

Abstract
Interleukin-6 (IL-6) is a pleiotropic cytokine responsible for many different processes including the regulation of cell growth, apoptosis, differentiation, and survival in various cell types and organs, including the heart. Recent studies have indicated that IL-6 is a critical component in the cell-cell communication between myocytes and cardiac fibroblasts. In this study, we examined the effects of IL-6 deficiency on the cardiac cell populations, cardiac function, and interactions between the cells of the heart, specifically cardiac fibroblasts and myocytes. To examine the effects of IL-6 loss on cardiac function, we used the IL-6−/− mouse. IL-6 deficiency caused severe cardiac dilatation, increased accumulation of interstitial collagen, and altered expression of the adhesion protein periostin. In addition, flow cytometric analyses demonstrated dramatic alterations in the cardiac cell populations of IL-6−/− mice compared with wild-type littermates. We observed a marked increase in the cardiac fibroblast population in IL-6−/− mice, whereas a concomitant decrease was observed in the other cardiac cell populations examined. Moreover, we observed increased cell proliferation and apoptosis in the developing IL-6−/− heart. Additionally, we observed a significant decrease in the capillary density of IL-6−/− hearts. To elucidate the role of IL-6 in the interactions between cardiac fibroblasts and myocytes, we performed in vitro studies and demonstrated that IL-6 deficiency attenuated the activation of the STAT3 pathway and VEGF production. Taken together, these data demonstrate that a loss of IL-6 causes cardiac dysfunction by shifting the cardiac cell populations, altering the extracellular matrix, and disrupting critical cell-cell interactions.
Keywords: cytokines, interleukin-6, cardiac fibroblasts, myocytes
heart disease is the leading cause of morbidity and mortality in the United States with ∼11% of the adult population reporting some type of heart disease (36). Interactions between cardiac myocytes, fibroblasts, and the extracellular matrix (ECM) maintain the function and form of the organ from both a biochemical and a biomechanical standpoint through autocrine/paracrine signaling and direct cell-cell interactions (4, 11, 12, 21, 22, 31, 33, 34, 41, 44). Alterations in these signals or biomechanical input can bring about deleterious, adaptive, and/or compensatory changes in the heart.
IL-6 is a pleiotropic cytokine that regulates numerous biological functions in a cell-specific manner in various organs, including the heart (28). The IL-6 family of cytokines includes IL-6, IL-11, IL-27, leukemia-inhibitory factor-1, oncostatin-M, ciliary neurotrophic factor, and cardiotrophin-1 (1, 10, 14, 28, 43, 45). The biological activity of IL-6 is regulated by binding to the IL-6Rα/gp130 signal transduction complex (28). Upon binding and homodimerization, the gp130 subunit triggers the activation of several signal transduction pathways, including the Janus-activated kinase-signal transducer and activator of transcription (JAK-STAT) pathway (28).
Accumulating evidence from several studies suggests that the IL-6-gp130-JAK-STAT pathway is critical in myocardial function and cardioprotection (9, 24, 25, 49). Cardiac hypertrophy results in an increase in IL-6 expression (3, 8, 37). In addition, in vivo studies have demonstrated that the continuous activation of the gp130 receptor via constant IL-6/soluble (s)IL-6Rα stimulation causes myocardial hypertrophy (26). Moreover, studies in rats have demonstrated that constant IL-6/sIL-6Rα stimulation can reduce infarct size and protect against myocyte apoptosis (35). Previous studies have also demonstrated that IL-6 plays an important role in cardiac myocyte function and gene expression (2, 5, 15, 19, 33, 47, 50, 51). However, despite all of these studies, the functional effects of IL-6 loss on cardiac fibroblast-myocyte interactions remain largely unknown.
Recent data from our laboratory have demonstrated that the cell populations of the heart fluctuate during development and progression to the adult (4). Thus alterations in autocrine/paracrine factors that affect the structure and function of the myocardium could also cause changes in the cardiac cell populations. Therefore, the aim of this study was to 1) characterize IL-6 deficiency on cardiac function; 2) study the resultant cell population changes; and 3) more specifically, characterize the effects of IL-6 on cardiac fibroblast function and fibroblast-myocyte interactions. Here we demonstrate that IL-6 deficiency has a deleterious effect on the heart, alters the cardiac cell populations, capillary density, and cardiac fibroblast-myocyte interactions, and acts via the STAT3 signaling pathway to regulate vascular endothelial growth factor (VEGF) levels.
MATERIALS AND METHODS
Animal procedures.
Animals were euthanized via cervical dislocation. The experiments detailed in these studies involving animals were submitted and approved by the University of South Carolina School of Medicine's Institutional Animal Care and Use Committee. This investigation conforms to the Guide for the Care Use of Laboratory Animals, published by the National Institutes of Health (NIH Publication No. 85-23, Revised 1996). For this study we used commercially available IL-6−/− mice (Jackson) on a C57BL/6 background and aged-matched wild-type (WT) littermates.
Echocardiographic analysis.
Transthoracic echocardiography was preformed on mice at neonatal days 5 and 15 (D5 and D15, respectively) and 12–14 wk (adult). Briefly, mice were anesthetized using 3% isoflurane and then moved to a biofeedback warming station that measured heart rate, maintained core body temperature, and had nose cone anesthesia (3% isoflurane). Ultrasound gel was placed on the chest of the animal, and echocardiography measurements were obtained using a 15-mHz probe for adults, a 45-mHz probe for neonates, and the VEVO 770 software package (Visual Sonics). M-mode tracings were taken and measured for posterior and anterior wall thicknesses, as well as the interdimensional space for both systole and diastole using the VEVO 770 software package. Ejection fraction and fractional shortening were calculated as previously described (36, 48). An average of three to five cardiac cycles per animal were analyzed.
Western blot analysis.
Whole hearts or in vitro coculture samples were isolated, snap frozen, and homogenized in 25 mM HEPES, 300 mM NaCl, 0.2 M EDTA, 0.1% Triton X-100, and 50 μM Na-orthovanadate, supplemented with a protease inhibitor cocktail (Sigma, St. Louis, MO). Samples were centrifuged for 5 min at 14,000 g, supernatants were isolated, and protein concentration was determined. The protein was loaded and separated electrophoretically using 7.5 to 12% polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% BSA and then blotted with a rabbit anti-periostin IgG (a gift from Dr. Roger Markwald), anti-STAT3 (06-596, Upstate), anti-phospho-STAT3 (9131S, Cell Signaling), anti-MAPK 42/44 (9102, Cell Signaling), anti-phospho-MAPK 42/44 (9106S, Cell Signaling), anti-AKT (9272, Cell Signaling), anti-phospho-AKT (9271S, Cell Signaling), and anti-GAPDH (MAB374, Chemicon), all at 1:1,000. Kodak Molecular Imaging software (Kodak, Rochester, NY) was used to analyze blots.
