

Abstract
Reduced availability of reactive oxygen species is a key component of neuroprotection against various toxic stimuli. Recently we showed that the hydrogen peroxide scavenger catalase plays a central role in delayed preconditioning induced by the mitochondrial ATP-sensitive potassium channel opener BMS-191095. The purpose of the experiments discussed here was to investigate the neuroprotective effect of catalase in vitro using a recombinant adenoviral catalase gene transfer protocol. To induce catalase overexpression, cultured rat cortical neurons were infected with the adenoviral vector Ad5CMVcatalase and control cells were incubated with Ad5CMVntLacZ for 24h. Gene transfer effectively increased catalase protein levels and activity, but did not influence other antioxidants tested. Ad5CMVcatalase, with up to 10 plaque forming units (pfu) per neuron, did not affect cell viability under control conditions and did not protect against glutamate excitotoxicity or oxygen-glucose deprivation. In contrast, catalase overexpression conferred a dose-dependent protection against exposure to hydrogen peroxide (viability: control, 33.02±1.09%; LacZ 10 pfu/cell, 32.85±1.51%; catalase 1 pfu/cell, 62.09±4.17%*; catalase 2 pfu/cell, 98.71±3.35%*; catalase 10 pfu/cell, 99.68±1.99%*; *p<0.05 vs. control; mean±SEM). Finally, the protection could be antagonized using the catalase inhibitor 3-aminotriazole. Our results support the view that enhancing cellular antioxidant capacity may play a crucial role in neuroprotective strategies.
Keywords: neuroprotection, neuronal culture, catalase, 3-aminotriazole, adenoviral gene transfer
1. Introduction
Reactive oxygen species (ROS) play a dual role in living cells. Whereas these molecules are generated continuously in small quantities during aerobic metabolism and are necessary components of signal transduction, large amounts resulting from increased generation and/or decreased elimination induce oxidative stress and contribute to the pathogenesis of various disorders of the nervous system, including stroke and neurodegenerative diseases such as Parkinson's, Alzheimer's, and amyotrophic lateral sclerosis [7,31,32,34]. ROS are mainly generated as a byproduct of the mitochondrial electron transport chain when electrons leak to molecular oxygen resulting in the formation of superoxide anion which is then converted to hydrogen peroxide by superoxide dismutases. Hydrogen peroxide is also produced by several enzymes, including xanthine oxidase and monoamine oxidases, during their normal activity. H2O2 belongs to a special group of ROS, because it is neither a radical (it does not have unpaired electrons) nor an ion (it is not charged). Thus, it can cross cell membranes easily and can affect cellular structures distant from its place of origin. We and others have shown that neurons are particularly sensitive to H2O2 induced toxicity [11,18]. If not converted to water by catalase or the glutathione system, H2O2 can form hydroxyl radicals via the Fenton reaction, which is the most damaging form of ROS, and which can directly attack lipid membranes, proteins, and nucleic acids ultimately resulting in cell death.
The neuroprotective effect of cellular antioxidants against oxidative stress is well established. In a recent paper, for example, we demonstrated that delayed neuronal preconditioning with the selective mitochondrial ATP-sensitive potassium (mitoKATP) channel opener BMS-191095 results in increased protein levels and activity of catalase [14]. In addition, BMS-191095 induced protection could be partially antagonized using the catalase inhibitor 3-aminotriazole (3AT) suggesting that catalase was partly responsible for the neuroprotective effect of BMS-191095. Others have shown the beneficial effects of increased expression of cellular antioxidants by gene therapy in the central nervous system [3-5,19,38,42]. Whereas most of these studies used adenoviral vectors, catalase gene transfer to cultured neurons using adenoviral vectors has not yet been reported. On the other hand, adenovirus mediated catalase gene transfer was reported to protect other cell and tissue types from oxidative stress [10,25]. Nonetheless, neurons have unique features which makes extrapolation of results from other cell types questionable.
In this study we evaluated the effects of catalase gene transfer on cultured cortical neurons using an adenoviral vector. We examined whether infection with the catalase gene-containing adenoviral vector affected other cellular antioxidant systems. Finally, we tested the protective effect of catalase overexpression against various toxic stimuli.
2. Results
Preliminary experiments showed that viral infection of cultured cortical neurons using the Ad5CMVntLacZ control vector with a titer of 1 pfu/cell resulted in about 30% of cells expressing β-galactosidase, whereas more than 95% of neurons expressed this enzyme after incubation with 10 plaque forming units (pfu) per cell (Fig. 1). Catalase gene transfer by the Ad5CMVcatalase vector caused a dose-dependent increase in both catalase activity (Fig. 2A) and protein levels (Fig. 2B) although the increase in activity was not directly proportionate to protein levels, suggesting that some of the catalase protein was enzymatically inactive. Infection of cultured neurons using the Ad5CMVntLacZ control vector did not influence the expression (Fig. 2B) and activity of catalase (data not shown). We also tested the expression of other antioxidants and found that manganese dependent superoxide dismutase (MnSOD), copper and zinc dependent superoxide dismutase (CuZnSOD), and peroxiredoxin III (Prx III) were not affected by the infection procedure (Fig. 2C). In addition, catalase gene transfer had no effect on total SOD activity (non-infected control, 100.00±1.52%; Ad5CMVcatalase 1 pfu/cell, 102.92±1.52%; Ad5CMVcatalase 2 pfu/cell, 99.84±1.83%; Ad5CMVcatalase 5 pfu/cell, 101.12±2.04%; Ad5CMVcatalase 10 pfu/cell, 103.65±1.68%; mean±SEM; n=16-32 wells). Glutathione peroxidase (GPX) was barely detectable in our neuronal cultures and catalase gene transfer did not increase its expression or activity (data not shown).
