

Abstract
Inorganic arsenic that is ingested through drinking water or inhalation is metabolized by biological methylation pathways into organoarsenical metabolites. It is now becoming understood that this metabolism that was formerly considered to be detoxification may contribute as much or more to increasing the toxicity of arsenic. One proposed mode of the toxic action of arsenic and its organoarsenic metabolites is through its binding to proteins and inactivating their enzymatic activity. The classic case has been considered the affinity of the proximal 1,3 sulfhydryl groups of the lipoic acid cofactor of the pyruvate dehydrogenase complex for arsenic. A 2:1 stoichiometry of sulfhydryl to arsenic groups has been measured in proteins and arsenical complexes can be synthesized using free D,L-lipoic acid. The relative importance of this site for arsenic binding has come in to question through the use of methylating bifunctional arsenic complexes that suggested the methylation of an active site histidine may also be important, and the suggestion that arsenic inhibits the pyruvate dehydrogenase complex indirectly by elevating mitochondrial hydrogen peroxide generation. In order to separate the effects of direct trivalent arsenite toxicity from that of hydrogen peroxide and activated oxygen, we studied the inhibition of the PDH complex under conditions that did not generate hydrogen peroxide but did expose the lipoic acid group in its reduced state to arsenicals. We also studied the effects of arsenicals in the inhibition of the α-ketoglutarate dehydrogenase complex. We found that only trivalent arsenical compounds inhibited the activity of both dehydrogenase complexes and only when the lipoic acid was in its reduced form. Arsenite inhibited both enzyme complexes approximately equivalently while monomethylarsenite inhibited the PDH complex to a greater extent than the KGDH complex – although both complexes were very sensitive to inhibition by this complex. Dimethylarsenite inhibition of both complexes was only observed with longer pre-incubation periods. Cumulative inhibition by the reduced arsenical was observed for all complexes indicating a binding mode of inhibition that is dependent upon lipoic acid being in its reduced state.
Keywords: Arsenic, pyruvate dehydrogenase, alpha ketoglutarate dehydrogenase, lipoic acid, methyl arsenic
Introduction
Arsenic is a well-established toxic and carcinogenic environmental contaminant with the majority of non-industrial human exposure occurring as a result of contaminated drinking water [1]. While exposure is principally to the inorganic forms of arsenic; arsenite, (As III) and arsenate (AsV), these are metabolized upon ingestion to form a number of organo-arsenic species, including methyarsonic acid (MMAsV),dimethylarsinic acid – also called cacodylic acid – (DMAsV), and their reduced counterparts, monomethylarsonous acid (MMAsIII) and dimethylarsonous acid (DMAsIII) along a pathway that has been described as much for its activation of arsenic as its detoxification [2]. Arsenic ingestion has been associated with diverse health effects including cardiovascular and peripheral vascular diseases, haematologic effects and diabetes, adverse reproductive outcomes, adverse neurological effects, respiratory system dysfunction and lung cancer, and dermatological effects as well as cancer of the skin, kidney, urinary bladder, colon and liver [reviewed in 2,3]. Mechanistic events described in arsenic exposed tissues and cells include oxidative stress [4], enhanced cell proliferation and transformation [reviewed in 5] and changes in DNA methylation patterns [6]. The diversity of these effects can be ascribed in part to inter-individual differences in the metabolic activation of inorganic arsenic into organoarsenic as well as to a complicated non-linear exposure and toxicity profile for the element [7,8]. The toxic mode of action may depend not only upon the form of arsenic but also may be tissue and cell dependent or even protein specific as suggested by Hu et al. [9]. Arsenic inhibits the actions of numerous enzymes [9-14] and it has been proposed that there may be ‘several hundred good binding sites for trivalent arsenicals’ in each organ [15]. Very early studies into the mechanistic toxicology of arsenic observed that trivalent arsenicals inhibited cellular respiration and the oxidation of pyruvic acid [16,17]. It was subsequently decided that sulfhydryl groups were the probable arsenite reactive moiety (or arsenic “chemo-receptor” as it was called at the time) and studies with ‘kerateine’ (sic) determined that about 75% of the arsenic combined with sulfhydryl groups in a one to two ratio. As only dithiol and not monothiol reagents were able to mitigate arsenic-induced inhibition of pyruvate oxidation, these studies resulted in the discovery of the dithiol arsenic chelating agent British anti-lewiste (or BAL) [17]. It was further proposed that some dithiol moiety of the pyruvate oxidase enzyme would be the site of arsenic interaction and inhibition. Pyruvate oxidation is carried out by a multi-enzyme complex, the pyruvate dehydrogenase (PDH) complex. This is a nuclear-encoded mitochondrial enzyme complex that contains a 1,3-dithiol complex, lipoic acid as a post-translational modification on one of its subunits - the acetyltransferase or E2 subunit. As this satisfied the contemporary ‘ring hypothesis’ of arsenic binding [18], as lipoic acid out-competed monothiols for arsenical chelation [19], and as free D,L- lipoic acid complexed arsenicals well enough for some products to be structurally characterized [18,20,21], the lipoic acid moiety was the proposed dithiol site of arsenic binding and enzyme inhibition.
There are a small number of enzymes – three in mammals - that are post-translationally modified by the addition of the lipoic acid co-factor and all are mitochondrial and important for respiration [22]. In addition to the PDH complex we were also interested in studying a second one of these, the α–ketoglutarate dehydrogenase (KGDH) complex. Like PDH, this is a massive multienzyme enzyme complex and the E3 lipoamide dehydrogenase subunit is the identical, nuclear-encoded protein in the two enzyme complexes. Important differences also occur between the two enzymes, as the PDH complex also includes a fourth lipoic-acid containing regulatory protein (Protein X) within the complex that KGDH does not have. Additionally the E2 subunit of PDH is modified with an additional lipoic acid, whereas the KGDH contains only one. All of the lipoic acid enzymes generate acetyl CoA and are important for energy generation. However, if the ability of glucose to provide acetyl CoA is inhibited through the diminished activity of the PDH complex, this can be provided through the β-oxidation of fatty acids or amino acid catabolism. On the other hand, KGDH is not only an integral enzyme within the citric acid cycle, it is a control point for the operation of the citric acid cycle and therefore respiration. We were also interested in studying KGDH as KGDH deficiency is observed in patients with numerous neurodegenerative diseases [22-26] and neurodegeneration is one of the health outcomes observed following arsenic poisoning [2]. As susceptibility is pronounced in individuals with deficiencies in the one carbon recycling system (MTFTR) [26] it is highly plausible that intermediately metabolized organoarsenicals may contribute to neurotoxicity.
The relative importance of the lipoic acid site for arsenite binding and enzyme inhibition has recently been questioned. Samikannu and co-workers [27] suggested that reactive oxygen species may play more of a role specifically in PDH inhibition in cells than arsenic binding, which they stated only occurred at levels well above toxicity for the arsenic compound they studied. Ramanathan et al. [28] previously observed that co-administration of dietary antioxidants with arsenite restored the arsenite-induced depletion of mitochondrial enzyme activity although they ascribed this to restoration of the arsenite-depleted thiol reserves of the mitochondria. We were therefore interested in whether we could establish direct evidence for the binding of arsenicals to the lipoic acid moiety within the PDH (and the KGDH) complex in a cell free (and therefore hydrogen peroxide free) system, particularly the organoarsenicals that have been identified as mammalian metabolites of ingested inorganic arsenic and potentially more toxic than arsenite or arsenate [29-30,12].