Morphological and histological analysis.
Whole hearts were isolated from aged-matched WT and IL-6−/− mice (12 wk). Hearts were rinsed in phosphate-buffered saline (PBS) and imaged using a Leica S8Apo dissecting microscope with a Nikon Coolpix digital camera. Following the imaging, the hearts were fixed in fresh 4% paraformaldehyde supplemented with 50 mM potassium for 60 min and placed into either paraffin or acrylamide blocks. From acrylamide blocks, 100-μm sections were cut and imaged on a Leica S8Apo dissecting scope as above. From paraffin blocks, 5-μM sections were cut and stained according to Masson's trichrome standard protocol (42). From control and experimental groups, 10 images (×40) from the right ventricle, interventricular septum, and left ventricle were collected and analyzed for the relative amount and location of collagen in each heart section. Color (24 bit) images were analyzed using MetaMorph 6.1 (Molecular Devices, Downingtown, PA) software. The blue-staining collagen was thresholded as a region of interest (ROI) using the hue-saturation intensity color model in the Set Color Threshold subroutine to establish the hue range of blue-stained collagen to be accepted for analysis. The area of the thresholded collagen ROI was defined as the inclusive region for measurement. The area of the remaining cardiac tissue (all red-stained tissue with holes in the tissue was excluded) was measured as the exclusive ROI in the tissue section. The area of collagen and area of cardiac tissue in the section were recorded using the Show Regions Statistics subroutine. The percentage of collagen present in the section was determined by the ratio of inclusive area (I) of the blue-stained ROI and the exclusive area (E) of the red stained cardiac tissue ROI times 100 (%collagen = [I/E] × 100). The ratios representing the percentage of collagen in each section were analyzed and compared using SigmaStat analytic software (SSPS).
Real-time PCR analysis.
Total RNA was isolated from IL-6−/− or WT age-matched (12 wk) littermates using TRIzol reagent (Invitrogen) as described by the manufacturer. Real-time PCR analyses were performed using a Bio-Rad RT-PCR kit for probes and the Bio-Rad IQ5 Real Time Thermocycler as described by the manufacturer. We used primer/probe sets that were specific for murine periostin and acidic ribosomal phosphoprotein P0, that latter of which served as our internal control for normalization. Primer and probe sequences are available upon request from T. A. Baudino (troy.baudino@uscmed.sc.edu).
Hydroxyproline assay.
A modified hydroxyproline assay was performed as previously described (4, 16). Briefly, samples were isolated, weighed, and freeze dried overnight in a Labconco Freeze Dryer 3. Samples were hydrolyzed overnight in 6 N HCl at 120°C, washed, and incubated with Ehrlich reagent for 30 min at 80°C. A standard curve was generated using known samples ranging from 2 and 100 μg. Samples were read using a Bio-Rad Benchmark Plus plate reader set at 557 nm. Concentrations of isolated samples were calculated against known standards. Total hydroxyproline per milligram of total heart was then calculated.
Flow cytometric analysis.
WT and IL-6−/− hearts were prepared and labeled as previously described (4). Briefly, hearts were isolated and dispersed into heterogeneous single-cell suspensions. Cells were then immunolabeled for each cell type with known molecular markers: discoidin domain receptor (N-20, Santa Cruz) for cardiac fibroblasts, α-myosin heavy chain (ab-15, AbCam) for cardiac myocytes, CD31 (37-0700, Zymed) for endothelial cells, and α-smooth muscle actin (MAB1420, R&D Systems) for vascular smooth muscle cells. All immunolabels were conjugated to quantum dots as described by the manufacturer (Invitrogen).
Cardiac cell isolation for in vitro studies.
Hearts were isolated from neonatal IL-6−/− or WT mice (1 to 3 days old) and rinsed in ice-cold Moscona's solution. Hearts were placed into Krebs-Ringer buffer I (KRB-I), containing 136 mM NaCl, 28.6 mM KCl, 1.9 mM NaHCO3, 0.08 mM NaHPO4, 1 mg/ml BSA, 2 mg/ml glucose, and 20 U/ml penicillin-streptomycin (pH 7.4,) and minced into small pieces using scissors. Minced tissue was then placed into a sterile 150-ml Erlenmeyer flask and incubated with gentle agitation at 37°C for 10 min. The supernatant was collected, and the remaining tissue was immersed in KRB-II solution, consisting of KRB-I plus 120 U/ml collagenase type 2 and 20 mg/ml BSA, for subsequent digestions. The supernatants were pooled and filtered through a 40-μm nylon mesh filter. The filtered supernatant was then centrifuged at 1,000 g for 5 min at 4°C, the supernatant was removed, and the cells were placed into myocyte media (DMEM, 5% normal bovine serum and 8% horse serum). Cells were plated for 40 min at 37°C and 5% CO2-95% room air. The media containing nonadherent cells was then removed, and the adherent cells were considered to be pure cardiac fibroblasts. Cardiac fibroblasts were then grown to 80% confluence in fibroblast medium (DMEM, 20% fetal bovine serum) at 37°C and 5% CO2-95% room air. Nonadherent cells were then considered to be pure cardiac myocytes and plated as needed for subsequent experiments.
In vitro adhesion assay.
Cardiac myocytes were plated at 1 × 106 cells onto aligned collagen on a 60-mm tissue culture dish and allowed to adhere for 48 h at 37°C and 5% CO2-95% room air. Cardiac fibroblasts were then plated onto cardiac myocytes at 5 × 105 cells per plate. Cells were then returned to the incubator and allowed to adhere for 4 or 8 h. At each time point, the nonadherent cells were counted and quantified. Periostin-conditioned media was a kind gift from Dr. Chip Norris at the Medical University of South Carolina.
In vitro signaling assays.
Cells were cocultured as previously described (7). Briefly, 1 × 106 myocytes were plated onto aligned collagen and allowed to adhere for 48 h. Fibroblasts, 5 × 105 IL-6 deficient or WT, were then plated onto cardiac myocytes and allowed to adhere for 24 h. Cocultures were serum starved for 24 h and then stimulated with IL-6 (R&D Systems) and sIL-6Rα (Prospecbio), both at 25 ng/ml or platelet-derived growth factor (PDGF) (Sigma) at 10 ng/ml for 15 min. Cell lysates were subjected to Western blot analyses as listed above (see Western blot analysis).
In vitro VEGF ELISA.
Cells were cocultured and stimulated as described above for 24 h, and the media was collected. The samples were analyzed using a VEGF Duoset ELISA kit (R&D Systems), as per the manufacturer's instructions. The limits of detection for this kit were between 16 and 1,000 pg/ml, with a linear range of 125–750 pg/ml, and our samples fell within this range (250–500 pg/ml).