Figure 1. Photomicrographs of cultured cortical neurons before and after infection with the adenoviral vector Ad5CMVntLacZ.
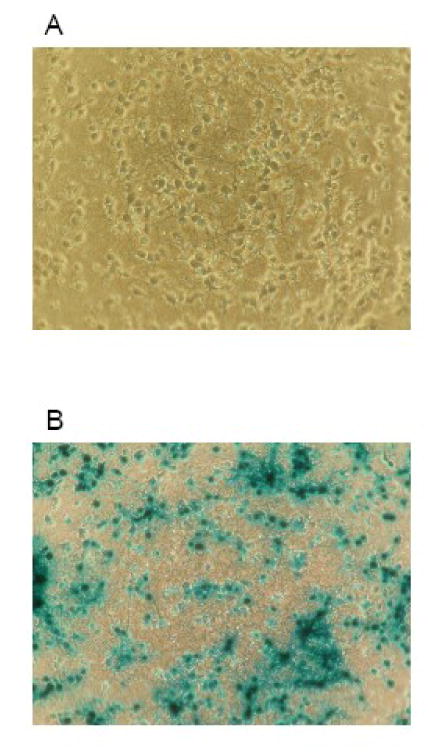
Panel A shows cortical neurons in culture on day 9 in vitro. On panel B, cells were incubated with the Ad5CMVntLacZ vector in the regular culture medium for 24 h using a titer of 10 plaque forming units per cell. Thereafter, the cells were fixed and stained for β-galactosidase using a commercially available kit. Cells were counted in 5 randomly selected fields in each sample using a phase contrast microscope to calculate the ratio of cells expressing the desired protein.
Figure 2. Adenoviral catalase gene transfer enhanced neuronal catalase activity and expression but did not influence the expression of other antioxidants.
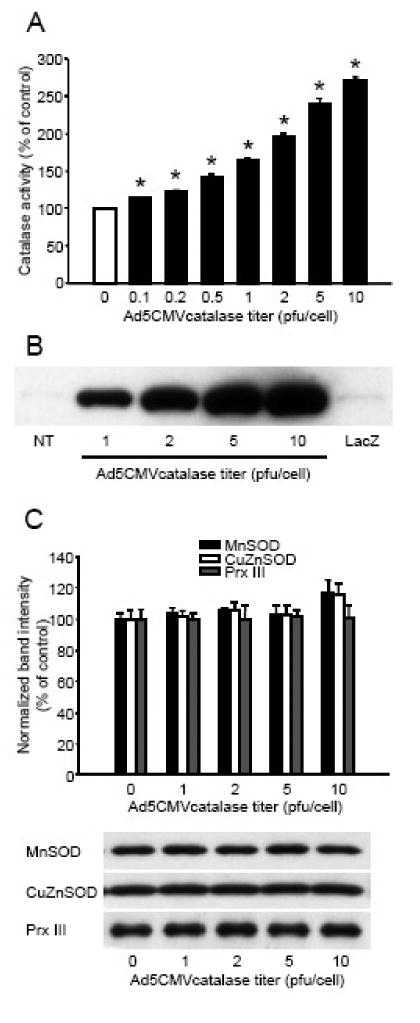
Cultured cortical neurons were incubated with different titers of the Ad5CMVcatalase vector or 10 plaque forming units/cell titer of the Ad5CMVntLacZ (LacZ) vector for 24 h in the regular cell culture medium. The cultures were rinsed and catalase enzyme activity was measured using a commercially available kit (Panel A). Enzyme activity was expressed as percent of non-infected control. *Significant difference (p<0.05) compared with untreated control. Data are expressed as mean ± SEM; n = 16 in each group. In other experiments, 24 h after infection, the cultures were rinsed and protein was extracted and subjected to western blot analysis for catalase (Panel B), or manganese dependent superoxide dismutase (MnSOD), copper and zinc dependent superoxide dismutase (CuZnSOD), and peroxiredoxin III (Prx III) (Panel C). Bands were normalized to the intensity of β-actin, and were expressed as percent of non-infected control. Representative blots are shown below the graph. Data are expressed as mean ± SEM; n = 4 in each group; NT, non-treated; pfu, plaque forming unit.
Vector titers up to 10 pfu/cell did not influence the viability of quiescent neurons (Fig. 3A). This was verified by the measurement of cellular ATP content (in fmol/cell: non-infected control, 3059±16; Ad5CMVcatalase 1 pfu/cell, 3006±26; Ad5CMVcatalase 2 pfu/cell, 2997±27; Ad5CMVcatalase 5 pfu/cell, 3027±31; Ad5CMVcatalase 10 pfu/cell, 3033±24; mean±SEM; n=16 wells in each group, 312 500 cells/well). One day after the gene transfer, we exposed our cultures to glutamate (200 μM for 60 min) or combined oxygen and glucose deprivation (OGD; 180 min). Both lethal challenges resulted in about 50% cell loss as measured by a decrease in the reducing capacity of living cells (Figs. 3B and 3C). Incubation with catalase gene-containing or control vectors did not affect the viability of neurons exposed to glutamate or OGD. In contrast, adenoviral catalase gene transfer with 2 pfu/cell resulted in complete protection against a H2O2 challenge even if almost 70% of control neurons were lost (Fig. 3D). Thus, titers of 5 and 10 pfu/cell provided no further protection. To verify that the protective effect of adenoviral gene transfer was a direct result of increased catalase activity, we infected neuronal cultures with 2 pfu/cell catalase gene-containing vectors and treated them with the catalase inhibitor 3AT during exposure to H2O2. These experiments showed that blocking catalase activity counteracted the protection against H2O2 (Fig. 4).