Materials and Methods
Sodium arsenate was purchased from J.T. Baker. Sodium arsenite, and potassium iodide were purchased from Fischer Scientific. Hydrochloric acid was purchased from EMD Chemicals, USA. Disodium methylarsonate was purchased from Chem Service (West Chester, PA). The sodium salt of dimethylarsinic acid, or cacodylic acid, was purchased from J.T. Baker. Purified enzyme preparations of the porcine pyruvate dehydrogenase (PDH) complex and the α-ketoglutarate dehydrogenase (KGDH) complex were purchased from Sigma.
Synthesis of reduced arsenicals
Hazardous Materials
Caution! Arsenite is a known human carcinogen and toxicant. Studies – including these presented here – indicate that the toxic effects of methylarsenite may exceed those known for arsenite. Methylated arsenic compounds may also be teratogens. Proper care and caution should be taken in handling these materials.
Methylarsonous diiodide (CH3)As I2
Methylarsonous diiodide (MMA) was prepared by a scaled down methods adapted from Goddard [31] and Styblo et al. [11], as the large quantities of those syntheses were not required for these studies and to minimize risks associated with storage and handling of excess toxic compound. Amounts and relative amounts differ between the two protocols and are listed consecutively. Briefly, the sodium salt of methylarsonic acid (2 g, Goddard or 0.44 g Styblo et al.) was placed into a one-neck pear shaped flask and nanopure water (5 ml or 1.7 mL) and potassium iodide (2.5 g or 0.876 g) added, the mixture acidified with the addition of 1.5 g or 0.3 mL HCl and the contents swirled until all the components were dissolved. Sulfur dioxide was generated by heating sodium metabisulfite and the generated gas bubbled through the reaction mixture by introduction through a Pasteur pipette. For the Goddard-derived preparation the diiodomethylarsenite precipitated out of solution directly. For the Styblo et al. preparation, after about 9 minutes an oily yellow orange precipitate begins to accumulate on the bottom of the pear shaped flask. After one hour the pipette is removed, the flask capped and reaction vessel is placed on ice whereupon the oily precipitate crashes out of solution as a solid precipitate which was collected by vacuum filtration. The precipitate was washed twice with ice-cold nanopure water, returned to a vacuum flask and dried in vacuo for one hour. 1H-NMR analysis of the reduced species gave a peak at δ 3.10 in CDCl3 and at δ 1.24-1.25 in D2O for the methyl peak as has been observed previously [11,12]. Following synthesis the sample was removed to an ampoule and stored under argon or nitrogen, in a desiccator at -20 °C until use.
Dimethylarsonous iodide (CH3)2As I
DMAsIII was prepared as the iodine salt according to a scaled down method of Goddard [31] that was adapted by Styblo et al. [11]. Cacodylic acid (dimethylarsinic acid, 4.14 g) was dissolved in nanopure water (30 mL) in a 100 mL round bottom flask. Potassium iodide (5.48 g) was added and swirled until dissolved. The solution was acidified by the addition of concentrated sulfuric acid (H2SO4, 1.7 mL) and sulfur dioxide generated as described above was bubbled through the solution. After one hour, the pipette was removed and the yellow oily precipitate was removed by separating funnel. The sulfur dioxide bubbler was then returned to the reaction mixture for an hour, the oily precipitate again removed and the two fractions were pooled together and dried with anhydrous sodium sulfate (Na2SO4), rinsed with chloroform (HCCl3) and stored at -20 °C overnight. Dimethylarsonous iodide was then purified by vacuum distillation with mild heating. Arsenic stock solutions were prepared immediately before use. 1H-NMR analysis of the reduced species gave a peak at δ 2.0 in CDCl3 as has been observed previously [11].
Purified Enzyme Assays
The purified pyruvate dehydrogenase complex (PDH) and the α-ketoglutarate dehydrogenase (KGDH) complex from porcine heart were obtained from Sigma in a solution containing 50% glycerol, 10 mg/mL bovine serum albumin, 30% sucrose, 2.5 mM EDTA, 2.5 mM β-mercaptoethanol, 0.5% Triton X-100, 0.005% sodium azide, and 25 mM potassium phosphate, (pH 6.8). An aliquot of enzyme (7.5 mU) was centrifuged for 30 minutes at 14,000 g at 4 °C through a Microcon YM −30 Centrifugal Filter. The enzyme was resuspended in 1.8 mL 120 mM KCl, 5.0 mM KH2PO4, and 5.0 mM MOPS at pH 7.40 (Buffer 1). This was repeated twice more. The enzyme preparation was then added to a reaction buffer containing thiamin pyrophosphate (TPP, 400 μM, also called thiamin diphosphate) and calcium chloride (CaCl2, 20 μM). Where indicated the reaction buffer also contained substrate at 1 mM (pyruvate for PDH and α-ketoglutarate for KGDH), Coenzyme A (200 μM) and the indicated amounts and types of arsenic. This reaction mixture was incubated for 30 minutes at 30 °C. To begin the reaction oxidized nicotinamide adenine dinucleotide (NAD+,0.5 mM) was added and NADH formation was recorded at 340 nm in a 96 well Molecular Devices Spectromax II spectrophotometer. Kinetic analysis was performed by measuring the initial rate (NADH/min) of NADH formation at 340 nm then converted to relative activity compared to that of the arsenic-free control conditions. Inhibition graphs were plotted in Kaleidagraph 3.5.0.0 (Synergy Software) and logarithmic curve fits plotted through the 50% inhibition region. IC50 were calculated and given along with R2 values for each fit in Table1.
Table 1.
IC50 for the inhibition of the pyruvate dehydrogenase (PDH) and the α-ketogluratate dehydrogenase (KGDH) complexes after a 30 min or one hour preincubation with arsenic, substrate, TPP, and Coenzyme A. None of the pentavalent complexes inhibited either enzyme complex. *ND – not determined. **calculated.