In vivo proliferation and apoptosis assays.
Hearts from D5 WT and IL-6−/− mice were isolated and fixed, and 7-μm paraffin sections were cut. For proliferative analyses, D5 sections were stained for phospho-histone H3 (1:50, SC8656R; Santa Cruz) and for apoptosis sections were stained for cleaved-poly(ADP-ribose) polymerase (1:50, 9544S, Cell Signaling). The sections were counterstained with tropomyosin (1:50, T9283, Sigma) and wheat germ agglutinin-Alexa 488 (1:200, W11261, Invitrogen) and 4,6-diamidino-2-phenylindole. The sections were then imaged using a Zeiss LSM 510 META microscope and analyzed using MetaMorph. Tropomyosin-positive cells were counted as myocytes.
Cardioangiography.
Adult mice were anesthetized using a ketamine and xylazine cocktail. Hearts were perfused with saline-heparin to clear the vasculature and then with FluroSpheres red (580/605 nm) (F8801; Molecular Probes). Sections (50 μm) were cut and imaged using a Zeiss LSM 510 META microscope. The images were analyzed using MetaMorph 6.1 software. Briefly, the red channel was separated and the threshold was set as the ROI. The area analysis was then compared with the total area in the image, and the percentage was determined.
Statistical analysis.
The data obtained from all analyses were measured for significance using Student's t-test or ANOVA with a Mann-Whitney test. Analysis was performed on SigmaStat software (SYSTAT Software).
RESULTS
IL-6−/− hearts display gross cardiac abnormalities and reduced cardiac function.
We observed gross anatomical differences in the IL-6−/− hearts compared with WT hearts (Fig. 1, A and B). Whereas the IL-6-deficient hearts appeared larger in size, in actuality the heart weight-to-body weight ratio (HW/BW) and heart weight-to-tibia length ratio (HW/TL) in these animals were significantly decreased (Fig. 1, E and F, and Table 1). No changes in total body weight or tibia length were observed between WT and IL-6−/− animals (Table 1). Upon sectioning the hearts, we observed a significant thinning of the left ventricular wall and dilated chamber size in the IL-6−/− hearts (Fig. 1, C and D). To quantify wall thickness and examine cardiac function in these animals, we performed a transthoracic echocardiography M-mode image analysis. An echocardiographic analysis revealed that IL-6−/− mice had a dramatic thinning in both the anterior and posterior walls of the left ventricle during the diastolic and systolic phases compared with WT mice (P < 0.001, Table 2). Additionally, IL-6−/− mice were observed to have a significant decrease in both fractional shortening and ejection fraction compared with WT littermates (P < 0.001, Table 2). To examine the effects of IL-6 deficiency in the developing hearts, we carried out ultrasound analysis on D5 and D15 hearts. We observed that the cardiac dilatation was reduced in D5 IL-6−/− neonates but was not significantly different from WT counterparts. However, D15 IL-6−/− neonates were observed to have cardiac dysfunction similar to that observed in adult animals (Table 2). Furthermore, at D5 and D15, we observed decreases in cardiac ejection fraction and fractional shortening similar to those observed in adult animals. These findings demonstrate that IL-6−/− mice have a progressively reduced cardiac function compared with normal animals.
Fig. 1.

Interleukin-6 (IL-6) loss causes cardiac abnormities. A and B: representative images of adult wild-type (WT) and IL-6−/− hearts, respectively. C and D: representative 100-μm sections of left (LV) and right ventricular (RV) chambers of WT and IL-6−/− identically fixed hearts, respectively. E: analysis of heart weight (HW) normalized to body weight (BW). F: analysis of HW compared with tibia length (TL). Scale bars set to 1 mm. N = 5 animals per condition. *P < 0.05 vs. WT.
Table 1.
HW, BW, and TL analyses: WT vs. IL-6−/− mice
| Genotype | BW, g | HW, mg | TL, mm | HW/BW, mg/g | HW/TL, mg/mm |
|---|---|---|---|---|---|
| WT | 22.8±1.48 | 137.2±6.53 | 16.6±0.13 | 6.0±0.30 | 8.3±0.44 |
| IL-6−/− | 21.6±1.39 | 101.6±5.77*† | 17.2±0.70 | 4.6±0.19*† | 5.9±0.52*† |
Values are means ± SD; N = 5 animals per condition. Heart weight (HW), body weight (BW), tibia length (TL), and corresponding ratio for adult age-matched animals are shown. *P < 0.05, †P < 0.01, and *†P < 0.001 vs. wild-type (WT).
Table 2.
Cardiac ultrasound analyses: WT vs. IL-6−/− mice
| Age/Genotype | LVAWd, mm | LVAWs, mm | LVPWd, mm | LVPWs, mm | %FS | %EF |
|---|---|---|---|---|---|---|
| Day 5 | ||||||
| WT | 0.44±0.11 | 0.76±0.22 | 0.47±0.03 | 0.68±0.08 | 45.98±5.22 | 80.03±5.08 |
| IL-6−/− | 0.38±0.04 | 0.68±0.05 | 0.42±0.05 | 0.60±0.09 | 37.78±3.28* | 71.01±4.56* |
| Day 15 | ||||||
| WT | 0.54±0.05 | 0.92±0.07 | 0.59±0.04 | 0.92±0.12 | 48.50±8.47 | 80.03±5.08 |
| IL-6−/− | 0.43±0.09* | 0.70±0.11*† | 0.54±0.04* | 0.71±0.07† | 31.50±5.81*† | 61.15±7.93*† |
| Adult | ||||||
| WT | 0.91±0.08 | 1.52±0.22 | 0.96±0.02 | 1.39±0.19 | 43.36±7.80 | 74.70±7.96 |
| IL-6−/− | 0.64±0.11*† | 1.02±0.19*† | 0.74±0.10*† | 1.01±0.12*† | 31.79±5.10*† | 60.95±7.03*† |
Values are means ± SD; N = 7–10 animals per condition. Echocardiographic analyses of IL-6−/− vs. WT mice are shown: M-mode image analysis for neonatal day-5, day-15, and adult animals. LVAWd and LVAWs, left ventricular anterior walls during diastolic and systolic phases, respectively; LVPWd and LVPWs, left ventricular posterior walls during diastolic and systolic phases, respectively; FS, fractional shortening; EF, ejection franction.
P < 0.05,
P < 0.01, and
P < 0.001 vs. WT.
IL-6−/− hearts have increased interstitial collagen deposition.
