showed partial OxyS induction (Fig. 1). In general, the level of OxyS expression in the strains with mutations in the thioredoxin and glutaredoxin systems correlated with the level of cytoplasmic disulfide bond formation as measured by alkaline phosphatase activity (Fig. 1).
Figure 1.

OxyR activity in thioredoxin and glutaredoxin mutants. (A) Total RNA was isolated from untreated and hydrogen peroxide (200 μM)-treated wild-type (DHB4), trxA− (WP570), trxA− gorA− (FÅ378), trxA− gshA− (WP612), trxA− grxA− (WP813), and katG− ahpCF− (FÅ369) cells, and the levels of the OxyS RNA were assayed by primer extension. (B) The levels of alkaline phosphatase activity in midlogarithmically growing cells were determined as described (7).
These findings suggested that OxyR might be activated by a change in the cellular redox status in the trxA− gorA− and trxA− gshA− mutants. To determine whether the thiol—disulfide status was altered, we measured the levels of reduced (GSH) and oxidized (GSSG) glutathione in the wild-type and trxA− gorA− strains. We also measured the GSH and GSSG levels in a katG− ahpCF− mutant. This strain has increased intracellular levels of peroxides because of the lack of the hydroperoxidase I and alkyl hydroperoxide reductase activities and was shown previously to have constitutive OxyR activation (ref. 11; see also Fig. 1). Although GSH/GSSG ratios for the wild-type strain (223 ± 35) and the katG− ahpCF− mutant (291 ± 30) were similar, the ratio was decreased substantially in the trxA− gorA− mutant (18 ± 7). Thus, the constitutive activation of OxyR in the trxA− gorA− strain could be due directly to the altered redox state of the cell.§ Alternatively, the activation might be the result of increased levels of hydrogen peroxide. Therefore, we measured the levels of hydrogen peroxide in the wild-type, trxA− gorA−, and katG− ahpCF− strains. Whereas the wild-type and trxA− gorA− strains had equally low levels of hydrogen peroxide, the levels were increased at least 2-fold in the katG− ahpCF− strain. Together, the measurements of the OxyS RNA, alkaline phosphatase, GSH, GSSG, and hydrogen peroxide levels are consistent with two different mechanisms of OxyR activation within the cell: by decreases in the thiol–disulfide ratio or by increases in the levels of hydrogen peroxide.
Transient Activation of OxyR by Hydrogen Peroxide in Vivo.
We showed previously that the OxyR-regulated oxyS gene is induced within 1 min after cells are treated with 200 μM hydrogen peroxide (ref. 14; see also Fig. 2A). OxyS RNA levels are elevated for approximately 20 min in wild-type cells (ref. 1, see also Fig. 2A). Because oxyS transcription is a downstream event of OxyR activation, we sought to establish a direct method of monitoring the kinetics of OxyR oxidation. This was achieved by alkylating the C199 and C208 thiol groups, present in reduced but not oxidized OxyR, with AMS. The addition of the high-molecular-mass (2 × 500 Da) AMS moiety to the reduced but not the oxidized protein allowed separation of the two forms by gel electrophoresis. To simplify our analysis we studied the OxyR4C→A derivative, in which all cysteine residues except C199 and C208 are substituted with alanine. Previous studies have shown that the activity of the OxyR4C→A protein is identical to wild type in vivo and in vitro (1). Cells expressing OxyR4C→A first were treated with 10% TCA to protonate all thiols and to precipitate the cellular proteins. The thiols subsequently were alkylated by AMS. The proteins then were separated by gel electrophoresis, and OxyR4C→A was detected by immunoblot analysis. As expected, we observed differently migrating forms of OxyR4C→A before and after treatment with hydrogen peroxide (Fig. 2B). Because purified reduced and oxidized OxyR4C→A treated with AMS comigrated with the top and bottom bands, respectively (data not shown), the top band corresponds to OxyR4C→A with C199 and C208 as AMS-alkylated thiols and the bottom band corresponds to the C199-C208 disulfide form of OxyR4C→A. To verify that the TCA precipitation and AMS alkylation did not alter the oxidation state of OxyR4C→A, we mixed purified oxidized and reduced OxyR4C→A with cells from an oxyR− strain before treatment. We did not observe any change in the oxidized and reduced proteins (data not shown), confirming that the TCA and AMS treatments do not perturb the redox status of OxyR4C→A.