Figure 3. Catalase overexpression protected cultured cortical neurons against hydrogen peroxide, but not against oxygen-glucose deprivation (OGD) and glutamate excitotoxicity.
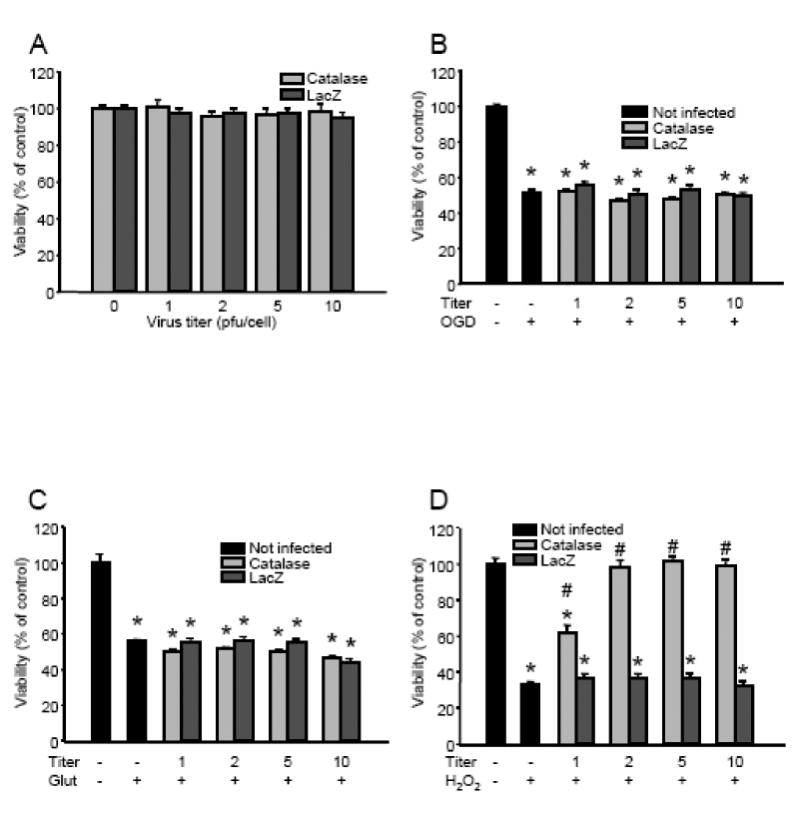
Cultured cortical neurons were incubated with different titers of either the Ad5CMVcatalase (Catalase) or the Ad5CMVntLacZ (LacZ) vector for 24 h in the regular cell culture medium. Then the cultures were rinsed, and viability was measured (Panel A), or the cells were exposed to OGD for 180 min in glucose-free Earle's balanced salt solution (Panel B), 200 μM of glutamate for 60 min in the regular culture medium (Panel C), or 50 μM H2O2 and 1 U/ml glucose oxidase for 60 min in Neurobasal medium (Panel D). Cell viability was assessed 24 h after the toxic challenges and was expressed as percent of the viability of control cultures which were not infected with the vectors and were not exposed to any lethal stimuli. *Significant difference (p < 0.05) compared with untreated control. #Significant difference (p < 0.05) compared with the corresponding cultures infected using the Ad5CMVntLacZ vector. Data are expressed as mean ± SEM; cells from at least two individual cultures; n = 16–32 wells; Glut, glutamate; OGD, oxygen-glucose deprivation; pfu, plaque forming unit.
Figure 4. The catalase inhibitor 3-aminotriazole (3AT) inhibits the protective effect of adenoviral catalase gene transfer.
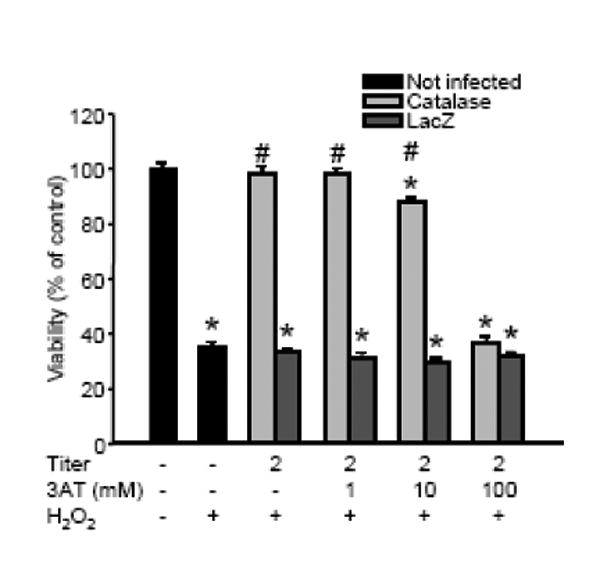
Cultured cortical neurons were incubated with either the Ad5CMVcatalase (Catalase) or with the Ad5CMVntLacZ (LacZ) vector (2 plaque forming units/cell) in the regular cell culture medium. After 24 h the cultures were rinsed and the cells were exposed to50 μM H2O2 and 1 U/ml glucose oxidase for 60 min in Neurobasal medium. To block catalase activity, the cultures were treated with increasing doses of 3AT 10 min prior to and during H2O2 exposure. Cell viability was measured 24 h after the challenge and was expressed as percent of the viability of control cultures which were not infected with the any of the vectors and were not exposed to H2O2. *Significant difference (p < 0.05) compared with untreated control. #Significant difference (p < 0.05) compared with the corresponding cultures infected using the Ad5CMVntLacZ. Data are expressed as mean ± SEM; cells from at least two individual cultures; n = 16 in each group.