| ENZYME: Arsenical 50% enzyme inhibition | |||||||
|---|---|---|---|---|---|---|---|
| Half hour | incubation | One hour | incubation | ||||
| PDH: | (ppb) | (μM) | R2 | (ppb) | (μM) | R2 | |
| As 3+ | 704 | 9.39 | 0.998 | 267 | 3.56 | 0.98 | |
| MMA 3+ | 10.5 | 0.140 | 0.95 | 6.9 | 0.0921 | 0.96 | |
| DMA 3+ | ND* | 0.76 | 786.55 | 10.498 | 0.97 | ||
| KGDH: | |||||||
| As3+ | 733 | 9.78 | 0.96 | 488 | 6.51 | 0.98 | |
| MMA 3+ | 35 | 0.467 | 0.99 | 21 | 0.280 | 0.93 | |
| DMA 3+ | ND* | 0.71 | 7535** | 100.6 | 0.95 | ||
Slot blot analysis
Solutions of each arsenic solution were made up to a high concentration of 2 mM (as indicated) in Tris buffered saline (50 mM Tris-HCl, 150 mM NaCl, pH 7.6; TBS). PDH (2 μL of 13 mg/mL) or KGDH (2 μL of 12.2 mg/mL) was added to 500 μL of each arsenic solution and each As - enzyme solution was incubated at 34°C for 2 hr. Each arsenic –enzyme preparation was pipetted into an individual slot of a prepared Bio-Rad slot blotter and the enzyme transferred to nitrocellulose membrane (Hybond – ECL, Amersham Bioscences) by applying a vacuum according to the manufacturers instructions. After rinsing each well with TBS, the nitrocellulose membrane was removed from the slot blot apparatus and blocked with TBS containing 3% bovine serum albumin (BSA) for one hour. The membrane was rinsed twice with TBS containing 0.5% Tween 20 (TBS-T) by rocking at room temperature for 5 min each, then incubated for 3 hr at room temperature with polyclonal anti-lipoic acid antibody (a generous donation from Drs. Luke and Pam Szweda, Case Western University) at 1:25,000 dilution. The membrane was washed twice with TBS-T and incubated with horseradish peroxidase conjugated goat anti-rabbit antibody (Bio-Rad) at 1:3000 in TBS-T for one hr at room temperature. The membrane was then rinsed twice with TBS-T, once with TBS and the antibody visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce) exposed to X-ray film (KODAK BioMax (MS). Normalized loading between the individual blots was visualized by Ponceau Red staining – a reversible non-specific azo-dye-based protein staining - of the membrane blots using standard techniques [32].
Results
Both the PDH complex and the KGDH complex go through a three step catalytic cycle (Figure 1). A detailed structure-function analysis of these enzyme complexes is reviewed by Hengeveld and de Kok [22]. Briefly, the initial substrate dependent step (1) is the α-keto acid dehydrogenase reaction whereby the enzyme uses thiamin to oxidatively decarboxylate pyruvate or α-ketoglutarate (dependent upon the dehydrogenase) to generate CO2 and an acylated lipoic acid. The second acyltransferase step (2) of the catalytic cycle is carried out by the lipolylated E2 subunit that transfers the acyl group to coenzyme A, generating a reduced lipoic cofactor on the E2 enzyme subunit. This step is obviously dependent upon the presence of the cofactor to proceed. The third step is catalyzed by the dihydrolipoamide dehydrogenase subunit (3) that is common to all of the α-ketoacid dehydrogenase complexes and is dependent upon NAD+ to proceed. In the α-ketoglutarate dehydrogenase complex this constitutes a point of control for the whole citric acid cycle as this catalytic cycle is inhibited by the presence of excess NADH.
Figure 1.
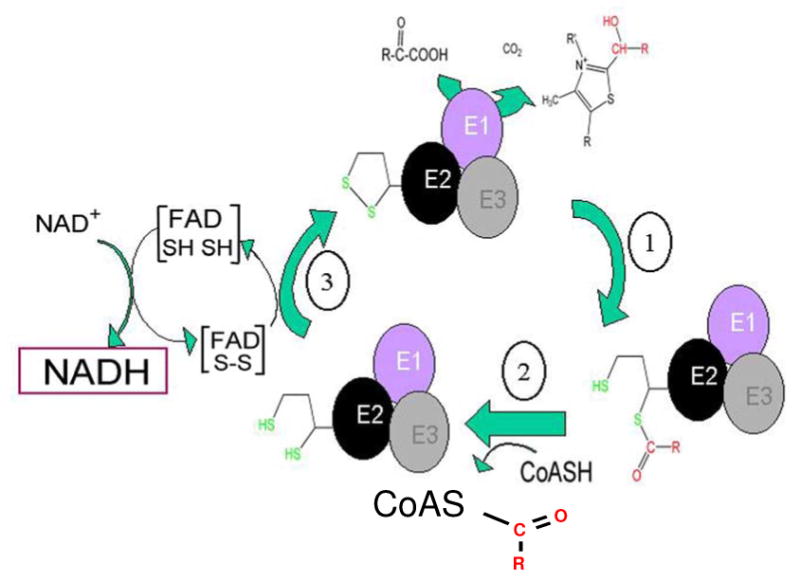
Reaction scheme for the α-ketoacid dehydrogenase complexes, where R=CH3 for the pyruvate dehydrogenase complex or R = HOOC-CH2-CH2 for the α-ketoglutarate dehydrogenase complex. The substrate specificity is conferred by the individual EI α-keto acid dehydrogenase subunit of each multienzyme complex. The second step in the enzymatic cycle is catalyzed by the lipoate-modified E2 enzyme, the acyl transferase and uses coenzyme A as a substrate. The third, dihydrolipoamide dehydrogenase, step regenerates the starting state of the multienzyme complex and generates NADH.
The lipoic acid group must be in the reduced state for effective enzyme inhibition by arsenite
With the commercially obtained enzymes we could hope at best for a mixture of states upon opening the bottle but in all probability the enzyme in its resting state – particularly once the buffer exchange has been completed – almost certainly contains oxidized lipoate group(s). We originally began these studies when we noticed that both dehydrogenase complexes exhibited a lag period before arsenite began to inhibit the enzyme, effectively not changing the initial reaction rate (v0) for the enzymes. In order to determine with which state of the enzyme arsenic reacts, we stalled the enzyme in state 1 by withholding substrate (pyruvate or α-ketoglutarate ) during the incubation period; in state 2 by withholding coenzyme A during the incubation period and in state 3 (with reduced lipoate group) by incubating the enzyme with arsenic as well as substrate and coenzyme A. In all cases the reaction began with the addition of NAD+, and the missing reactive component where appropriate. We determined that both enzymes needed to be in the enzymatic state that held the lipoate in the reduced state in order for arsenic to inhibit the initial reaction rate (Figure 2).
Figure 2.
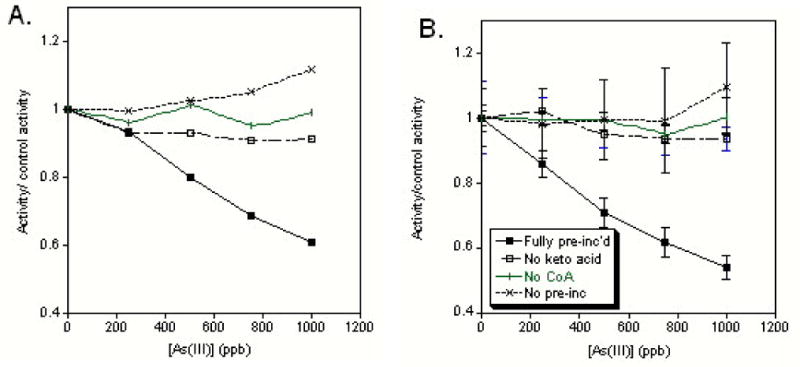
Variations on incubation conditions of (A) PDH or (B) KGDH with inorganic arsenite. Only when substrate (pyruvate or α-ketoglutarate) and CoASH (solid squares) were added during the pre-incubation period and the dehydrogenase complexes were stalled in the reduced lipoic acid form during incubation was inhibition observed.
Arsenite gives greater inhibition than any arsenate compound with both purified PDH and KGDH
Analysis of inhibition under these pre-incubation conditions with varying concentrations of all three arsenate compounds and arsenite found that only arsenite inhibited the initial rate of enzymatic NADH production when the enzyme complex is paused in state 3 with reduced lipoic acid groups (Figure 3).
Figure 3.