Figure 2.

OxyS RNA expression, redox status of OxyR, and GSH/GSSG ratio after treatment with hydrogen peroxide. (A) Midlogarithmically growing wild-type cells (FÅ371) were treated with 200 μM hydrogen peroxide, and then aliquots were taken at 0.5, 2, 4, 6, 10, 20, and 30 min. Total RNA was isolated and the levels of OxyS expression were analyzed by primer extension. (B) Aliquots of the above cells also were precipitated with TCA, treated with AMS, and subjected to SDS/PAGE and immunoblot analysis. (C) The GSH and GSSG levels in aliquots of the indicated cultures were determined by the DTNB-glutathione reductase recycling assay (8).
Using the AMS alkylation method, we examined the in vivo kinetics of OxyR4C→A oxidation and reduction after treatment with 200 μM hydrogen peroxide. For wild-type cells in midlogarithmic growth (OD600 = 0.4), OxyR4C→A was reduced completely in the absence of hydrogen peroxide and was oxidized fully within 30 sec after hydrogen peroxide was added (Fig. 2B). Five minutes after the treatment, half of the OxyR4C→A protein was reduced. The thioredoxin protein was oxidized and reduced with nearly the same kinetics as OxyR (data not shown). In contrast, we did not observed a significant change in the GSH/GSSG ratio or the levels of either compound (Fig. 2C). Interestingly, the deactivation profiles varied depending on the density of the cultures. The half-time of OxyR deactivation in cells at OD600 = 0.1 was 17 min, whereas the half-time of deactivation in cells at OD600 = 1.6 was 2 min (data not shown). This result likely reflects the greater capacity of a dense culture to metabolize hydrogen peroxide.
Transient Activation of OxyR by Hydrogen Peroxide in Vitro.
Because we did not observe a drop in the GSH/GSSG ratio after wild-type cells were treated with hydrogen peroxide, we propose that OxyR reacts directly with hydrogen peroxide and that oxidized OxyR is metastable with respect to the glutathione buffer of the cytosol. To test this hypothesis in vitro, we incubated 1 μM purified OxyR4C→A with 10 μM purified glutaredoxin 1, 25 mM GSH, and 0.1 mM GSSG (equivalent to a thiol–disulfide redox potential of −263 mV) under anaerobic conditions for a minimum of 24 hr. OxyR4C→A was reduced completely under these conditions. After the addition of 2 μM hydrogen peroxide, we were able to achieve complete oxidation of OxyR4C→A within 30 sec (Fig. 3). This demonstrates that in an environment containing 25 mM GSH, the C199 and C208 thiols of OxyR4C→A, which are present at micromolar concentrations, are still preferentially oxidized by hydrogen peroxide. To approximate the rate of the reaction between hydrogen peroxide and reduced OxyR, we performed a titration experiment in which 0.01 μM reduced OxyR4C→A was incubated with various concentrations of hydrogen peroxide (see below). In these experiments, 0.1 μM hydrogen peroxide led to 50% OxyR4C→A oxidation within 30 sec, allowing us to calculate an apparent second-order rate constant of ≈107 M−1⋅min−1. This reaction rate is approaching the rate of ≈108 M−1⋅min−1 that was determined for the reaction of hydrogen peroxide with Enterococcus faecalis NADH peroxidase (15, 16). We also followed OxyR4C→A reduction over time and found that 50% of the protein was reduced between 10 and 30 min (Fig. 3). Although the kinetics were slower than observed in vivo, that OxyR4C→A is ultimately fully reduced demonstrates that the C199-C208 disulfide bond in OxyR4C→A is metastable relative to the surrounding GSH/GSSG buffer.
Figure 3.

Transient OxyR oxidation by hydrogen peroxide in vitro. OxyR4C→A (1 μM final concentration) was reduced fully by incubation with a buffer containing 25 mM GSH, 0.1 mM GSSG, and 10 μM Grx1. Hydrogen peroxide (2 μM) was added, and the redox status of OxyR was assayed at 0.5, 10, 30, 60, 120, 180, and 240 min. Samples were mixed with a 1/10 vol of 100% TCA and then treated with AMS. Separation and detection of OxyR were achieved by SDS/PAGE and immunoblot analysis.