To further evaluate the cardiac dysfunction observed in IL-6−/− mice, we analyzed the interstitial collagen content using both histological and biochemical approaches. Masson's trichrome staining revealed that interstitial collagen was markedly increased in IL-6−/− mice relative to WT littermates (Fig. 2, A and B). When these images were further examined using morphometric analysis, we observed a significant increase in collagen deposition in the left and right ventricular free walls of the IL-6−/− heart compared with the WT heart (P < 0.001, Fig. 2C). We also observed a significant increase in interstitial collagen in the interventricular septa of the IL-6−/− animals; however, this increase was less dramatic than those observed in the right and left ventricular free walls. To confirm the increase in interstitial collagen observed by histology, we performed hydroxyproline analyses. IL-6−/− mice were observed to have significantly increased levels of collagen (1.19 ± 0.26 μg collagen/mg tissue) compared with WT animals (0.95 ± 0.08 μg collagen/mg tissue) (Fig. 2D).
Fig. 2.

Ventricular collagen levels in IL-6−/− mice. A and B: representative Masson's trichrome-stained sections from WT and IL-6−/− LV chambers, respectively. C: quantification of collagen content in the LV and RV free walls and intraventricular septa (IVS). D: quantification of cardiac collagen content via hydroxyproline analysis in adult mice. N = 4–6 animals per condition. #P < 0.001 and *P < 0.05 vs. WT.
Increased periostin expression in IL-6−/− mice.
Previous studies have indicated that periostin is a critical factor in ventricular dilation and collagen fibrillogenesis (30, 38, 39). Having observed similar defects in IL-6−/− mice, we examined their hearts for periostin expression. Western blot analysis revealed that IL-6−/− hearts displayed significantly increased periostin expression relative to WT hearts (Fig. 3, A and B). We also examined periostin expression by real-time PCR (Fig. 3C). As was observed at the protein level, we observed that IL-6−/− mice displayed an increased periostin expression (Fig. 3C). These data are congruent with our data that IL-6−/− mice display an increase in fibrosis.
Fig. 3.

Periostin levels in IL-6−/− mice. A and B: total heart periostin levels as determined by Western blot analysis and densitometry. Representative blot shown. C: total heart periostin mRNA levels as determined by real-time PCR analyses. D and E: analysis of cardiac fibroblast-myocyte adhesion in vitro. The total number of adherent cardiac fibroblasts plated onto myocytes (Myo) is shown. Cardiac fibroblasts or myocytes were isolated from neonatal WT and IL-6−/− hearts, and different combinations were plated as indicated. Con, conditioned. N = 5–7 animals per condition. *P < 0.05 vs. WT.
IL-6 loss alters cardiac fibroblast function in vitro.
Katsuragi and colleagues (30) have suggested that periostin plays a role in attenuating myocyte-fibroblast interactions by promoting side-to-side slippage of cells observed in cardiac dilatation. Given the observed decrease in cardiac function in IL-6−/− animals, we decided to characterize the effects of IL-6 loss on cardiac fibroblast-myocyte interactions. The loss of IL-6 in both myocytes and fibroblasts abrogated cellular adhesion completely throughout the entire time course (P < 0.001, Fig. 3D). However, the presence of either WT myocytes or fibroblasts with IL-6 null cells recovered this phenotype, suggesting that IL-6 deficiency is required in both cell types for the attenuation of cell-cell interactions (Fig. 5D). These results were similar to the disruption of the cell-cell interactions observed in cocultures of WT myocytes and cardiac fibroblasts in the presence of periostin-conditioned media (P < 0.001, Fig. 3E).
Loss of IL-6 alters cell populations in the adult heart.
Previous work from our laboratory has confirmed a direct link between an increase in the number of cardiac fibroblasts and an increase in collagen deposition (4). To define the cell population changes in the heart caused by an IL-6 loss, we performed flow cytometry analyses on age-matched adult hearts. Composite flow cytometry data are shown in Fig. 4A. The percentage of cardiac fibroblasts in IL-6−/− hearts was observed to be significantly higher relative to the levels observed in WT hearts (31 ± 4.7 vs. 24 ± 2.3%, P < 0.001). Subsequently, the percentage of myocytes decreased in IL-6−/− compared with WT hearts (51 ± 4.5 vs. 59 ± 5.3%, P < 0.01), as did endothelial cells (2.1 ± 1.2 vs. 5.3 ± 3.1%, P < 0.01) and vascular smooth muscle cells (3.5 + 1.2 vs. 5.9 ± 2.5%) (Fig. 4B).
Fig. 4.

Cardiac cell populations of IL-6−/− mice. A: representative flow cytometry experiment. B: cell populations of the WT and IL-6−/− heart as determined by flow cytometry analysis of cells labeled with immunospecific markers conjugated to quantum dot. #P < 0.001 and *P < 0.05 vs. WT. N = 3 to 4 animals per condition. DDR2, discoidin domain receptor-2; α-MHC, α-myosin heavy chain; VSMC, vascular smooth muscle cell; α-SMA, α-smooth muscle actin.
Loss of IL-6 alters proliferation and apoptosis in the heart.
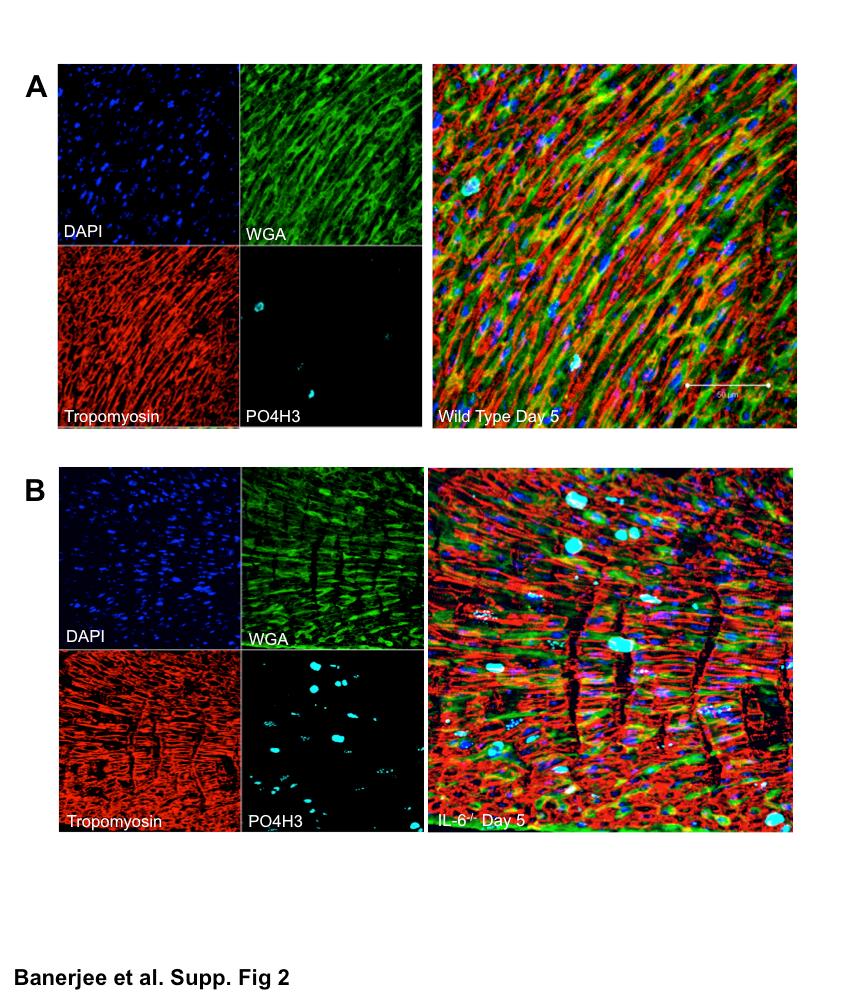
Having observed dramatic changes in the cardiac cell populations, we next examined the effects of IL-6 deficiency on the proliferation and apoptosis in the neonatal heart. IL-6−/− D5 hearts displayed a marked increase in proliferation relative to WT hearts (P < 0.001, Fig. 5A, and supplemental Figs. 1 and 2; note: all supplemental material may be found posted with the online version of this article). Interestingly, the nonmyocyte populations were observed to be the cells undergoing increased proliferation, whereas no changes were observed in the myocyte populations (Fig. 5A). No changes in proliferation were observed between D15 WT and IL-6−/− neonatal hearts (supplemental Fig. 1). In addition, we observed a significant increase in apoptosis in D5 IL-6−/− hearts compared with WT hearts (P < 0.01, Fig. 5B, and supplemental Figs. 1 and 3); however, by D15, the difference in apoptotic levels was no longer significant (supplemental Fig. 1). This observed increase in apoptosis was localized to the myocyte population (Fig. 5B).
