

Abstract
Allosteric regulation of nitrite reduction by deoxyhemoglobin has been proposed to mediate nitric oxide (NO) formation during hypoxia. Nitrite is predominantly an anion at physiological pH, raising questions about the mechanism by which it enters the red blood cell (RBC) and whether this is regulated and coupled to deoxyhemoglobin-mediated reduction. We tested the hypothesis that nitrite transport by RBCs is regulated by fractional saturation. Using human RBCs, nitrite consumption was faster at lower fractional saturations, consistent with faster reactions with deoxyheme. A membrane-based regulation was suggested by slower nitrite consumption with intact versus lysed RBCs. Interestingly, upon nitrite addition, intracellular nitrite concentrations attained a steady state that, despite increased rates of consumption, did not change with decreasing oxygen tensions, suggesting a deoxygenation-sensitive step that either increases nitrite import or decreases the rate of nitrite export. A role for anion exchanger (AE)-1 in the control of nitrite export was suggested by increased intracellular nitrite concentrations in RBCs treated with DIDS. Moreover, deoxygenation decreased steady-state levels of intracellular nitrite in AE-1-inhibited RBCs. Based on these data, we propose a model in which deoxyhemoglobin binding to AE-1 inhibits nitrite export under low oxygen tensions allowing for the coupling between deoxygenation and nitrite reduction to NO along the arterial-to-venous gradient.
Keywords: oxygen, anion exchanger-1, hypoxic vasodilation
nitrite (NO2−) was, for a long time, considered a relatively inert end product and marker of nitric oxide (NO) metabolism (23). More recently, however, this paradigm has been challenged by multiple studies highlighting the potential for nitrite as an endogenous and therapeutic mediator of NO homeostasis during hypoxia (for a review, see Ref. 41). This concept is derived from studies using low physiological concentrations of nitrite (nM–μM) that are also safe from a therapeutic perspective (35). A key element of the proposal that nitrite is a mediator of NO function is the elucidation of mechanisms that operate under biological conditions to reduce nitrite to NO. These include reactions between nitrite and xanthine oxidoreductase and/or deoxyheme proteins including neuroglobin, cytoglobin, myoglobin, and hemoglobin (Hb) (2, 5, 11, 13, 18, 26–29, 31, 42, 46, 48, 56, 62, 63, 65). With the latter, the general model proposed links heme deoxygenation with a reactivity that results in the one-electron reduction of nitrite to NO. In the case of Hb, we and others (13, 28) have shown that nitrite reduction occurs by an allosterically regulated nitrite reductase activity of deoxyHb that is kinetically maximal at oxygen tensions that result in ∼50% Hb-oxygen fractional saturation, i.e., the Hb P50 (13, 28). Desaturation of Hb close to its P50 may occur during transit of red blood cells (RBCs) through the resistance (arteriolar) vasculature under physiological and hypoxic stress conditions (6, 59), leading to the proposal that NO derived from nitrite reduction by the RBC is a mediator of hypoxic vasodilation (11, 22, 31, 38, 42, 44, 50, 51).
Most of the insights with respect to deoxyHb-nitrite reactions have been gleaned from cell free-based studies with key observations including maximal kinetics at the Hb P50 being verified with RBCs also (13, 24, 27, 28). Nitrite is predominantly an anion at physiological pH (pKa = 3.2), raising the question of what role the RBC membrane plays in the regulation of nitrite movement and subsequent reactions with Hb. Under basal conditions, significant concentrations of nitrite exist in the RBC (∼300 nM) (15). Given that both oxyHb and deoxyHb consume nitrite, these data suggest a continual import of nitrite into the RBC. As RBCs deoxygenate, the rate of nitrite-heme reactions increases (27, 28, 46). Moreover, transit of RBCs across one circuit of the coronary microcirculation results in an increase in intraerythrocytic markers of nitrite-deoxyHb reactivity (52), further underscoring the requirement for movement of nitrite from the plasma into the RBC compartment during hypoxemia. How nitrite movement across membranes is controlled remains unclear. In this study, we hypothesized that during hypoxia, net nitrite transport into RBCs also increases to match accelerated nitrite-deoxyheme reactions and, moreover, that transport of nitrite is also regulated by Hb fractional saturation.