3. Discussion
Numerous studies have demonstrated that ROS generated during neurotoxic insults play a central role in subsequent neuronal cell death. Therefore, strategies that enhance cellular defense mechanisms against oxidative stress are of great importance. One possible way to induce the overexpression of antioxidant enzymes is gene therapy. Adenoviral vectors are a particularly useful means of neuronal gene transfer because they are capable of infecting differentiated cells [37]. In addition, catalase gene transfer using adenoviral vectors has been shown to be protective in a lens epithelial cell line, in the optic nerve, and in large arteries [10,17,25]. In this study, we utilized an adenoviral catalase gene transfer protocol in cultured cortical neurons and successfully protected them from the damaging effects of peroxidative stress. Infecting neurons with as few as two viral particles per cell provided complete protection, even though this titer was not enough to induce catalase overexpression in all the cells, suggesting that not all neurons have to overexpress catalase to survive the damaging effect of H2O2. Similar findings have been reported by others [25].
Catalase is a very common enzyme found in the peroxisomes of all animal species and in most plants. It is one of the most efficient enzymes and is able to decompose millions of hydrogen peroxide molecules per second. In addition to H2O2, catalase can metabolize a variety of substrates, including ethanol, methanol, phenols, and nitrites [30]. The enzyme has four subunits each containing a heme group that allows the enzyme to react with H2O2. Thus, its activity is dependent on the capability of cells to produce not only the protein framework, but also the iron-containing heme groups which are partially assembled in mitochondria. This complexity of catalase production may explain the disproportionate increase in activity when compared with protein levels in our experiments. Nevertheless, we found a similar disconnection between catalase protein levels and activity in previous studies [12,14].
In addition to catalase, neurons have other enzymes to eliminate H2O2. The glutathione system is present in the cytosol and in mitochondria, and the activity of GPX in the brain is greater than that of catalase suggesting that GPX may be the more important enzyme to eliminate H2O2 [26]. However, most of this activity is found in astrocytes and inhibition of catalase in neurons dramatically decreases the clearance rate of exogenous H2O2 [9]. Furthermore, GPX is dependent on reduced glutathione which limits its capacity. Therefore, during sustained oxidative stress or when reduced glutathione stores are depleted, neuronal catalase may play an even more important role in the decomposition of H2O2. In addition to the glutathione system, neurons and other cells in the central nervous system also contain peroxiredoxins, a recently characterized family of antioxidants [22,41]. In addition to detoxifying H2O2, peroxiredoxins are also involved in reducing alkyl hydroperoxides and peroxynitrite, as well as in the regulation of H2O2 mediated signal transduction [41]. Prx III is a mitochondrial-specific peroxiredoxin, which scavenges H2O2 and regulates apoptotic signaling in mitochondria [6].
Infection of neuronal cells with adenoviral particles might also induce a generalized stress response and activate pro-survival mechanisms, as in preconditioning where a sublethal stress renders cells tolerant to a subsequent lethal insult [15]. Therefore, we also tested whether catalase gene transfer would affect the expression of other antioxidants. This was particularly important because overexpression of these systems has been demonstrated to be neuroprotective [19,33,38,42]. Thus, we performed western blot analysis of MnSOD, CuZnSOD, Prx III, and GPX after infection with different titers of the catalase gene-containing adenoviral vector, but found no increase in their protein levels.
Next, we examined the neuroprotective effect of catalase gene transfer against various toxic models and found that it provided complete protection against peroxidative stress. The protection was due to catalase overexpression confirmed by using the catalase inhibitor 3AT after gene transfer and during H2O2 challenge. 3AT, in a concentration known to inhibit catalase [8], virtually abolished the protection, providing evidence for the explicit role of increased catalase activity in protecting neurons against peroxidative damage in our model.
Others have demonstrated the protective effect of catalase overexpression using a herpes simplex-1 amplicon vector in hippocampal and cortical neuronal cultures against several cytotoxic models, including glutamate excitotoxicity, but not against OGD [38]. This is in partial agreement with our results because we also found that catalase overexpressing cortical neurons were not protected against OGD. In our gene transfer model, however, catalase did not protect cortical neuronal cultures against glutamate. Glutamate is an excitatory neurotransmitter which exerts its action via ionotropic and metabotropic receptors in the central nervous system. During ischemia, the extracellular level of glutamate increases to the point where it induces excitotoxic neuronal cell death as a combined effect of Ca2+ overload, increased generation and ineffective elimination of ROS, and energy failure. Glutamate excitotoxicity mainly results in necrosis, but depending on concentration, exposure time, receptor expression, cell density, and several other factors, glutamate may also induce apoptosis [29,40]. In our experimental model, high concentrations of glutamate result in a rapid loss of mitochondrial function, including ATP production and a consequent necrosis which is almost exclusively dependent on Ca2+ influx through NMDA receptors [11,24]. Lower glutamate concentrations such as used in the study of Wang et al. [38], however, may induce only a moderate Ca2+ influx which allows mitochondria to maintain ATP production, but Ca2+ entry would still trigger ROS generation [23] with a consequent depletion of GSH [13,35]. In the latter case, increased cellular catalase levels may take over the role of the glutathione system and prevent ROS-induced cell death.