The hydrogen peroxide added to the reaction in Fig. 3 appears to be consumed because OxyR returns to its reduced form in the course of the experiment. Thus, we propose that OxyR is acting as a peroxidase in our in vitro reactions. This proposition is supported by the finding that significant NADPH consumption, in samples containing hydrogen peroxide, glutathione, glutaredoxin 1, and glutathione reductase under anaerobic conditions, is observed only upon the addition of OxyR (data not shown). Although the rate of the reaction of hydrogen peroxide with OxyR approaches that of the NADH peroxidase, the overall rate of peroxidase activity is slow, which is likely to be caused by the slow rate of OxyR reduction.
Exquisite Hydrogen Peroxide Sensitivity of OxyR.
To determine the minimum concentrations of hydrogen peroxide required to rapidly oxidize OxyR in vivo and in vitro, we again monitored the redox status of OxyR4C→A. For aerobically growing wild-type cells in exponential phase, the lowest concentration of exogenously added hydrogen peroxide able to oxidize 50% of OxyR4C→A within 30 sec was 5 μM (Fig. 4A). We also carried out several hydrogen peroxide titration experiments in vitro. Submicromolar concentrations of OxyR4C→A (0.001, 0.01, and 0.1 μM) were incubated with different concentrations of hydrogen peroxide in the presence of 10 μM glutaredoxin 1, 5 and 25 mM GSH, and 0.1 mM GSSG. Under all of these conditions, between 0.05 and 0.2 μM hydrogen peroxide was required to oxidize 50% of the OxyR protein within 30 sec (Fig. 4B), illustrating the exquisite hydrogen peroxide sensitivity of OxyR. The difference in the minimum hydrogen peroxide concentration required to rapidly oxidize OxyR in vivo and in vitro is probably caused by the fact that much of the exogenously added hydrogen peroxide is degraded by bacterial enzymes.
Figure 4.

Minimum concentrations of hydrogen peroxide required to oxidized OxyR. (A) Midlogarithmically growing wild-type cells (FÅ371) were treated with 0, 0.5, 1, 2, 5, and 10 μM hydrogen peroxide. (B) OxyR4C→A (0.01 μM final concentration) was reduced fully by incubation with a buffer containing 10 μM glutaredoxin 1, 25 mM GSH, and 0.1 mM GSSG. Aliquots were removed and treated with hydrogen peroxide to achieve 0, 0.025, 0.05, 0.075, 0.1, 0.2, 0.5, 1, 2, and 5 μM final concentrations. After 30 sec, the samples in both A and B were acidified with TCA and then treated with AMS. Again, separation and detection of OxyR was achieved by SDS/PAGE and immunoblot analysis.
DISCUSSION
Here we show that OxyR oxidation and activation can be achieved in two possible ways: by a shift of the redox status of the cell or through the high reactivity of OxyR with hydrogen peroxide (Fig. 5). In wild-type cells, the thiol–disulfide redox potential of the E. coli cytosol (−280 mV) is −100 mV lower than the redox potential of OxyR (−185 mV). We propose that OxyR is constitutively activated in the trxA− gorA− mutant strain because the thermodynamic barrier of 100 mV is lowered. In contrast, in wild-type cells, the high thermodynamic barrier is overcome by the high reactivity of OxyR with hydrogen peroxide; however, the oxidized protein is in a metastable state.
Figure 5.

Pathways of OxyR oxidation and reduction. Two different reactions can determine the redox status of OxyR. During normal growth, when hydrogen peroxide concentrations are low, the reaction rate for the second pathway is low, so the first pathway dominates. Under these conditions in wild-type strains, the cellular GSH/GSSG ratio favors reduced OxyR. However, in mutant strains with a low GSH/GSSG ratio, oxidized OxyR is formed. After exposure to ≥5 μM external hydrogen peroxide, the second pathway dominates and oxidized OxyR becomes the major species.
The finding that OxyR can be activated by a change in the thiol–disulfide redox status in the absence of hydrogen peroxide has two important implications. First, mutants compromised in the thiol–disulfide reducing systems actually may be more resistant to peroxide stress. The observation that OxyR is constitutively active in the mutants defective in the disulfide bond reducing systems might help explain the result that trxB− strains are less susceptible to the lethal effects of hydrogen peroxide than wild-type cells (17) and is consistent with the observation that trxA− gshA− mutants have 36-fold-higher levels of grxA expression (18). Further studies on the roles of thioredoxin and glutathione/glutaredoxin in other organisms should take into account the possibility that cells compromised in these reducing systems actually may have increased resistance to oxidative stress because of the activation of antioxidant pathways.