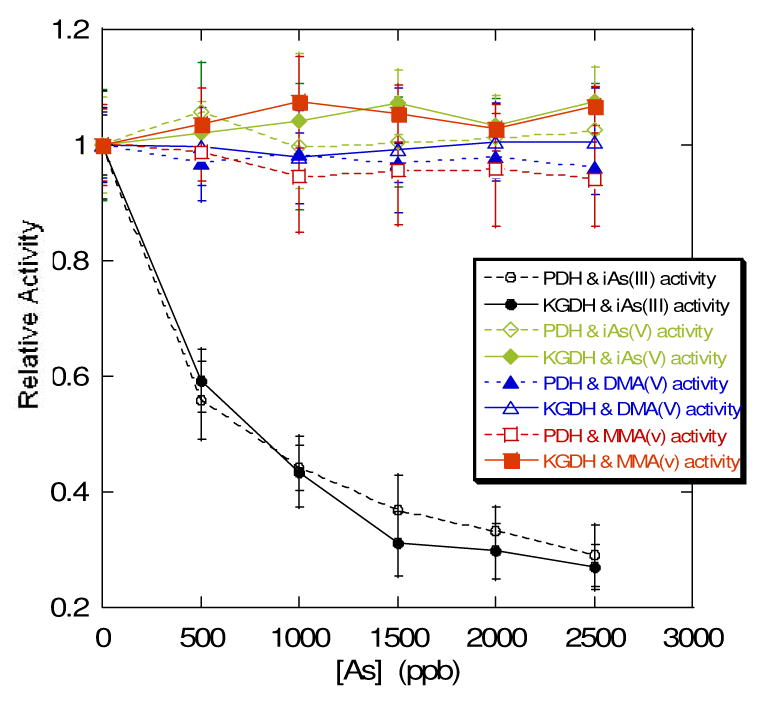
Comparison of initial enzyme velocity relative to control after a half hour incubation with the four different forms of arsenate and arsenite that were available commercially: sodium arsenite (circles), sodium arsenate (diamonds), cacodylic acid or dimethylarsenate (triangles), and monomethylarsenate (squares). PDH – hollow symbols, KGDH – solid symbols.
Arsenite inhibition was dose and time-dependent for both enzyme complexes. PDH was more sensitive to arsenicals than KGDH
Both enzymes were very strongly inhibited by the reduced monomethylarsenite species. While the inorganic arsenite complex inhibited both dehydrogenases approximately equivalently, the pyruvate dehydrogenase complex was more sensitive to the reduced monomethylarsenite than the α-ketoglutarate dehydrogenase complex (Figure 4A). Control reactions with potassium iodide (not shown) determined that enzyme inhibition was caused by the arsenite complex and not by the iodide counter-ion that was also introduced into the solutions with the synthetic monomethylarsenite diiodide complex. Initially we observed that the reduced dimethyl species inhibited neither enzyme particularly strongly although the pyruvate dehydrogenase complex was inhibited slightly at higher concentrations. However when we extended the pre-incubation period to an hour we found measurable differences in enzyme activity compared to non-arsenic treated controls and that the pyruvate dehydrogenase complex was far more sensitive to arsenite and dimethylarsenite than the α-ketoglutarate dehydrogenase complex (Figure 4B). We found that the level of inhibition caused by 250 ppb of sodium arsenite increased up to the longest time period measured of 165 minutes (not shown). In order to compare the enzymes we measured the amount of arsenical required for 50% inhibition of the initial reaction rate following an 30 min or a one hour pre-incubation (Table 1).
Figure 4.
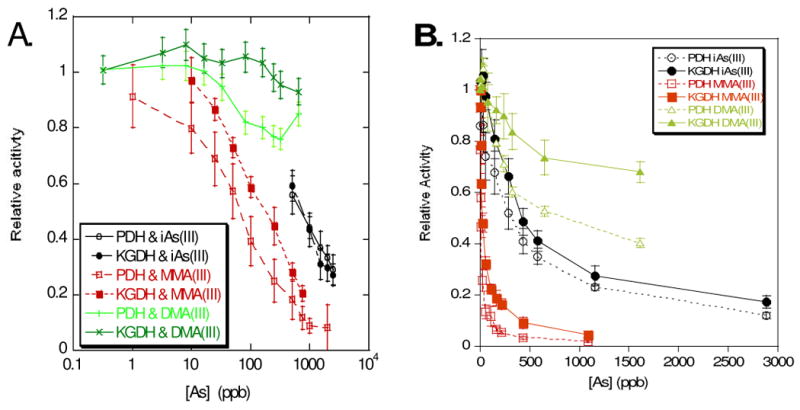
Comparison of relative enzyme velocity at (A) 30 min and (B) 60 min preincubation with substrate, coenzyme A and the arsenical indicated. PDH – hollow symbols, KGDH – solid symbols.
Loss of recognition by a lipoic acid-specific antibody indicates binding at the lipoic acid moiety of PDH
Increased enzyme inhibition with time is indicative of a binding mode of inhibition [33]. As the lipoic acid groups have historically been considered the target of arsenical binding to these enzymes we wanted to see if this binding would lead to the loss of the ability of the anti-lipoic acid antibody to recognize the moiety as it does when lipoic acid is modified by the addition of hydroxynonenol [34]. We did not observe any loss of lipoate recognition in the arsenite treated samples. Because of the nature of the experiment, this does not necessarily imply lack of arsenite binding at this site as the arsenite group may be too small to affect recognition by the comparatively large antibody. We did observe loss of recognition of the lipoic acid groups of the pyruvate dehydrogenase complex that were treated with monomethyl and dimethylarsenite, indicating that the presence of the methyl groups on the arsenical species is enough to alter antibody recognition at this site.
Discussion
The oxidation state of arsenic will determine the extent of inhibition of the respiratory 2-oxo-keto acid dehydrogenases as no pentavalent form of arsenate inhibited the purified enzyme complexes. Varying substrates in the incubation mixture to stall the dehydrogenase complexes at various stages of lipoic acid cycling established that the lipoic acid groups needed to be in the reduced form during incubation for inhibition of both PDH and KGDH by arsenite to be observed. This implies that KGDH and the homologous PDH are inhibited by arsenite modification of reduced lipoic acid. Stevenson et al. [35] also observed in passing that co-incubation the PDH complex with substrate (pyruvate) and coenzyme A had to occur for the enzyme to be inhibited by high concentrations of a synthetic p-amino-phenyl arsenoxide. While that study concluded that arsenical inhibition was reversible, our study using arsenite and the synthetic analogs of the biological metabolites of arsenic indicated a cumulative effects of the toxicity of arsenite that is kinetically interpreted as a binding and therefore irreversible mode of inhibition under the time scale of these experiments. For reduced arsenicals the enzyme must be in the third state of its catalytic cycle that contains the reduced lipoic acid groups that favor arsenic chelation. The enzyme active site in this state also contains a pair of reactive oxidized thiols that participate in the electron transfer of the next phase of the catalytic cycle, moving themselves through a reduced state [22]. Kitchen and Wallace observed that cysteines placed five or nine amino acids apart in a peptide also provide a high affinity arsenite binding site provided the cysteines are in the reduced state [36]. It is difficult to compare the flexible peptides of that study with the more fixed architecture of a protein active site. The spacing of seven amino acids between the two active cysteines would a priori indicate a potential high affinity binding site were the cysteines in the reduced state. As the cysteines share a dithiol bond throughout the catalytic cycle a better comparision may be with the highest affinity site reported, that of adjacent cysteines [36]. As the reduction of these cysteines requires catalytic turnover through this state it is unlikely that this site contains a high percentage population of reduced cysteines under our conditions. In order to test this hypothesis in our system however we would have to had pre-incubated the enzyme complex with NADH to drive this reaction step backwards and this would then prevent us measuring enzyme activity which we accomplished by monitoring NADH production. The enhanced sensitivity of PDH over KGDH to reduced arsenical complexes (vide infra) led us to propose the lipoic acid group as a primary binding site, while noting the possibility that these cysteines of subunit one provide a potential alternate site that we cannot formally rule out under the conditions reported here.