Fig. 5.

Analysis of proliferation and apoptosis in IL-6−/− mice. A: analyses of proliferation by phospho-histone H3, tropomyosin, and wheat germ agglutinin staining in neonatal day-5 WT and IL-6−/− hearts. B: analyses of apoptosis via cleaved poly(ADP-ribose) polymerase (cleaved PARP), tropomyosin, and wheat germ agglutinin staining in day-5 hearts from WT and IL-6−/− mice. N = 5 experiments. #P < 0.01.
IL-6 deficiency causes a decrease in capillary density.
Given our observed decrease in endothelial cells and vascular smooth muscle cells (Fig. 4, A and B), we next performed fluorescent cardioangiography to assess the capillary density in the hearts of IL-6−/− and WT mice (Fig. 6, A–C, and supplemental videos 1–4). Using MetaMorph image analysis software, we calculated a marked decrease in capillary density in IL-6−/− mice compared with WT mice (Fig. 6C, P < 0.05).
Fig. 6.

Cardioangiography analysis. A and B: representative cardiac microsphere angiography images from WT and IL-6−/− mice, respectively. C: capillary density for WT and IL-6−/− hearts. N = 3 to 4 experiments. *P < 0.05. Scale bar equals 50 μm.
IL-6 signals via the STAT3 pathway and alters VEGF expression in myocyte-fibroblast cocultures.
To gain a mechanistic insight of the signaling pathway of IL-6 stimulation in the heart, we used an in vitro coculture system previously described by our laboratory to recapitulate the in vivo cardiac environment (7). Upon stimulation of cocultures of myocytes and WT cardiac fibroblasts with recombinant IL-6 alone or IL-6 + sIL-6Rα, we observed a robust STAT3 activation (Fig. 7A). p42/44 MAPK and AKT pathways were not activated by IL-6 or IL-6 + sIL-6Rα at the 15-min time point (Fig. 7A). We also examined the cultures after 60 min of stimulation, but no activation was observed (data not shown). Cocultures of IL-6-deficient fibroblasts in the presence of IL-6 did not display STAT3 activation. However, STAT3 activation was observed when both IL-6 and sIL-6Rα were added to the culture medium (Fig. 7A). PDGF was used as a positive control for the activation of AKT, p42/44 MAPK, and STAT3.
Fig. 7.

Analysis of fibroblast-myocyte signaling interactions. A: representative Western blot analyses of STAT3, MAPK p42/44, and AKT activation of in vitro coculture analyses. B: ELISA analyses of fold increase of VEGF levels secreted in the media of cardiac fibroblasts-myocyte cocultures at 24 h. KO, knockout; Rc, receptor. N = 3 experiments. *P < 0.05.
IL-6 via STAT3 activation has been shown to regulate VEGF (18, 40). This prompted us to examine VEGF expression in our coculture models. We observed a significant attenuation of VEGF expression from IL-6-null cardiac fibroblasts cocultured with WT myocytes when compared with WT controls (Fig. 7B, P < 0.05). Indeed, VEGF was significantly increased in WT cardiac fibroblast-myocyte cocultures compared with nonstimulated controls (Fig. 7B, P < 0.01). Moreover, VEGF deficiency in IL-6-null cardiac fibroblast WT-myocyte cocultures was recovered following the stimulation with IL-6 or when both IL-6 and sIL-6Rα were added (Fig. 7B, P < 0.05).
DISCUSSION
In the present study we have examined the effects of IL-6 loss on the murine heart. Previous studies have demonstrated that IL-6 is upregulated during chronic hypertension and that constitutive IL-6/sIL-6Rα stimulation can cause hypertrophy of the heart, as well as a reduction in infarct size in the mouse (3, 8, 26, 35, 37). Ischemia preconditioning studies have demonstrated that IL-6 can regulate the expression of cardioprotective factors such as inducible nitric oxide synthase and cyclooxygenase-2 (14, 43). These data have led to speculation from several groups, including our own, that IL-6 can be cardioprotective; therefore, we hypothesized that an IL-6 loss would be deleterious to the heart (32).
IL-6-loss causes ventricular dysfunction and increases periostin expression.
Ventricular dilation and increased myocardial fibrosis are two hallmarks of heart disease and indicators of a poor prognosis (30, 32, 40). Initial observations revealed that the IL-6−/− heart had distinct morphological differences relative to the WT heart (Fig. 1, A–D). IL-6−/− animals also displayed a decrease in HW/BW and HW/TL (Fig. 1, E and F, and Table 1). Surprisingly, our data contradict studies from Coles et al. (13) and Kamiñski et al. (29), as these authors did not observe a significant decrease in HW/BW; however, a close examination of these studies reveals a downward trend in the HW/BW in the IL-6−/− mouse. These data suggest that an IL-6 loss causes cardiovascular dilatation and/or a change in pump function. To confirm our initial observations, we performed an echocardiographic analysis on adult and neonatal D5 and D15 hearts. Progressive cardiac dilatation is observed in IL-6−/− animals relative to WT animals, indicating an altered cardiac function and possibly reduced cell-cell interactions due to a loss of IL-6 starting at neonatal D5 (Table 2). These data are congruent with observations from Faldt et al. (17) that IL-6−/− mice have an age-related reduction in exercise capacity at 4 and 8 mo of age.