Second, the constitutive OxyR activity seen in mutants allowing disulfide bond formation in the cytoplasm suggests that OxyR could be activated by “disulfide stress” as a result of a drop in the cellular thiol–disulfide ratio. This might occur under certain growth conditions and on exposure to certain oxidants. Diamide, which shows high reactivity with glutathione, activates OxyR (1). However, because relatively high concentrations of diamide are required to activate OxyR, diamide oxidation of OxyR may not be direct but rather be a consequence of the lower GSH/GSSG ratio observed after diamide treatment (data not shown). OxyR has been reported to be required for the expression of grxA and gorA after several days in stationary phase (19), and we found OxyR to accumulate progressively in the oxidized form after 2 days in stationary phase (data not shown). Possibly, changes in the thiol–disulfide status in stationary-phase cells could lead to some OxyR activation. Interestingly, the activities of two other proteins have been proposed recently to be modulated by disulfide bond formation in the cell. The σR sigma factor of Streptomyces coelicolor induces the expression of the thioredoxin operon in response to diamide treatment (20), and the activity of the E. coli Hsp33 chaperone is activated by hydrogen peroxide (21). As additional redox-active proteins are characterized, it will be interesting to see whether some proteins predominantly respond to “peroxide stress” whereas others sense “disulfide stress.”
The AMS alkylation method we developed for OxyR allowed us to monitor directly the oxidation state of the transcription factor. We thus were able to examine the kinetics of OxyR oxidation and reduction in vivo and in vitro. Whereas OxyR oxidation by hydrogen peroxide was complete within 30 sec in logarithmically growing wild-type cells, the half-time of the deactivation process was 5 min. The in vitro kinetics were similar to the in vivo kinetics, with complete oxidation observed in 30 sec and a half-time of deactivation of 10–30 min. Given the faster kinetics of OxyR oxidation by hydrogen peroxide compared with the kinetics of rereduction by the glutaredoxin and thioredoxin systems, OxyR remains oxidized and activated for a defined period of time. We showed previously that OxyR is preferentially reduced by glutaredoxin 1 in vivo and that the expression of both glutaredoxin 1 (grxA) and glutathione reductase (gorA) is induced by OxyR. In this regard, it is interesting that we did not detect a change in the GSH/GSSG ratio after cells were treated with hydrogen peroxide, whereas thioredoxin was oxidized with nearly the same kinetics as OxyR. The difference in the glutathione and thioredoxin reactivities with hydrogen peroxide may be one reason why the glutaredoxin system predominates over the thioredoxin system in reducing OxyR in vivo.
We also were able to show that the minimum hydrogen peroxide concentrations required to completely oxidize OxyR within 30 sec are 5 μM in vivo and between 0.05 and 0.2 μM in vitro. These concentrations agree with the submicromolar concentrations required for the in vivo activation of an OxyR-regulated katG-lacZ fusion (22). The findings suggest that OxyR protein has evolved to sense levels of hydrogen peroxide that just exceed the tolerable, normal intracellular concentrations.
In summary, we propose that the level of OxyR activation is determined by the balance between multiple processes: the generation and degradation of hydrogen peroxide, both internal and external to the cell, and OxyR reduction by glutaredoxin 1 and, possibly, other reductases and other, as yet unidentified, factors in the cell. Whether OxyR is activated under specific hydrogen peroxide concentrations and specific redox conditions is influenced by several important parameters of OxyR. These include the redox potential and the rates of OxyR reaction with hydrogen peroxide and glutaredoxin 1, which reflect the thermodynamic and kinetic limitations on OxyR oxidation, respectively. Measurements of these parameters should provide a better understanding of the nature of constitutive OxyR mutants. A mutant may be constitutively active because of a lower redox potential or because of altered reactivity toward hydrogen peroxide or glutaredoxin 1. The methods that we have developed will allow us to compare OxyR sensitivity with other oxidizing compounds and to understand the chemical basis for the exquisite OxyR sensitivity to hydrogen peroxide.