Our results suggest that H2O2 may not be the most relevant ROS during OGD and glutamate excitotoxicity and it was shown that superoxide anion may play a more important role in these challenges [1,29]. However, Hyslop et al. reported extracellular H2O2 concentrations of up to 100 μM after 4-vessel occlusion in the rat [21]. They also reported that this dose of H2O2 applied for 30 min was sufficient to kill more than 50% of cortical neurons in culture. These findings, in combination with others demonstrating the in vivo protective effect of H2O2 scavengers [39], also imply that in vitro cytotoxicity models may not necessarily show a straightforward correlation with in vivo observations and may underestimate the role of H2O2 in ischemia/reperfusion injury.
We previously suggested that catalase overexpression may be partially responsible for the protection against glutamate in delayed neuronal preconditioning induced by the mitoKATP opener BMS-191095 [14]. In that study, we found a robust increase in catalase expression and activity following treatment with BMS-191095. Additionally, the catalase inhibitor 3AT partially antagonized the neuroprotective effect of BMS-191095 against glutamate. However, the fact that 3AT could not completely inhibit the protection implies that increased catalase activity was not the only protective mechanism. We also demonstrated other beneficial effects of mitoKATP channel activation, including sustained mitochondrial depolarization which was shown to reduce mitochondrial Ca2+ accumulation [20], and increased ATP levels that were better preserved during glutamate exposure, suggesting that BMS-191095 treated neurons could preserve mitochondrial integrity during a glutamate challenge. In such a case, a glutamate induced increase in ROS production may play a more important role in cell death; therefore, enhanced cellular antioxidant activity may become a crucial part of the protection. Hence, whether neurons overexpressing catalase survive a glutamate challenge may depend not only on the enzymatic activity of catalase, but also on other factors such as mitochondrial function, concentration of glutamate, and exposure time.
In summary, our results indicate that infection of primary cultured cortical neurons with a catalase gene-containing recombinant adenoviral vector is capable of significantly increasing catalase activity and protecting these cells from the damaging effects of hydrogen peroxide even using a low titer of 2 pfu/cell. In contrast, catalase overexpression did not protect neurons against glutamate excitotoxicity and OGD. Our study also demonstrates that gene transfer using adenoviral vectors is a useful model for studying the effects of selective overexpression of proteins such as antioxidants in cortical neuronal cultures and supports the view that gene therapy targeting ROS generation is a promising neuroprotective approach.
4. Experimental Procedures
Materials
Cell culture plastics were obtained from Becton-Dickinson (San Jose, CA, USA). Adenoviral vectors Ad5CMVntLacZ and Ad5CMVcatalase were purchased from the Gene Transfer Vector Core, University of Iowa (Iowa City, IA, USA). Dulbecco's modified Eagle medium (DMEM), Neurobasal medium, B27 Supplement, 2-mercaptoethanol, and horse serum were from Gibco BRL (Grand Island, NY, USA). Dispase I was purchased from Roche (Mannheim, Germany). CellTiter 96 AQueous One Solution Assay was procured from Promega (Madison, WI, USA). Amplex Red Catalase Assay Kit was obtained from Molecular Probes (Eugene, OR, USA) and the SOD Assay Kit from Fluka (Buchs, Switzerland). The Bio-Rad DC Protein Assay was procured from Bio-Rad (Hercules, CA, USA) and the β-Gal Staining Kit from Invitrogen (Carlsbad, CA, USA). All other chemicals were from Sigma-Aldrich (St. Louis, MO, USA). Antibodies were obtained from the following sources: anti-glial fibrillary acidic protein antibody from Chemicon (Temecula, CA, USA); anti-microtubule associated protein-2 and anti-MnSOD antibodies from Becton-Dickinson; anti-catalase and anti-CuZnSOD antibodies from Calbiochem (Gibbstown, NJ, USA), anti-GPX from Abcam (Cambridge, MA, USA), anti-Prx III from Sigma-Aldrich, anti-rabbit IgG and anti-mouse IgG from Jackson Immuno-Research (West Grove, PA, USA).
Primary rat cortical neuronal culture
Timed pregnant Sprague-Dawley (SD) rats were obtained from Harlan (Indianapolis, IN, USA) and were maintained and used in compliance with the principles set forth by the Animal Care and Use Committee of Wake Forest University Health Sciences. Primary rat cortical neurons were isolated from embryonic day 18 SD rat fetuses as described elsewhere [13]. Briefly, the cells were plated at a density of 2×105 cells/cm2 onto poly-D-lysine coated glass coverslips for confocal microscopic analysis and 106 cells/cm2 were placed onto poly-D-lysine coated plates or dishes for other experiments in plating medium consisting of 60% DMEM, 20% Ham's F-12 Nutrient Mixture, 20% horse serum, and L-glutamine (0.5 mM). After cell attachment, the plating medium was replaced with regular cell culture medium [feeding medium (FM)] consisting of Neurobasal medium supplemented with B27 (2%), L-glutamine (0.5 mM), 2-mercaptoethanol (55 μM), and KCl (25 mM). Positive immunostaining for microtubule-associated protein-2 and negative immunostaining for glial fibrillary acidic protein verified that the cultures were composed of more than 98% of neurons on day 7 in vitro (DIV 7). Experiments were carried out on 7–9 day old cultures, during which neurons expressed N-methyl-d-aspartate, α-amino-3-hydroxy-5-methylisoxazole-4-propionate, and kainate receptors and were vulnerable to glucose deprivation [27,28].