We further analyzed cardiovascular dysfunction in IL-6−/− mice by exploring the interstitial fibrosis in the heart via Masson's trichrome staining and hydroxyproline analyses. Morphometric Masson's trichrome analysis revealed that IL-6 deficiency caused a significant increase in the amount of collagen deposition in the heart as did hydroxyproline analyses (Fig. 2, A–D). Another interesting finding is that an IL-6 loss results in an increase in the expression of periostin. Periostin is an ECM factor that plays a critical role in cardiovascular function, fibrosis, hypertrophy, myocardial infarction, and dilatation (23, 30, 38, 39, 52). Thus one can conclude that an increase in periostin would have a profound effect on cardiovascular dysfunction resulting in increases in collagen deposition and a decrease in myocyte-fibroblast interactions resulting in ventricular dilatation. Indeed, Western blot and real-time PCR analyses demonstrated a marked increase in the amount of periostin in IL-6−/− hearts compared with those of WT hearts (Fig. 3, A–C).
IL-6 loss alters fibroblast-myocyte interactions in vitro.
We next examined the effect of IL-6 loss on cardiac fibroblast-myocyte interactions. Interactions between cardiac fibroblasts and myocytes are critical for proper maintenance of the heart (4, 11, 12, 21, 22, 31, 33, 34, 41, 44). Several groups, including our own, have shown that IL-6 is increased in cocultures of cardiac myocytes and fibroblasts (5, 19, 20, 33). Studies have also examined the role and function of IL-6 in cardiac myocytes, with data suggesting regulatory roles in hypertrophy and atrial natriuretic peptide expression (1, 2, 33, 47). Despite these suggested roles of IL-6, little has been explored in terms of the role of IL-6 on fibroblast-myocyte interactions. Our in vivo data suggest that fibroblast-myocyte function is altered, given that IL-6 deficiency caused cardiac dilatation, increased collagen deposition and periostin expression, and altered fibroblast and myocyte populations in the IL-6−/− heart.
Our in vitro data demonstrate that an IL-6 loss in both myocytes and fibroblasts caused a marked decrease in fibroblast-myocyte interactions compared with WT myocytes and fibroblasts (Fig. 3D). These data regarding cell-cell interactions were similar to those observed by Katsuragi et al. (30) in periostin-overexpressing cells. Their report suggested that periostin has an inhibitory effect on integrin activity, thus altering the adhesion between cardiac myocytes and fibroblasts. Indeed, our observations using periostin-conditioned media showed an inhibitory effect on cell-cell interactions similar to our IL-6 null experiments (Fig. 3, D and E). Given that our in vivo observations revealed an increase in periostin, it is not surprising that we observed a decrease in cellular adhesion in our IL-6−/− cocultures. These data suggest that an IL-6 loss leads to an increased expression of periostin, such that the cell-cell interactions are altered. The reduced adhesion between myocytes and fibroblasts subsequently results in a deregulated myocardium, decreased cardiac output, and ventricular dilatation. The increase in fibroblasts and periostin may also cause the increased collagen content and fibrillogenesis, which is impacted in a positive manner by an increase in periostin expression (38, 39). The present study enforces these findings and suggests that IL-6 plays a role in the regulation of periostin and cardiac dilatation and dysfunction.
IL-6 loss alters the cardiac cell populations, capillary density, and developmental cardiac cell proliferation and apoptosis in vivo.
The communication and relative numbers of the cellular constituents is critical both in the pathogenesis of heart disease and in the homeostatic heart (4, 11, 12, 21, 22, 31, 33, 34, 41, 44). Our observed cardiovascular dysfunction, altered cell-cell interactions, and increases in fibrosis in the IL-6−/− heart prompted us to examine the cardiac cell populations of the IL-6−/− mouse. Flow cytometry analyses of adult IL-6−/− and WT mice demonstrated that an IL-6 loss resulted in a significant increase in the cardiac fibroblast population from ∼23% in WT to 31% in IL-6−/− mice (Fig. 4). These data are not surprising, as the majority of the literature supports cardiac fibroblasts as the primary producers of interstitial collagen and periostin (1, 11, 21, 30, 34, 39, 52). We also observed that the myocyte population decreased in IL-6−/− animals from ∼59% in WT to 50% in knockout mice (Fig. 4). These data are not unexpected, as studies have demonstrated that as cardiac dilatation progresses, myocytes lengthen and decrease in number (1, 52).
We also observed a decrease in the endothelial and vascular smooth muscle cell populations (Fig. 4). One explanation for the decrease in cells that make up the vasculature could be due to the downstream effects of IL-6. Cancer studies have indicated that IL-6 can regulate VEGF, a potent factor involved in vasculogenesis (18, 23). Moreover, the cardiac-specific STAT3 mouse displayed a marked decrease in capillary density (24). Several studies have shown that IL-6 is upstream of STAT3 activation and VEGF expression (18, 40). Thus a loss of IL-6 may contribute to an immature vascular formation in the heart and cause an alteration in the cell populations of the heart (32, 40). This vascular deficit would in turn result in a dysfunctional myocardium and lead to the promotion of fibrosis and ventricular dilation.
We observed that an IL-6 loss caused a significant decrease in the density of the myocardial vasculature (Fig. 6, and supplemental videos 1–4). These data suggest that the age-related dilatation observed in IL-6−/− mice is due to an inhibition of vasculature remodeling, resulting in an increase in fibrosis, and myocyte apoptosis, similar to that of the VEGF blockade model (27). Several groups, including our own, have observed that the cardiac cell populations fluctuate and the vasculature increases during neonatal development (4–5, 10, 21, 27, 31, 34).
We hypothesized that there would be changes in developmental cell proliferation and/or apoptosis given our observed cell population differences and developmental cardiac output dysfunction. Indeed, we observed an increase in the proliferation of cardiac cells, primarily the nonmyocyte population at D5 (Fig. 5A, and supplemental Figs. 1 and 2). It is possible that the small increase in phospho-histone H3 myocyte-positive cells in the IL-6−/− D5 neonates is due to binucleation, as observed in previous studies of stress-driven cardiac growth (1). Furthermore, we observed an increase in apoptosis in the myocytes in the D5 neonatal IL-6−/− heart (Fig. 5B, and supplemental Figs. 1 and 3). Collectively, these data support our observed increase in fibroblasts and decrease in myocytes in the IL-6−/− mice and progressive decrease in cardiac output between neonatal D5 to D15 (Fig. 4, A and B, and Table 2). Together, these data are similar to those observed with the VEGF blockade and the cardiac-specific Stat3−/− mouse (2, 27). Not surprisingly, these models also displayed an increased cardiac dilatation and fibrosis during pressure overload and development, respectively.