Acknowledgments
We thank W. Prinz, D. Smillie, R. Hayward, and D. Touati for strains, J. Bushweller for purified glutaredoxin 1, B. González-Flecha for advice on the hydrogen peroxide assays, B. Demple for use of an anaerobic chamber, L. Poole for helpful discussions, and J. Bardwell, R. Glockshuber, J. Imlay, and L. Poole for comments on the manuscript. This work was supported by the intramural program of the National Institute of Child Health and Human Development, National Institutes of Health Grants GM41883 and GM55090, and fellowships from the European Molecular Biology Organization (F.Å.) and the American Cancer Society (M.Z.). J.B. is an American Cancer Society Research Professor.
ABBREVIATIONS
- TCA
trichloroacetic acid
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- GSH
glutathione
- GSSG
glutathione disulfide
Footnotes
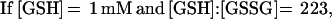 |
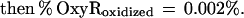 |
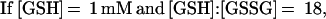 |
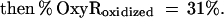 |
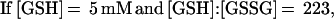 |
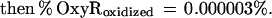 |
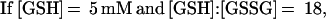 |
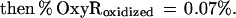 |
References
- 1.Zheng M, Åslund F, Storz G. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- 2.Gilbert H F. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 3.Holmgren A, Fagerstedt M. J Biol Chem. 1982;257:6926–6930. [PubMed] [Google Scholar]
- 4.Hwang C, Sinskey A J, Lodish H F. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 5.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 6.Storz G, Altuvia S. Methods Enzymol. 1994;234:217–223. doi: 10.1016/0076-6879(94)34088-9. [DOI] [PubMed] [Google Scholar]
- 7.Derman A I, Prinz W A, Belin D, Beckwith J. Science. 1993;262:1744–1747. doi: 10.1126/science.8259521. [DOI] [PubMed] [Google Scholar]
- 8.Anderson M E. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- 9.Storz G, Tartaglia L A, Ames B N. Science. 1990;248:189–194. doi: 10.1126/science.2183352. [DOI] [PubMed] [Google Scholar]
- 10.Prinz W A, Åslund F, Holmgren A, Beckwith J. J Biol Chem. 1997;272:15661–15667. doi: 10.1074/jbc.272.25.15661. [DOI] [PubMed] [Google Scholar]
- 11.Rosner J L, Storz G. Antimicrob Agents Chemother. 1994;38:1829–1833. doi: 10.1128/aac.38.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miranda-Vizuete A, Rodríguez-Ariza A, Toribio F, Holmgren A, López-Barea J, Pueyo C. J Biol Chem. 1996;271:19099–19103. doi: 10.1074/jbc.271.32.19099. [DOI] [PubMed] [Google Scholar]
- 13.Newton G L, Arnold K, Price M S, Sherrill C, Delcardayre S B, Aharonowitz Y, Cohen G, Davies J, Fahey R C, Davis C. J Bacteriol. 1996;178:1990–1995. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altuvia S, Weinstein-Fischer D, Zhang A, Postow L, Storz G. Cell. 1997;90:43–53. doi: 10.1016/s0092-8674(00)80312-8. [DOI] [PubMed] [Google Scholar]
- 15.Poole L B, Claiborne A. J Biol Chem. 1989;264:12330–12338. [PubMed] [Google Scholar]
- 16.Crane E J, III, Parsonage D, Poole L B, Claiborne A. Biochemistry. 1995;34:14114–14124. doi: 10.1021/bi00043a016. [DOI] [PubMed] [Google Scholar]
- 17.Takemoto T, Zhang Q-M, Yonei S. Free Radical Biol Med. 1998;24:556–562. doi: 10.1016/s0891-5849(97)00287-6. [DOI] [PubMed] [Google Scholar]
- 18.Gallardo-Madueño R, Leal J F M, Dorado G, Holmgren A, López-Barea J, Pueyo C. J Biol Chem. 1998;273:18382–18388. doi: 10.1074/jbc.273.29.18382. [DOI] [PubMed] [Google Scholar]
- 19.Dukan S, Nyström T. Genes Dev. 1998;12:3431–3441. doi: 10.1101/gad.12.21.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paget M S B, Kang J-G, Roe J-H, Buttner M J. EMBO J. 1998;17:5776–5782. doi: 10.1093/emboj/17.19.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakob U, Muse W, Eser M, Bardwell J C A. Cell. 1999;96:341–352. doi: 10.1016/s0092-8674(00)80547-4. [DOI] [PubMed] [Google Scholar]
- 22.González-Flecha B, Demple B. J Bacteriol. 1997;179:382–388. doi: 10.1128/jb.179.2.382-388.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]