Adenoviral gene transfer
Adenoviral vectors were stored in 3% sucrose/phosphate buffered saline (PBS) at -80°C. Aliquots of adenovirus expressing either β-galactosidase (Ad5CMVntLacZ) or catalase (Ad5CMVcatalase) were thawed on ice and diluted with FM to the desired titer. Neuronal cultures on DIV 9 were incubated with the catalase gene-containing vector at titers of 3 × 106 − 3 × 107 pfu/ml (1 – 10 pfu/cell) in FM at 37 °C for 24 h, then the cultures were rinsed with PBS and transferred to FM. Control cultures were exposed to the Ad5CMVntLacZ vector in FM as well and similarly incubated at 37°C for 24 h.
Determination of the efficiency of gene transfer
To determine whether the gene transfer protocol resulted in the production of the desired protein, β-galactosidase expression was evaluated 24 h after incubation with different titers of Ad5CMVntLacZ using the β-Gal Staining Kit (Invitrogen). Briefly, neuronal cultures were rinsed in PBS and then were fixed with Fixative Solution (2% formaldehyde and 0.2% glutaraldehyde in PBS) for 10 min at room temperature (21 °C). After fixation, the cultures were rinsed twice with PBS, were incubated in Staining Solution for 60 min at 37°C, which was followed by phase contrast microscopy to determine the ratio of cells expressing β-galactosidase. Cells were counted in 5 randomly selected fields in each sample and the ratio of cells expressing the desired protein was calculated.
Combined oxygen and glucose deprivation
Neurons in 96-well plates were exposed to OGD for 180 min at 37 °C using a protocol described previously [16,24]. Briefly, 24 h after the initiation of adenoviral gene transfer, the cells were rinsed, the medium was replaced with glucose-free Earle's balanced salt solution (EBSS), and the cultures were placed in a ShelLab Bactron Anaerobic Chamber (Sheldon Manufacturing Inc., Cornelius, OR, USA) filled with a humidified anaerobic gas mixture (5% CO2, 5% H2 and 90% N2) at 37 °C. The hydrogen content of the gas mixture is used by a catalyst to convert remaining traces of oxygen into water, thus keeping oxygen levels typically below 0.1% in the chamber. Control cell cultures were incubated in glucose-containing (5.5 mM) EBSS in a regular 5% CO2 cell culture incubator for 180 min. OGD was terminated by replacing the glucose-free EBSS with FM. Thereafter the cultures were maintained in the regular 5% CO2 incubator within normoxic conditions. These experimental conditions usually result in about 50% decrease in neuronal viability.
Glutamate excitotoxicity
After 24 h incubation with the adenoviral vectors, cell cultures in 96-well plates were washed with PBS and then exposed to glutamate (200 μM) for 60 min in FM at 37°C in the 5% CO2 incubator. Afterward the cells were rinsed and returned to the 5% CO2 incubator in FM. This protocol usually results in approximately 40-50% cell death.
Exogenous hydrogen peroxide toxicity
Neuronal cultures in 96-well plates were rinsed and the cells were incubated in Neurobasal medium containing H2O2 (50 μM) and glucose oxidase (1 U/ml) at 37°C in the 5% CO2 incubator for 1 h. The continuous generation of H2O2 by glucose oxidase was shown to compensate for the consumption of the initial dose of H2O2 by neurons [2,36]. After the challenge, the cultures were washed, FM was restored, and the cells were returned to the 5% CO2 incubator. In preliminary experiments, we found that this regimen caused about 50-60% cell loss. In some experiments, different concentrations of the catalase inhibitor 3AT (1, 10, 100 mM) were applied to the cultures 10 minutes before and during H2O2 toxicity.
Quantification of neuronal survival
The cell viability in neuronal cultures was determined 24 h after the neurotoxic insults using the tetrazolium-based CellTiter 96 AQueous One Solution assay. Absorbance at 492 nm was measured with a microplate reader (FLUOstar OPTIMA, BMG Labtech GmbH, Offenburg, Germany). Comparisons were always made in the same manner, between sister cultures exposed to the neurotoxic stimulus on the same day, with cell viability expressed as a percentage of the corresponding control culture (untreated, and not exposed to the lethal insult).
ATP Assay
The ATP level of neurons was measured with the glowtype CellTiter-Glo Luminescent Assay, as directed by the manufacturer. Cortical neurons cultured in opaque-walled 96-well plates were equilibrated to room temperature (21 °C) for 30 mins. CellTiter-Glo was added to each well and the plates were incubated at room temperature for 10 mins to stabilize the luminescent signal, which was then measured with a FLUOstar OPTIMA microplate reader. An ATP standard curve was generated for each measurement to calculate the ATP contents of cells.
Western blotting for catalase, MnSOD, CuZnSOD, GPX, and Prx III
Cultured cells were washed twice in ice-cold PBS and were harvested by scraping in ice-cold Nonidet P40 lysis buffer supplemented with a protease inhibitor cocktail and a phosphatase inhibitor cocktail (all from Sigma-Aldrich). Equal amounts of protein for each sample were separated by 4-20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto a polyvinylidine difluoride sheet (Polyscreen PVDF; Perkin Elmer Life Sciences, Boston, MA, USA). Membranes were incubated in a blocking buffer (Tris-buffered saline, 0.05% Tween 20, and 1% bovine serum albumin) for 1 h at room temperature after which the blots were incubated with polyclonal anti-catalase (1:5000), monoclonal anti-MnSOD (1:5000), polyclonal anti-CuZnSOD (1:5000), polyclonal anti-GPX (1:10 000), and monoclonal anti-Prx III (1:10 000) antibodies overnight at 4 °C. The membranes were then washed three times in Tris-buffered saline with 0.05% Tween 20 and incubated for 1 h in the blocking buffer with anti-rabbit IgG (1:100 000) or anti-mouse IgG (1:100 000) conjugated to horseradish peroxidase. The final reaction products were visualized using enhanced chemiluminescence (SuperSignal West Pico; Pierce, Rockford, IL, USA) and recorded on X-ray film. Films were digitalized using a FOTO/Analyst® Investigator-PC Electronic Documentation System with the provided Image v5.00 software (FOTODYNE Inc., Hartland, WI, USA). Densitometry of digitalized blots was performed with the ImageJ 1.30v software (http://rsb.info.nih.gov/ij; National Institutes of Health, USA).