IL-6 deficiency results in altered STAT3 signaling via IL-6/sIL-6Rα transsignaling and altered VEGF expression.
We hypothesized that loss of IL-6 signaling would alter STAT3 activation and VEGF signaling given our observed in vivo data. Previous studies in our laboratory established that the cardiac fibroblasts were the primary producers of IL-6 in coculture models (5). Therefore, to recapitulate the in vivo effects of IL-6 loss on the heart, we cocultured both WT and IL-6-deficient fibroblasts onto neonatal rat cardiac myocytes. We observed in WT fibroblast cultures that only the STAT3 pathway was activated in our coculture system upon stimulation with IL-6 at 15 min (Fig. 7A). We also analyzed the samples following 60 min of stimulation; however, no activation of STAT3, MAPK42/44, or AKT was observed (data not shown). The activation of STAT3 was observed in both IL-6 and IL-6/sIL-6Rα cultures, suggesting that both transsignaling and IL-6 alone stimulate this pathway. Indeed, Hirota and colleagues (26) demonstrated, in studies involving transgenic mice, that both IL-6 and IL-6Rα were required to induce left ventricular hypertrophy, whereas alone, IL-6 or IL-6Rα transgenic mice did not produce any detectable cardiac phenotype.
One possibility for the robust response is endogenous IL-6/sIL-6Rα signaling in these cultures, given our previous data showing IL-6 increases in myocyte-fibroblast cocultures (5). IL-6-deficient cultures upon stimulation with both IL-6 and sIL-6Rα did show an activation of the STAT3 pathway, suggesting that transsignaling may be critical for IL-6-STAT3 stimulation. Indeed, Coles et al. (13) observed that IL-6−/− mice had less circulating sIL-6Rα than WT mice (13). The activation of the STAT3 pathway is not surprising, given our observed phenotypic similarities between the IL-6-deficient mouse and the cardiac-specific STAT3 mouse. These results suggest a signaling mechanism for IL-6 function in cardiac myocyte-fibroblast communication via the STAT3 pathway.
VEGF is a potent mediator of vasculogenesis, which given our observations of an attenuated capillary density in IL-6−/− mice and previous studies demonstrating IL-6 and STAT3 regulation of VEGF expression, we examined VEGF levels in our coculture model (18, 40). We observed that a loss of IL-6 in the cardiac fibroblasts abrogated VEGF expression. However, upon exogenous stimulation with IL-6 and/or IL-6/sIL-6Rα, VEGF levels were recovered in our IL-6-deficient cardiac fibroblast-WT myocyte cocultures. These data are surprising, given that STAT3 activation was only recovered in IL-6/sIL-6Rα-stimulated IL-6-deficient cocultures (Fig. 7). It is possible that there is a secondary mechanism involved here or that the increased VEGF expression which we are observing in the IL-6-deficient cultures is not occurring through the STAT3 pathway; however, further biochemical studies are clearly needed to examine these data. VEGF levels were also significantly increased in response to IL-6 or IL-6/sIL-6Rα stimulation in our WT fibroblast-myocyte cocultures relative to WT controls. Taken together, these data suggest that IL-6 regulates VEGF via STAT3 and that this pathway can alter the formation of the vasculature. Overall, these studies provide important evidence that IL-6 contributes to the formation of a healthy myocardium via IL-6/sIL-6Rα transsignaling, activation of STAT3, and VEGF expression. Furthermore, these data suggest that IL-6 and its targets can possibly be used therapeutically in the heart and that IL-6 plays a role in cardiac development by regulating fibroblast-myocyte interactions (32).
GRANTS
This research was funded by National Heart, Lung, and Blood Institute Grant 1RO1-HL-85847.
Supplementary Material
Acknowledgments
We thank Drs. Marj Pena and Thomas K. Borg for helpful suggestions and for technical support and also Valerie Kennedy, Dr. Christopher Wilson, Adam Bedenbaugh, Colby Souders, Stephanie Bowers, Sadiye Rieder, and Dr. Edie Goldsmith for technical support in the assays for the study.
REFERENCES
- 1.Ahuja P, Sdek P, Maclellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 87: 521–544, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ancey C, Menet E, Corbi P, Fredj S, Garcia M, Rücker-Martin C, Bescond J, Morel F, Wijdenes J, Lecron JC, Potreau D. Human cardiomyocyte hypertrophy induced in vitro by gp130 stimulation. Cardiovasc Res 59: 78–85, 2003. [DOI] [PubMed] [Google Scholar]
- 3.Aukrust P, Ueland T, Lien E, Bendtzen K, Müller F, Andreassen AK, Nordøy I, Aass H, Espevik T, Simonsen S, Frøland SS, Gullestad L. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol 83: 376–382, 1999. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA. Determination of cell type, and number during cardiac development in the neonatal and adult rat and mouse. Am J Physiol Heart Circ Physiol 293: H1883–H1891, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee I, Yekkala K, Borg TK, Baudino TA. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann NY Acad Sci 1080: 76–84, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Baudino TA, McFadden A, Fix C, Hastings J, Price R, Borg TK. Cell patterning: interaction of cardiac myocytes and fibroblasts in three-dimensional culture. Microsc Microanal 14: 117–125, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Baumgarten G, Knuefermann P, Kalra D, Gao F, Taffet GE, Michael L, Blackshear PJ, Carballo E, Sivasubramanian N, Mann DL. Load-dependent and -independent regulation of proinflammatory cytokine and cytokine receptor gene expression in the adult mammalian heart. Circulation 105: 2192–2197, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Betz UA, Bloch W, van den Broek M, Yoshida K, Taga T, Kishimoto Addicks KT, Rajewsky K, Müller W. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. J Exp Med 188: 1955–1965, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Booz GW, Day JN, Baker KM. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol 34: 1443–1453, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res 65: 40–51, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res 94: 828–835, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Coles B, Fielding CA, Rose-John S, Scheller J, Jones SA, O'Donnell VB. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am J Pathol 171: 315–325, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawn B, Xuan YT, Guo Y, Rezazadeh A, Stein AB, Hunt G, Wu WJ, Tan W, Bolli R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res 64: 61–71, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du JH, Xu N, Song Y, Xu M, Lu ZZ, Han C, Zhang YY. AICAR stimulates IL-6 production via p38 MAPK in cardiac fibroblasts in adult mice: a possible role for AMPK. Biochem Biophys Res Commun 337: 1139–1144, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Edwards CA, O'Brien WD. Modified assay for determination of hydroxyproline in a tissue hydrolyzate. Clin Chim Acta 104: 161–167, 1980. [DOI] [PubMed] [Google Scholar]
- 17.Fäldt J, Wernstedt I, Fitzgerald SM, Wallenius K, Bergström G, Jannsson JO. Reduced exercise endurance in interleukin-6 deficient mice. Endocrinology 145: 2680–2686, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Feurino LW, Zhang Y, Bharadwaj U, Zhang R, Li F, Fisher WE, Brunicardi FC, Chen C, Yao Q, Li M. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol Ther 6: 1096–1100, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Fredj S, Bescond J, Louault C, Delwail A, Lecron JC, Potreau D. Role of interleukin-6 in cardiomyocyte/cardiac fibroblast interactions during myocyte hypertrophy and fibroblast proliferation. J Cell Physiol 204: 428–436, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol 202: 891–899, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Gaudesius G, Miragoli M, Thomas SP, Rohr S. Coupling of cardiac electrical activity over extended distances by fibroblasts of cardiac origin. Circ Res 93: 421–428, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Harvey PR, Rosenthal N. Heart Development. San Diego, Academic, 1999.