The RBC contains multiple membrane proteins that regulate ion movement, enabling the appropriate transport of metabolic products such as carbon dioxide and protons, regulation of blood pH, adequate oxygen delivery, and modulation of erythrocytic volume (4, 20). Key among these is anion exchanger (AE-1), which represents ∼25% of RBC membrane protein (9). Both the COOH- and NH2-termini of AE-1 are cytoplasmic and have been shown to selectively interact and hence modulate cytosolic protein function (e.g., glycotlyic enzymes) (9). Interestingly, deoxyHb has higher affinity than oxyHb for the NH2-terminus of AE-1 (61). This specific binding has been shown to inhibit the movement of sulfate through this channel at low oxygen tensions (19). Thus, AE-1 represents a possible candidate to link Hb oxygen sensing with nitrite metabolism by the RBC. A previous study (55) using RBC ghosts suggested a role for AE-1 in the control of nitrite efflux. However, this and other studies (1, 43, 60, 64) evaluating nitrite-RBC interactions have only been performed under oxygenated conditions. Studies evaluating the role of oxygen saturation on RBC nitrite metabolism are largely limited to teleost fish, in which case faster nitrite consumption rates at lower oxygen tensions have been observed (33). Interestingly, this effect was not observed with pig RBCs (34) or in rat RBCs (18). Herein, we provide evidence that Hb deoxygenation regulates nitrite transport in human RBCs and that this effect is mediated by deoxygenation-sensitive inhibition of nitrite export via AE-1. These data suggest the hypothesis that nitrite reduction to NO by RBCs occurs via a concerted mechanism that matches increased rates of nitrite reduction by deoxyHb with an inhibition of nitrite export across RBC membranes by AE-1.
EXPERIMENTAL PROCEDURES
Materials.
4,4′-Diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) was purchased from Sigma-Aldrich (St Louis. MO), N-ethylmaleimide (NEM) was from Pierce (Rockford, IL), and NG-nitro-l-arginine methyl ester (l-NAME) was from Alexis Biochemical (Lausen, Switzerland). All other reagents were of analytical grade and purchased from Sigma-Aldrich.
RBC preparation.
Blood was obtained from healthy volunteers, who gave written informed consent to participate in the study, according to protocols approved by the University of Alabama at Birmingham Institutional Review Board. Briefly, blood was collected into vacutainers (BD, Franklin Lakes, NJ) containing anticoagulant citrate dextrose solution and centrifuged for 10 min at 1,000 g and 4°C. Both plasma and leukocyte coats were carefully removed, and the remaining RBC pellet was washed three times in Krebs-Henseleit (KH) buffer (pH 7.4) supplemented with 0.5% BSA (KH-BSA buffer). Under these conditions, hemolysis was typically <0.5%. For experiments in which nitrite consumption by hemolysates was studied, ice-cold RBC suspensions at ∼50% hematocrit in KH-BSA buffer were mechanically lysed using a tissue homogenizer (PowerGen 125, Fischer Scientific) at medium-high speed for up to 20 min. MetHb formation under these conditions did not significantly change compared with intact RBCs (not shown). Finally, we note that the absolute rate of nitrite consumption varied approximately two- to threefold between RBC preparations isolated from different donors (see Fig. 4). We have not systematically evaluated the nature of these differences and note that 1) times between venipuncture and experiment (<3 h), RBC P50 values, and protocols for the RBC preparation remained consistent for all presented experiments and 2) the relative effects of hypoxia on increasing rates of nitrite consumption were similar in all RBC preparations. Therefore, when data are presented from multiple experiments, nitrite consumption at different oxygen tensions is presented as the fold change as shown in the respective figures.
Determination of RBC Hb oxygen affinity.
Oxygen binding curves were measured by open-flow respirometry coupled with UV-Vis measurement of intraerythrocytic Hb as previously described (13).
Nitrite consumption by RBCs.