Enzyme activity assays
Catalase activity was assessed using the Amplex Red Catalase Activity Assay Kit. Enzyme activity was measured 24 h after the adenoviral gene transfer with the same microplate reader that was used for other fluorescent measurements (λex = 555 nm and λem = 590 nm). A standard curve was generated for each measurement to calculate catalase activity of wells. Data were expressed as a percentage of the activity of the non-infected control culture.
SOD activity was measured using the tetrazolium-based SOD Assay Kit as directed by the manufacturer. Twenty four hours after gene transfer, neurons on 96-well plates were rinsed with PBS, and enzyme activity was measured with the same microplate reader that was used for other measurements (λabs = 460 nm). Data were expressed as a percentage of the activity of the non-infected control culture.
Statistical analysis
Statistical analyses were performed with SigmaStat (SPSS, Chicago, IL, USA). Data are presented as means ± SEM. Differences between groups were assessed by one way ANOVA followed by Tukey comparison tests. A value of p < 0.05 was considered to be statistically significant.
Acknowledgments
The authors gratefully thank Nancy Busija, M.A., for editing the manuscript. This work was supported by the National Institutes of Health Grants (HL-030260, HL-065380, and HL-077731), and by the Hungarian Science Research Fund (OTKA K63401, K68976, and IN69967). Ferenc Domoki was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antunes F, Cadenas E. Cellular titration of apoptosis with steady state concentrations of H(2)O(2): submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic Biol Med. 2001;30:1008–1018. doi: 10.1016/s0891-5849(01)00493-2. [DOI] [PubMed] [Google Scholar]
- 3.Barkats M, Bemelmans AP, Geoffroy MC, Robert JJ, Loquet I, Horellou P, Revah F, Mallet J. An adenovirus encoding CuZnSOD protects cultured striatal neurones against glutamate toxicity. Neuroreport. 1996;7:497–501. doi: 10.1097/00001756-199601310-00029. [DOI] [PubMed] [Google Scholar]
- 4.Boulos S, Meloni BP, Arthur PG, Bojarski C, Knuckey NW. Peroxiredoxin 2 overexpression protects cortical neuronal cultures from ischemic and oxidative injury but not glutamate excitotoxicity, whereas Cu/Zn superoxide dismutase 1 overexpression protects only against oxidative injury. J Neurosci Res. 2007;85:3089–3097. doi: 10.1002/jnr.21429. [DOI] [PubMed] [Google Scholar]
- 5.Chang P, Cheng E, Brooke S, Sapolsky R. Marked differences in the efficacy of post-insult gene therapy with catalase versus glutathione peroxidase. Brain Res. 2005;1063:27–31. doi: 10.1016/j.brainres.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 6.Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem. 2004;279:41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 7.Chinta SJ, Andersen JK. Redox imbalance in Parkinson's disease. Biochim Biophys Acta. 2008;1780:1362–1367. doi: 10.1016/j.bbagen.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawson RMC, Elliott DC, Elliott WH, Jones KM. Data for Biochemical Research. Oxford University Press; Oxford: 1993. Inhibitors of mitochondrial and chloroplast function; pp. 302–303. [Google Scholar]
- 9.Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J Neurosci Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 10.Durand E, Al Haj Zen A, Addad F, Brasselet C, Caligiuri G, Vinchon F, Lemarchand P, Desnos M, Bruneval P, Lafont A. Adenovirus-mediated gene transfer of superoxide dismutase and catalase decreases restenosis after balloon angioplasty. J Vasc Res. 2005;42:255–265. doi: 10.1159/000085658. [DOI] [PubMed] [Google Scholar]
- 11.Gaspar T, Katakam P, Snipes JA, Kis B, Domoki F, Bari F, Busija DW. Delayed neuronal preconditioning by NS1619 is independent of calcium activated potassium channels. J Neurochem. 2008;105:1115–1128. doi: 10.1111/j.1471-4159.2007.05210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaspar T, Kis B, Snipes JA, Lenzser G, Mayanagi K, Bari F, Busija DW. Neuronal preconditioning with the antianginal drug, bepridil. J Neurochem. 2007;102:595–608. doi: 10.1111/j.1471-4159.2007.04501.x. [DOI] [PubMed] [Google Scholar]
- 13.Gaspar T, Kis B, Snipes JA, Lenzser G, Mayanagi K, Bari F, Busija DW. Transient glucose and amino acid deprivation induces delayed preconditioning in cultured rat cortical neurons. J Neurochem. 2006;98:555–565. doi: 10.1111/j.1471-4159.2006.03899.x. [DOI] [PubMed] [Google Scholar]
- 14.Gaspar T, Snipes JA, Busija AR, Kis B, Domoki F, Bari F, Busija DW. ROS-independent preconditioning in neurons via activation of mitoK(ATP) channels by BMS-191095. J Cereb Blood Flow Metab. 2008;28:1090–1103. doi: 10.1038/sj.jcbfm.9600611. [DOI] [PubMed] [Google Scholar]
- 15.Gidday JM. Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci. 2006;7:437–448. doi: 10.1038/nrn1927. [DOI] [PubMed] [Google Scholar]