MMAIII was the strongest inhibitor of both PDH and KGDH
Due to the cumulative nature of arsenite inhibition we reported our observed concentrations required for 50% enzyme inhibition - or IC50 – at 30 minutes and one hour in order that we would be able to compare our work with those components of it that have reference values in the literature. The value for which there is comparative literature data is the ability of arsenite to inhibit the PDH complex. For arsenite inhibition of the pyruvate dehydrogenase complex our numbers are comparable to those of Hu et al. [9] who determined an IC50 of 5.6 μM. That study did not indicate the total time period over which they monitored the reaction and our numbers at 30 and 60 minutes of 9.39 μM and 3.56 μM bracket theirs and can be considered comparable. Petrick et al. [12] indicated that they monitored the extent of reaction after 30 minutes by trapping a radioactive reaction product. That we detected much greater sensitivity than they did (IC50 of 115.7 μM) we attribute to the fact that we pre-incubated the enzyme complex in order to favor the reaction at the lipoic acid group. Under any other conditions arsenite also competes with the enzymes own catalytic cycle for the opportunity to react at the lipoic acid group. It is therefore not surprising that we observed much greater sensitivity.
This enhanced sensitivity of these enzymes towards reduced arsenicals under our experimental conditions is still more marked for the monomethylarsenite species where we observed an IC50 of 0.092 μM at 30 minutes compared to the 62 μM level Petrick et al.[12] observed, indicating the importance of the availability of the lipoic acid group for the arsenite reactivity and inhibition. Both enzymes are particularly susceptible to inhibition by the monomethylarsenite complex. This markedly enhanced affinity of monomethylarsenite for reduced thiol groups is also seen by Ramadan et al. [37] who determined dissociation constants (Kd) of 13-106 nM compared with 5-124 μM found by Kitchin and Wallace for arsenite [36]. We believe this susceptibility of both enzymes is caused in part by the defensive properties of the methyl group keeping external chelators from removing the arsenite moiety or alternately from preventing its interaction with the lipoic acid group.
PDH is more sensitive to reduced arsenicals than KGDH
While arsenite inhibition was comparable between the two enzymes, the pyruvate dehydrogenase complex was more sensitive to monomethylarsenite than the α-ketoglutarate dehydrogenase complex. This was also reflected in the ability of monomethylarsenite to prevent recognition of the lipoic acid antibody. After two hours, recognition of the lipoate groups of the KGDH complex is diminished but it is almost completely abolished in the PDH complex. After 30 minutes pre-incubation with inorganic arsenite we do not see a great deal of difference in inhibition between the two dehydrogenases but after one hour the PDH complex is inhibited to a greater extent than the KGDH complex. This could superficially be attributed to the greater number of lipoic acid groups carried by each PDH subunit – two per E2 subunit compared with one for KGDH. While this was believed to be a redundancy in the enzyme it may be that both lipoic acids participate in and are important for catalytic turnover and NADH production. An alternative explanation could include the fourth regulatory subunit, “Protein X” (also called the E2 binding protein, E2BP) within the PDH complex that is not found in the KGDH complex. This subunit also carries a lipoyl modification. This protein is thought to mediate the regulatory processes by binding and activating the starvation induced [38] regulatory kinases [39-41]. In addition to the ability of Protein X to bind the E1 subunit to the complex, removal of the lipoyl regions of the E2 protein indicated that Protein X also contributes residual acetyltransferase activity to the complex [41,42] indicating a role of the lipoate group of Protein X in overall catalytic function of the complex and not just in its structural integrity and regulation. It is therefore plausible that the increased sensitivity of the PDH complex to inhibition arsenical inhibition may be due to the involvement of up to three lipoyl residues per unit for effective functioning of the catalytic cycle compared with one for KGDH.
Dimethylarsenite (dimethylarsinous acid) may operate by a different mechanism that arsenite or monomethylarsenite
Our results for arsenite and monomethylarsenite (methylarsonous acid) indicate that the species are acting via the same mechanism, with the monomethylarsenite species creating a more stable binary complex in the enzyme active site than arsenite. In contrast the inhibitory action of the reduced DMAIII seems to present a much more complicated profile indicating differing or additional mechanistic considerations. The initial weak inhibitory action of this species observed at thirty minutes is replaced after an additional 30 minutes (one hour pre-incubation) by a more enhanced inhibition that approaches number seen for arsenite at thirty minutes. After two hours we seen strong interference in the ability of the lipoic acid antibody to recognize the lipoic acid groups on both enzymes. One concern with any studies involving DMAIII is that it is fairly unstable towards re-oxidation to the DMAV, cacodylic acid (dimethylarsinic acid). We have observed a maximum useful lifetime of the reduced species of only a few hours in aqueous solutions (Martin, Fischer and Rauk – unpublished observations) and it would be expected that after one and two hour pre-incubations we would expect some population of the oxidized dimethylarsenic acid (DMAV) species. As we do not observe any inhibition of either enzyme complex by this species we can rule out any direct effect due to the formation of the oxidized species, particularly given that the period in which we see the least effect i.e. the first 30 minutes, is also the period during which little to no dimethylarsenic acid (DMAV) is formed.
Two further explanations arise for our observations. The first is that dimethylarsinic acid binds proteins via a different mechanism and a different site than either arsenite or monomethylarsenite. It is known that DMAIII is rapidly taken up and retained in the red blood cells of rats and that this is due to the increased sulfhydryl content of rat hemoglobin compared with many other species [reviewed in 47]. Not surprisingly DMAIII is bound to monothiols as the additional methyl groups preclude chelated binding. The affinity is sufficient for cysteinyl-arsenic compounds to be identified by collision-induced dissociation tandem mass spectrometry (CID MS/MS) after a one hour incubation [48]. It may therefore be possible that single cysteine groups of catalytic importance within these enormous dehydrogenase complexes bind DMAIII in a mechanism distinct from that of the other two reduced arsenic compounds. Such binding sites could be identified by similar CID MS/MS techniques.