Experiments under different oxygen tensions were performed in a controlled-atmosphere chamber (Plas Labs, Lansing, MI) using gases containing different oxygen tensions balanced with N2 and 5% CO2 at either 22 or 37°C. For these experiments, KH buffer was incubated overnight under a 5% CO2 atmosphere to ensure full equilibration before the addition of BSA and RBCs. RBC suspensions at 5% hematocrit in KH-BSA buffer were equilibrated in six-well tissue culture plates with gentle rocking. We used 15 min of equilibration time for experiments performed at 10% oxygen (75 mmHg) and 30 min for experiments performed at 4% oxygen (30 mmHg). Preliminary observations indicated that these times were sufficient for oxygen fractional saturations to equilibrate. For the data shown in Fig. 2B, however, a 30-min equilibration time was used for all oxygen tensions tested. The pH of the RBC suspensions was adjusted to 7.4 after equilibration with desired oxygen tension and immediately before the addition of nitrite (final concentration of 100 μM in the RBC suspension unless otherwise specified). To ensure that pH was not a variable, pH was measured at the end of experiments and also remained constant during the course of the incubations. In some experiments, RBCs were preequilibrated with l-NAME or DIDS for 15–30 min before equilibration with oxygen tension. Appropriate vehicle control incubations were also included. To initiate experiments, nitrite was added [nitrite stock solutions made in PBS + 100 μM diethylene triamine pentaacetic acid (DTPA) also preequilibrated at respective oxygen tension], and aliquots were then removed at the indicated times. Aliquots were taken out of the chamber and immediately centrifuged 2,000 g for 30 s. The extracellular fraction (supernatant) was collected, and the RBC pellet was washed three times with ice-cold PBS containing 100 μM DTPA at pH 7.4. After three washes (∼3–4 min total), pellets were processed for the determination of nitrite, S-nitrosothiols (SNO), or metHb as described below.
Alternatively, unwashed pellets were collected for the measurement of iron-nitrosyl species. In all experiments, parallel incubations of nitrite alone in KH-BSA buffer were included and aliquots were collected. Nitrite in the extracellular fraction was measured by triiodide-mediated reduction followed by chemiluminescent detection using a NO analyzer (Sievers NOA model 280i, Boulder, CO) as previously described (39). Nitrite consumption from the extracellular compartment is defined as the nitrite concentration difference between nitrite alone versus RBC + nitrite conditions.
Intracellular nitrite and SNO determination.
Washed RBC pellets were immediately lysed in a stabilization solution containing 10 mM K3Fe(CN)6, 20 mM NEM, 1% Igepal, and 100 μM DTPA in PBS and then frozen at −80°C, resulting in the stabilization of RBC pellets within 5 min of sample collection. Nitrite or SNOs in RBC samples were measured within 4 days of collection, and freezing was not found to affect measured concentrations of these NO species (not shown). For SNO determination, lysates were treated with acid sulfanilamide [1.5% (wt/vol) in 2 M HCl for 5 min at room temperature] in the presence or absence of mercuric chloride (50 mM) and measured by reductive chemiluminescence as previously described (39). For the measurement of intracellular nitrite levels, samples were deproteinated by the addition of an equal volume of ice-cold methanol followed by centrifugation at 15,000 g for 2 min. Nitrite in the resulting supernatants was measured as described above. Background nitrite levels in stock solutions were <125 nM and <500 nM for DIDS and l-NAME, respectively.
MetHb determination.
Washed RBC pellets were resuspended in four volumes of KH-BSA buffer, transferred to electron paramagnetic resonance (EPR) tubes, and frozen in liquid nitrogen. MetHb was measured in an X-band EPR Spectrometer E Lexsys E500 (Bruker Biospin) with the following settings: modulation amplitude 15 G, microwave power 100.4 mW, temperature 120 K, frequency 9.437 GHz, time constant 10.24 ms, and conversion time 40.96 ms. Each spectrum was the average of eight scans, and the signal at g = 6 was quantified by a comparison to metHb standards.
Intracellular nitrosylHb determination.
RBC pellets were lysed in three volumes of high purity water containing 50 μM DTPA and measured on the same day or flash frozen in liquid nitrogen and thawed immediately before measurement. NitrosylHb (HbNO) was measured by chemiluminesence using 50 mM K3Fe(CN)6 in PBS supplemented with 1% SE-15 antifoam (Sigma) at 70°C and pH 7.4 as previously described (8, 39).
Carbon monoxide treatment.
RBCs were resuspended at 5% hematocrit in a carbon monoxide (CO)-saturated Tris buffer (140.5 mM NaCl, 21 mM Tris, 4.7 mM KCl, 1.2 mM MgSO4, 2 mM CaCl2, and 0.1% glucose containing 100 μM DTPA and 0.5% BSA). The resulting suspension was then exposed to a gentle stream of CO for a further 10–15 min before aliquots were removed for experiments in the controlled atmosphere chamber. This protocol yielded ≥90% conversion to carboxyHb as determined by the spectral measurement of Hb after RBC lysis in four volumes of high purity water.