- 16.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guy J, Qi X, Wang H, Hauswirth WW. Adenoviral gene therapy with catalase suppresses experimental optic neuritis. Arch Ophthalmol. 1999;117:1533–1539. doi: 10.1001/archopht.117.11.1533. [DOI] [PubMed] [Google Scholar]
- 18.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 19.Hattori F, Murayama N, Noshita T, Oikawa S. Mitochondrial peroxiredoxin-3 protects hippocampal neurons from excitotoxic injury in vivo. J Neurochem. 2003;86:860–868. doi: 10.1046/j.1471-4159.2003.01918.x. [DOI] [PubMed] [Google Scholar]
- 20.Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol. 1999;519(Pt 2):347–360. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995;671:181–186. doi: 10.1016/0006-8993(94)01291-o. [DOI] [PubMed] [Google Scholar]
- 22.Jin MH, Lee YH, Kim JM, Sun HN, Moon EY, Shong MH, Kim SU, Lee SH, Lee TH, Yu DY, Lee DS. Characterization of neural cell types expressing peroxiredoxins in mouse brain. Neurosci Lett. 2005;381:252–257. doi: 10.1016/j.neulet.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 23.Kahlert S, Zundorf G, Reiser G. Glutamate-mediated influx of extracellular Ca2+ is coupled with reactive oxygen species generation in cultured hippocampal neurons but not in astrocytes. J Neurosci Res. 2005;79:262–271. doi: 10.1002/jnr.20322. [DOI] [PubMed] [Google Scholar]
- 24.Kis B, Rajapakse NC, Snipes JA, Nagy K, Horiguchi T, Busija DW. Diazoxide induces delayed pre-conditioning in cultured rat cortical neurons. J Neurochem. 2003;87:969–980. doi: 10.1046/j.1471-4159.2003.02072.x. [DOI] [PubMed] [Google Scholar]
- 25.Ma W, Nunes I, Young CS, Spector A. Catalase enrichment using recombinant adenovirus protects alphaTN4-1 cells from H(2)O(2) Free Radic Biol Med. 2006;40:335–340. doi: 10.1016/j.freeradbiomed.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 26.Marklund SL, Westman NG, Lundgren E, Roos G. Copper- and zinc-containing superoxide dismutase, manganese-containing superoxide dismutase, catalase, and glutathione peroxidase in normal and neoplastic human cell lines and normal human tissues. Cancer Res. 1982;42:1955–1961. [PubMed] [Google Scholar]
- 27.Mattson MP, Wang H, Michaelis EK. Developmental expression, compartmentalization, and possible role in excitotoxicity of a putative NMDA receptor protein in cultured hippocampal neurons. Brain Res. 1991;565:94–108. doi: 10.1016/0006-8993(91)91740-r. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 30.Oshino N, Chance B, Sies H, Bucher T. The role of H 2 O 2 generation in perfused rat liver and the reaction of catalase compound I and hydrogen donors. Arch Biochem Biophys. 1973;154:117–131. doi: 10.1016/0003-9861(73)90040-4. [DOI] [PubMed] [Google Scholar]
- 31.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- 32.Saeed SA, Shad KF, Saleem T, Javed F, Khan MU. Some new prospects in the understanding of the molecular basis of the pathogenesis of stroke. Exp Brain Res. 2007;182:1–10. doi: 10.1007/s00221-007-1050-9. [DOI] [PubMed] [Google Scholar]
- 33.Shan X, Chi L, Ke Y, Luo C, Qian S, Gozal D, Liu R. Manganese superoxide dismutase protects mouse cortical neurons from chronic intermittent hypoxia-mediated oxidative damage. Neurobiol Dis. 2007;28:206–215. doi: 10.1016/j.nbd.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord. 2007;21:276–291. doi: 10.1097/WAD.0b013e31815721c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tirosh O, Sen CK, Roy S, Packer L. Cellular and mitochondrial changes in glutamate-induced HT4 neuronal cell death. Neuroscience. 2000;97:531–541. doi: 10.1016/s0306-4522(00)00028-2. [DOI] [PubMed] [Google Scholar]
- 36.Vauzour D, Vafeiadou K, Rice-Evans C, Williams RJ, Spencer JP. Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J Neurochem. 2007;103:1355–1367. doi: 10.1111/j.1471-4159.2007.04841.x. [DOI] [PubMed] [Google Scholar]
- 37.Volpers C, Kochanek S. Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004;6(Suppl 1):S164–171. doi: 10.1002/jgm.496. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Cheng E, Brooke S, Chang P, Sapolsky R. Over-expression of antioxidant enzymes protects cultured hippocampal and cortical neurons from necrotic insults. J Neurochem. 2003;87:1527–1534. doi: 10.1046/j.1471-4159.2003.02123.x. [DOI] [PubMed] [Google Scholar]
- 39.Weisbrot-Lefkowitz M, Reuhl K, Perry B, Chan PH, Inouye M, Mirochnitchenko O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res Mol Brain Res. 1998;53:333–338. doi: 10.1016/s0169-328x(97)00313-6. [DOI] [PubMed] [Google Scholar]
- 40.Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35:67–86. doi: 10.5483/bmbrep.2002.35.1.067. [DOI] [PubMed] [Google Scholar]
- 41.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 42.Zemlyak I, Nimon V, Brooke S, Moore T, McLaughlin J, Sapolsky R. Gene therapy in the nervous system with superoxide dismutase. Brain Res. 2006;1088:12–18. doi: 10.1016/j.brainres.2006.02.109. [DOI] [PubMed] [Google Scholar]