- 23.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat Rev Mol Cell Biol 7: 589–600, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Hilfiker-Kleiner D, Hilfiker A, Fuchs M, Kaminski K, Schaefer A, Schieffer B, Hillmer A, Schmiedl A, Ding Z, Podewski E, Podewski E, Poli V, Schneider MD, Schulz R, Park JK, Wollert KC, Drexler H. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ Res 95: 187–195, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr, Müller W, Chien KR. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell 97: 189–198, 1999. [DOI] [PubMed] [Google Scholar]
- 26.Hirota H, Yoshida K, Kishimoto T, Taga T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA 92: 4862–4866, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivey BL Microvascular plasticity and experimental heart failure. Hypertension 47: 1–3, 2006. [DOI] [PubMed] [Google Scholar]
- 28.Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol 203: 1–38, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Kamiñski KA, Oledzka E, Biaobrzewska K, Kozuch M, Musia WJ, Winnicka MM. The effects of moderate physical exercise on cardiac hypertrophy in interleukin 6 deficient mice. Adv Med Sci 52: 164–168, 2007. [PubMed] [Google Scholar]
- 30.Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, Kawashima K, Kaneda Y, Ogihara T, Sugimura K. Periostin as a novel factor responsible for ventricular dilation. Circulation 110: 1806–1813, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Kohl P Cardiac cellular heterogeneity and remodelling. Cardiovasc Res 64: 195–197, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Kurdi M, Booz GW. Can the protective actions of JAK-STAT in the heart be exploited therapeutically? Parsing the regulation of interleukin-6-type cytokine signaling. J Cardiovasc Pharmacol 50: 126–141, 2007. [DOI] [PubMed] [Google Scholar]
- 33.LaFramboise WA, Scalise D, Stoodley P, Graner SR, Guthrie RD, Magovern JA, Becich MJ. Cardiac fibroblasts influence cardiomyocyte phenotype in vitro. Am J Physiol Cell Physiol 292: C1799–C1808, 2007. [DOI] [PubMed] [Google Scholar]
- 34.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res 91: 1103–1113, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Matsushita K, Iwanaga S, Oda T, Kimura K, Shimada M, Sano M, Umezawa A, Hata J, Ogawa S. Interleukin-6/soluble interleukin-6 receptor complex reduces infarct size via inhibiting myocardial apoptosis. Lab Invest 85: 1210–1223, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Mensah GA, Mokdad AH, Ford ES, Greenlund KJ, Croft JB. State of disparities in cardiovascular health in the United States. Circulation 111: 1233–1241, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Niebauer J Inflammatory mediators in heart failure. Int J Cardiol 72: 209–213, 2000. [DOI] [PubMed] [Google Scholar]
- 38.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem 101: 695–711, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res 101: 313–321, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rega G, Kaun C, Demyanets S, Pfaffenberger S, Rychli K, Hohensinner PJ, Kastl SP, Speidl WS, Weiss TW, Breuss JM, Furnkranz A, Uhrin P, Zaujec J, Zilberfarb V, Frey M, Roehle R, Maurer G, Huber K, Wojta J. Vascular endothelial growth factor is induced by the inflammatory cytokines interleukin-6 and oncostatin m in human adipose tissue in vitro and in murine adipose tissue in vivo. Arterioscler Thromb Vasc Biol 27: 1587–1595, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol 292: H1561–H1567, 2007. [DOI] [PubMed] [Google Scholar]
- 42.Sheehan D, Hrapchak B. Theory and practice of histotechnology. Columbus, OH: Battelle, 1980, p. 189–190.
- 43.Smart N, Mojet MH, Latchman DS, Marber MS, Duchen MR, Heads RJ. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res 69: 164–177, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Sussman MA, McCulloch A, Borg TK. Dance band on the titanic: biomechanical signaling in cardiac hypertrophy. Circ Res 91: 888–898, 2002. [DOI] [PubMed] [Google Scholar]
- 45.Tone E, Kunisada K, Kumanogoh A, Negoro S, Funamoto M, Osugi T, Kishimoto T, Yamauchi-Takihara K. gp130-Dependent signalling pathway is not enhanced in gp130 transgenic heart after LIF stimulation. Cytokine 12: 1512–1518, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Yamauchi-Takihara K, Ihara Y, Ogata A, Yoshizaki K, Azuma J, Kishimoto T. Hypoxic stress induces cardiac myocyte-derived interleukin-6. Circulation 91: 1520–1524, 1995. [DOI] [PubMed] [Google Scholar]
- 48.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol Heart Circ Physiol 277: H1967–H1974, 1999. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida K, Taga T, Saito M, Suematsu S, Kumanogoh A, Tanaka T, Fujiwara H, Hirata M, Yamagami T, Nakahata T, Hirabayashi T, Yoneda Y, Tanaka K, Wang WZ, Mori C, Shiota K, Yoshida N, Kishimoto T. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc Natl Acad Sci USA 93: 407–411, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin F, Li P, Zheng M, Chen L, Xu Q, Chen K, Wang YY, Zhang YY, Han C. Interleukin-6 family of cytokines mediates isoproterenol-induced delayed STAT3 activation in mouse heart. J Biol Chem 278: 21070–21075, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Yin F, Wang YY, Du JH, Li C, Lu ZZ, Han C, Zhang YY. Noncanonical cAMP pathway and p38 MAPK mediate beta2-adrenergic receptor-induced IL-6 production in neonatal mouse cardiac fibroblasts. J Mol Cell Cardiol 40: 384–393, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Zhao M, Chow A, Powers J, Fajardo G, Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics 19: 93–105, 2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}