The results we observed indicate that there is an additional lag period associated with the toxicity of dimethylarsinous acid (DMAIII) that is not observed for arsenite nor methylarsenite, nor is it reported in the hemoglobin studies. This lag period may also be consistent with DMAIII undergoing further enzyme-mediated speciation. A transmethylation mechanism would give rise to a population of the highly toxic monomethylarsenite species. As the inhibition that we observe after one hour is still at fairly high concentrations (10.4 μM at one hour) compared to that of monomethylarsenite, a small percentage formation of this highly toxic compound could explain the results we observe. The ability of dimethylarsinic acid (DMAIII) to perform such transmethylation reaction was observed by Grüter et al. during a study of complicating side reactions during hydride generation at low pH [49]. As the reactions mixtures are buffered, low pH conditions are only likely to be generated by the microenvironment created by the proton shuffling in the active site. The enzyme active site for the lipoamide dehydrogenasae reaction lies in a deep cleft at the subunit interface [22]. The proposed mechanism is that the reduced lipoyl group transfers electrons and a proton to a reactive disulfide on subunit one with an active site histidine on subunit two also acting as a proton acceptor. Therefore any formation of demethylated DMAIII (namely MMAIII) would occur as a function of enzymatic activity over time.
It is intriguing in this context to speculate on the fate of any newly released methyl moiety. A potential explanation is suggested by the results of Stevenson et al. [35]. Using high concentrations of an artificial bifunctional arsenoxide, BrCH2CONH-PhAsO they observed that both the arsenic group and methylating functionality were required for rapid, complete and irreversible inactiviation of PDH. They were only able to restore functionality to PDH using a strong arsenic chelator 2,3-dithioproanol if they used a monofunctional arsenical reagent p-amino phenylarsenoxide and similarly the use of an analogous methylating agent bromoacetylaniline (BrCH2CONHPh) had little effect on enzyme activity. As they required the bifunctionality of both arsenic species and methylating ability they proposed a mechanism whereby arsenic was chelated by the lipoic acid and then the lipoic acid-arsenical complex delivered the methylating end of the reagent into the active site, irreversibly methylating one of the active site subunits. These amino acids were later identified as a histidine and to a lesser extent a cysteine which they attributed to being from the isolated E3 subunit [50] and are now known to be on subunits one and two [22].
Our conditions do not allow us to distinguish whether one or the other potential mechanism predominates – or whether both play a contributing role. Future mass spectrometric analysis of the inhibited enzyme complex will be required to provide some answers. It must however be emphasized that organoarsenicals arise as a consequence of intracellular speciation of ingested inorganic arsenic (arsenite and arsenite) and if DMAIII is in the presence of even low concentrations of arsenite and monomethylarsenite, its effect will be minor when compared with these two species. Unless these effects are truly irreversible compared with organoarsenic binding, any effects of DMAIII are likely to be minor players in terms of the biological inhibition of the PDH and KGDH complexes by trivalent arsenicals.
Conclusions
We reported our data as concentration, as has been the custom, but also as parts per billion of arsenic as the drinking water standards are referenced in parts per billion. Correlating the drinking water standards with tissue, cellular and even mitochondrial arsenic concentrations is at best descriptive, particularly once metabolism and speciation is taken into account. Nevertheless we feel that this conversion gives an important basis for interpretation and comparison that is relevant to the social discussion of arsenic poisoning and toxicity. This becomes particularly appropriate when considering that the numbers – including ours - are often reported as IC50 numbers and that some enzyme inhibition occurs at levels well below the 50% number reported. Even considering IC50, monomethylarsenite (methylarsonous acid, MMAIII) is able to inhibit both enzymes complexes at concentrations below the former US drinking water standard and for the case of the PDH complex below the current drinking water standards. The extent to which arsenic, and particularly methylated arsenicals, accumulate in the mitochondria is still being determined and may be tissue specific but is in the 15-30% range [42-45]. As only a component of ingested arsenic is retained in the tissues, intracellular levels of arsenic following speciation are unlikely to approach the regulatory levels in populations exposed to ‘safe’ drinking water. Populations exposed to elevated arsenic levels in their drinking water may well be another case and it is also important to note that we have also observed that toxicity toward these enzymes is cumulative. In addition, Aposhian et al. [46] found that that administration of arsenic chelators to humans who ingested high concentrations of arsenic in drinking water led to the urinary excretion of disproportionately high concentration of monomethylarsenite, indicating that the tissue concentration of this toxic metabolite may be a disproportionately higher component than previously appreciated. Samikkannu et al. [27] suggested that the inhibitory action of arsenic trioxide As2O3 toward PDH was greater in HL60 cells than in the purified enzyme because it was acting indirectly through the formation of oxidative stress and not directly by binding to enzymatic dithiols like lipoic acid. The role that intramitochondrial oxidative stress may play in arsenic toxicity cannot be under-estimated. However under the conditions we have used reactive oxygen species should not be generated in our system and we see enzyme inhibition measured as IC50 numbers far lower than the values of 182 μM than they observed in the purified complex. Monomethylarsenite (methylarsonous acid) – which they did not use in their studies - also inhibited the purified complexes at much lower an IC50 (submicromolar) than the 2 μM they observed in the cell extract preparations. The lag time we had initially observed prior to undertaking our pre-incubation studies indicated that enzyme turnover is required for arsenite inhibition, including monomethylarsenite and our conditions that poise the enzyme complex in the state containing reduced lipoic acid groups allowed us to see a direct effect of inhibition on enzyme activity giving us lower IC50 number than those observed by many others. The extent of the effect of the cellular milieu on these numbers can be debated, however it is clear from our studies that in vitro both arsenite and monomethylarsenite bind to and inhibit these enzyme complexes with great efficacy and that these effects are cumulative.
One of the consistent observations of toxicity in arsenic treated cells and arsenic exposed tissues is the measurement of many parameters of oxidative stress [4, 51-59]. This provides something of a conundrum for mechanistic toxicology, as arsenic will not be able to generate reactive oxygen species in any straightforward manner such as the Fenton and pseudo-Fenton metals do and an indirect mechanism needs to be determined. It is known that arsenite generates excessive oxygen radical formation through increasing the activity of the cells surface NADPH dehydrogenases [51,60]. While hydrogen peroxide is a fairly diffusible molecule in biology this mechanisms suggests more of an extracellular and cell signaling effect. Intracellularly it has been proposed that arsenite depletes intracellular glutathione stores [61] although oxidative stress can also do this. As arsenite is toxic to cells far below concentrations that would allow stoichiometric depletion of intracellular glutathione, it has also been suggested that arsenic acts indirectly by inhibiting the glutathione recycling enzyme glutathione reductase [11]. It has already been observed that it there are potentially several hundred good targets for arsenic binding and toxicity [15]. The appeal of the enzymes studied here is therefore that they are indeed mitochondrial enzymes that are involved in respiration and the mitochondrial metabolism of oxygen. Depletion of their activity would lead to depleted mitochondrial NADH reserves and the inefficient metabolism of oxygen at the inner mitochondrial membrane. As oxidative stress itself is also proposed to inhibit these enzymes [27,34] this then would lead to a spiraling effect on these enzymes systems following exposure to toxic arsenical compounds. Whether the initial inhibition by arsenic is the initiating event in biology is yet to be determined however the ability of arsenite and dimethylarsenite to inhibit these enzymes at very low concentrations in a cumulative manner suggests this as a good probability.
Figure 5.
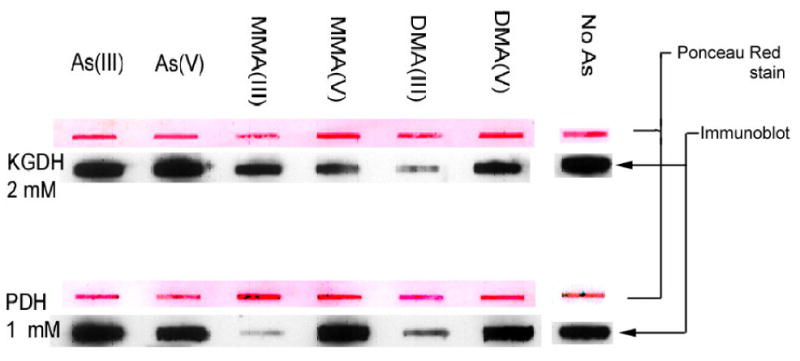
Slot blot of the α-ketoglutarate dehydrogenase complex or the pyruvate dehydrogenase complex, preincubated with TPP, CoA, substrate and the arsenical complex indicated. Ponceau red staining of each slot is also shown above the Western blotted band to indicate ∼equivalent loading.
Acknowledgments
This project was funded by NIH Grant Number RR-16455-01 from the BRIN Program of the National Center for Research Resources, Project IBS-CORE Undergraduate Research Fellowship provided by a grant from the Howard Hughes Medical Institute to the University of Montana, Centre for Biomedical Research Excellence (CoBRE) Grant 1P20RR017670, Program Project Three to Andrij Holian and Brooke Martin and National Institute of Environmental Health Sciences Grant No. ES10437 and ES014872 to Dr. Kent Sugden. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH. Thanks are also due to Drs Luke and Pam Szweda of Case Western University for the donation of anti-lipoic acid polyclonal antibody and to Dr. Bill Cullen for helpful advice regarding the synthesis of the reduced arsenical species.
Footnotes
Only reduced arsenic species were able to inhibit the enzymatic action of the pyruvate and α-ketoglutarate dehydrogenase complexes and only under condition that poised the enzyme complex in a catalytic state that contained reduced lipoic acid groups. Monomethylarsenite was by far the most potent compound toward enzyme inhibition.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abernathy CO, Liu Y, Longfellow D, Aposhian HV, Beck B, Fowler B, Goyer R, Menzer R, Rossman T, Thompson C, Waalkes M. Environ Health Perspect. 1999;107(7):593–597. doi: 10.1289/ehp.99107593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas DJ, Styblo M, Lin S. Toxicol Applied Pharmacol. 2001;176:127–144. doi: 10.1006/taap.2001.9258. [DOI] [PubMed] [Google Scholar]
- 3.Tchounwou PB, Patlolla AK, Centeno JA. Toxicol Pathol. 2003;31:575–588. doi: 10.1080/01926230390242007. [DOI] [PubMed] [Google Scholar]
- 4.Flora SJ. Clin Exp Pharmacol Physiol. 1999;26:865–869. doi: 10.1046/j.1440-1681.1999.03157.x. [DOI] [PubMed] [Google Scholar]
- 5.Rossman TG. Mutat Res. 2003;533:37–65. doi: 10.1016/j.mrfmmm.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Zhong CX, Mass MJ. Toxicol Letts. 2001;122:223–234. doi: 10.1016/s0378-4274(01)00365-4. [DOI] [PubMed] [Google Scholar]
- 7.Clewell HJ, Thomas RS, Gentry PR, Crump KS, Kenyon EM, El-Masri HA, Yager JW. Toxicol Applied Pharmacol. 2007;222:388–398. doi: 10.1016/j.taap.2007.03.021. [DOI] [PubMed] [Google Scholar]
- 8.Snow E, Sykora P, Durham TR, Klein CB. Toxicol Applied Pharmacol. 2005;207:S557–S564. doi: 10.1016/j.taap.2005.01.048. [DOI] [PubMed] [Google Scholar]
- 9.Hu Y, Su L, Snow ET. Mutat Res. 1998;408:203–218. doi: 10.1016/s0921-8777(98)00035-4. [DOI] [PubMed] [Google Scholar]
- 10.Chouane S, Snow ET. Chem Res Toxicol. 2001;14:517–522. doi: 10.1021/tx000123x. [DOI] [PubMed] [Google Scholar]
- 11.Styblo M, Serves SV, Cullen WR, Thomas DJ. Chem Res Toxicol. 1997;10:27–33. doi: 10.1021/tx960139g. [DOI] [PubMed] [Google Scholar]
- 12.Petrick JS, Jagadish B, Mash EA, Aposhian HV. Chem Res Toxicol. 2001;14:651–656. doi: 10.1021/tx000264z. [DOI] [PubMed] [Google Scholar]
- 13.Schiller CM, Fowler BA, Woods JS. Environ Health Perspect. 1977;19:205–207. doi: 10.1289/ehp.7719205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knowles FC. Arch Biochem Biophys. 1985;242(1):1–10. doi: 10.1016/0003-9861(85)90472-2. [DOI] [PubMed] [Google Scholar]
- 15.Kitchin K, Wallace J. J Inorg Biochem. 2008;102:532–539. doi: 10.1016/j.jinorgbio.2007.10.021. [DOI] [PubMed] [Google Scholar]
- 16.Peters RA, Sinclair HM, Thompson RHS. Biochem J. 1946;40:516–524. [PMC free article] [PubMed] [Google Scholar]
- 17.Stocken LA, Thompson RHS. Physiol Rev. 1949;29:168–194. doi: 10.1152/physrev.1949.29.2.168. [DOI] [PubMed] [Google Scholar]
- 18.Cullen WR, McBride BC, Reglinski J. J Inorg Biochem. 1984;21:179–194. [Google Scholar]
- 19.Carter DE, Aposhian HV, Gandolfi AJ. Toxicol Applied Pharmacol. 2003;193:309–334. doi: 10.1016/j.taap.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 20.Dill K, Adams ER, O'Conner RJ, McGowan EL. Chem Res Toxicol. 1989;2:181–185. doi: 10.1021/tx00009a010. [DOI] [PubMed] [Google Scholar]
- 21.von Döllen A, Strasdeit H. Eur J Inorg Chem. 1998:61–66. [Google Scholar]
- 22.Hengeveld AF, de Kok A. Current Med Chem. 2002;9:499–520. doi: 10.2174/0929867023370996. [DOI] [PubMed] [Google Scholar]
- 23.Mastrogiacomo F, Bergeron C, Kish SJ. J Neurochem. 1993;63(6):2007–2014. doi: 10.1111/j.1471-4159.1993.tb07436.x. [DOI] [PubMed] [Google Scholar]
- 24.Park LCH, Calingasan NY, Sheu KFR, Gibson GE. Anal Biochem. 2000;277:86–93. doi: 10.1006/abio.1999.4359. [DOI] [PubMed] [Google Scholar]
- 25.Gibson GE, Park LCH, Sheu KFR, Blass JP, Calingasan NY. Neurochem Int. 2000;36:97–112. doi: 10.1016/s0197-0186(99)00114-x. [DOI] [PubMed] [Google Scholar]
- 26.Brouwer OF, Onkenhout W, Edelbroek PM, de Kom JF, de Wolff FA, Peters AC. Clin Neurol Neurosurg. 1992;94(4):307–310. doi: 10.1016/0303-8467(92)90179-7. [DOI] [PubMed] [Google Scholar]
- 27.Samikkannu T, Chen CH, Yih LH, Wang ASS, Lin SY, Chen TS, Jan KJ. Chem Res Toxicol. 2003;16:409–414. doi: 10.1021/tx025615j. [DOI] [PubMed] [Google Scholar]
- 28.Ramanathan K, Shila S, Kumaran S, Panneerselvam J. Nutr Biochemistry. 2003;14:416–420. doi: 10.1016/s0955-2863(03)00076-7. [DOI] [PubMed] [Google Scholar]
- 29.Styblo M, del Razo LM, Vego L, Germolec DR, LeCluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ. Arch Toxicol. 2000;74:289–299. doi: 10.1007/s002040000134. [DOI] [PubMed] [Google Scholar]
- 30.Styblo M, del Razo LM, LeCluyse EL, Hamilton GA, Wang C, Cullen WR, Thomas DJ. Chem Res Toxicol. 1999;12:560–565. doi: 10.1021/tx990050l. [DOI] [PubMed] [Google Scholar]
- 31.Goddard AE. A Textbook of Inorganic Chemistry. In: Friend JN, editor. Organometallic compounds, Part II Derivatives of Arsenic. XI. Charles Griffin & Co.; London: [Google Scholar]
- 32.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning, a Laboratory Manual. 2nd. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 33.Palmer T. In: Understanding Enzymes. 2nd. Wiseman A, editor. Ellis Horwood Publishers; West Sussex, England: Series editor. [Google Scholar]
- 34.Humphries KM, Sweda LI. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 35.Stevenson KJ, Hale G, Perham RN. J Am Chem Soc. 1978;17(11):2189–2192. doi: 10.1021/bi00604a026. [DOI] [PubMed] [Google Scholar]
- 36.Kitchin KT, Wallace K. J Biochem Molecular Toxicology. 2006;20(1):35–38. doi: 10.1002/jbt.20112. [DOI] [PubMed] [Google Scholar]
- 37.Ramadan D, Cline DJ, Bai S, Thorpe C, Schneider JP. J Am Chem Soc. 2007;129:2981–2988. doi: 10.1021/ja067068k. [DOI] [PubMed] [Google Scholar]
- 38.Wu P, Blair PV, Sato J, Jaskiewicz J, Popov KM, Harris RA. Arch Biochem Biophys. 2000;381(1):1–7. doi: 10.1006/abbi.2000.1946. [DOI] [PubMed] [Google Scholar]
- 39.Rahmatullah M, Jilka JM, Radke GA, Roche TE. J Biol Chem. 1986;261(14):6515–6523. [PubMed] [Google Scholar]
- 40.Rahmatullah M, Gopalakrishnan S, Radke GA, Roche TE. J Biol Chem. 1989;264(2):1245–1251. [PubMed] [Google Scholar]
- 41.Rahmatullah M, Radke GA, Andrews PC, Roche TE. J Biol Chem. 1990;265(24):14512–14517. [PubMed] [Google Scholar]
- 42.Sanderson SJ, Miller C, Lindsay JG. Eur J Biochem. 1996;236:68–77. doi: 10.1111/j.1432-1033.1996.00068.x. [DOI] [PubMed] [Google Scholar]
- 43.Fowler BA, Woods JS, Schiller CM. Environ Health Perspec. 1977;19:197–204. doi: 10.1289/ehp.7719197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vahter M, Marafante E. Biol Trace Elem Res. 1989;21:233–239. doi: 10.1007/BF02917258. [DOI] [PubMed] [Google Scholar]
- 45.Dopp E, von Recklinghausen U, Hartman LM, Stueckradt I, Pollok I, Rabieh S, Hao L, Nussler A, Katier C, Hirner AV, Rettenmeier AW. Drug Metab Dispos. 2008;36(5):971–979. doi: 10.1124/dmd.107.019034. [DOI] [PubMed] [Google Scholar]
- 46.Aposhian HV, Zheng B, Aposhian MM, Le XC, Cebrian ME, Cullen W, Zakharyan RA, Ma M, Dart RC, Cheng Z, Andrewes P, Yip L, O'Malley GF, Maiorino RM, Van Voorhies W, Healy SM, Titcomb A. Toxicol Appl Pharmacol. 2000;165(1):74–83. doi: 10.1006/taap.2000.8922. [DOI] [PubMed] [Google Scholar]
- 47.Aposhian HV, Aposhian MM. Chem Res Toxicol. 2006;19(1):1–15. doi: 10.1021/tx050106d. [DOI] [PubMed] [Google Scholar]
- 48.Lu M, Wang H, Wang Z, Li XF, Le XC. J Proteome Res. 2008;7:3080–3090. doi: 10.1021/pr700662y. [DOI] [PubMed] [Google Scholar]
- 49.Grüter UM, Hitzke M, Kresimon J, Hirner AV. J Chromatog A. 2001;938:225–236. doi: 10.1016/s0021-9673(01)01342-5. [DOI] [PubMed] [Google Scholar]
- 50.Adamson SR, Robinson JA, Stevenson KJ. Biochemistry. 1984;23:1269–1274. doi: 10.1021/bi00301a039. [DOI] [PubMed] [Google Scholar]
- 51.Lynn S, Gurr JR, Lai HT, Jan KY. Circ Res. 2000;86(5):514–519. doi: 10.1161/01.res.86.5.514. [DOI] [PubMed] [Google Scholar]
- 52.Helleday T, Nilsson R, Jenssen D. Environ Mol Mutagen. 2000;35(2):114–122. doi: 10.1002/(sici)1098-2280(2000)35:2<114::aid-em6>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 53.Beckman G, Beckman L, Nordenson I. Environ Health Perspect. 1977;19:145–146. doi: 10.1289/ehp.7719145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matsui M, Nishigori C, Toyokuni S, Takada J, Akaboshi M, Ishikawa M, Imamura S, Miyachi Y. J Invest Dermatol. 1999;112(1):26–33. doi: 10.1046/j.1523-1747.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 55.Wang TS, Kuo CF, J KY, Huang H. J Cell Physiol. 1996;169:256–268. doi: 10.1002/(SICI)1097-4652(199611)169:2<256::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 56.Barchowsky A, Dudek EJ, Treadwell MD, Wetterhahn KE. Free Radic Biol Med. 1996;21(6):783–790. doi: 10.1016/0891-5849(96)00174-8. [DOI] [PubMed] [Google Scholar]
- 57.Lee TC, Ho Arch IC. Toxicol. 1995;69(7):498–504. doi: 10.1007/s002040050204. [DOI] [PubMed] [Google Scholar]
- 58.Samikkannu T, Chen CH, Yih LH, Wang ASS, Hei TK, Filipic M. Free Radical Biol Med. 2004;37(5):574–581. doi: 10.1016/j.freeradbiomed.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Huang C, Ke Q, Costa M, Shi X. Molec Cell Biochem. 2004;255:57–66. doi: 10.1023/b:mcbi.0000007261.04684.78. [DOI] [PubMed] [Google Scholar]
- 60.Smith KR, Klei LR, Barchowsky A. Am J Lung Cell Mol Physiol. 2001;280:L442–L449. doi: 10.1152/ajplung.2001.280.3.L442. [DOI] [PubMed] [Google Scholar]
- 61.Haugen AC, Kelley R, Collins JB, Tucker CJ, Deng C, Afshari CA, Brown JM, Ideker T, Van Houten B. Genome Biology. 2004;5:R95.1–R95.18. doi: 10.1186/gb-2004-5-12-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]