Data normalization.
Extracellular and intracellular nitrite concentrations were normalized to heme concentrations measured by the Drabkin's assay (17). Briefly, lysed RBCs were treated with excess K3Fe(CN)6 and KCN to quantitatively convert all Hb species into cyanometHb. The cyanometHb concentration was determined by measuring the resulting absorbance at 540 nm using an extinction coefficient of 11 mM−1·cm−1.
Vessel relaxation experiments.
Rat aortic rings were equilibrated under either 21% or 0% oxygen in KH buffer at 37°C in the presence of 5% CO2 as previously described (29). Vessels were pretreated with indomethacin (5 μM) and N-monomethyl-l-arginine (l-NMMA; 1 mM) and precontracted with phenylephrine (200 nM) before the addition of RBCs (0.3% hematocrit final concentration). Once vessels had reached a stable tone, vasorelaxation was elicited by the addition of a single dose of either sodium nitrite (final concentration: 10 μM) or the NO donor Mahma-NONOate (MNO; final concentration: 30 nM). The vasorelaxation response was determined in the absence and presence of RBCs, and the percent inhibition of nitrite- or MNO-dependent vasodilation by RBCs was calculated.
Statistical analysis.
All experiments were performed more than three times with more than three replicates within each experiment per condition unless otherwise specified. Data are expressed as means ± SE with statistical analysis as shown in the respective figures. Data were analyzed by performing unpaired and paired t-tests or one-way and two-way repeated-measures ANOVA followed by Bonferroni post test analysis using GraphPad Prism software. P values of ≤0.05 were considered significant.
RESULTS
Effects of RBC oxygen fractional saturation on nitrite- and NO-dependent vasodilation.
Previous studies (14, 29) using isolated aortic ring bioassays have shown that the effects of cell-free Hb on nitrite-dependent vasodilation depends on the oxygen fractional saturation. Cell-free oxyHb inhibits nitrite- and NO-dependent vasodilation, consistent with heme-based NO scavenging. Under deoxygenated conditions, a similar inhibition of NO-dependent vasodilation is observed; however, nitrite-dependent vasodilation is unaffected, consistent with a nitrite reductase activity of deoxyHb that generates NO and counters NO scavenging. The results shown in Fig. 1 extend this concept to intact RBCs and demonstrate that under the employed experimental conditions, both oxygenated or deoxygenated RBCs inhibit NO-dependent vasodilation to the same extent. In contrast, inhibition of nitrite-dependent vasodilation by RBCs was significantly attenuated at 0% compared with 21% oxygen.
Fig. 1.

Effects of red blood cells (RBCs) on nitrite- and nitric oxide (NO)-dependent vasodilation at high and low oxygen tension (Po2). Relaxation of aortic rings by bolus additions of either Mahma-NONOate (MNO; 30 nM) or nitrite (10 μM) in the absence or presence of RBCs [0.3% hematocrit (Hct)] was determined at 21% and 0% oxygen. Shown is the inhibition of MNO- or nitrite-dependent vasodilation by RBCs at 21% and 0% oxygen. Data are means ± SE; n = 4–6. P values were calculated by unpaired t-test.
Hb-oxygen fractional saturation regulates nitrite consumption by RBCs.
Previous studies (27, 28) have shown that the nitrite-deoxyHb (nitrite reduction) reaction is faster compared with the nitrite-oxyHb (nitrite oxidation) reaction. To test if this occurs with intact RBCs and if this effect is mediated by lower dissolved oxygen concentrations or by different Hb-oxygen saturations, RBC-dependent nitrite consumption was evaluated at 22 and 37°C. These two temperatures were used to modulate oxygen affinity (being higher at lower temperatures; Fig. 2A) and, hence, Hb fractional saturation at a given oxygen tension. The results shown in Fig. 2B demonstrate that, regardless of the temperature used in the assay, nitrite consumption increased as Hb fractional saturation decreased, reaching a maximum at saturation values close to the Hb P50 and then decreasing as Hb-oxygen saturation decreased beyond the P50. Note that maximal nitrite consumption over 15 min was different at the two temperatures studied, being 106.1 nmol/μmol heme at 28.8 mmHg O2 and 28 nmol/μmol heme at 7.8 mmHg O2 at 37 and 22°C, respectively. Taken together, these data demonstrate that increased nitrite consumption is related to the oxygen fractional saturation of Hb in a bimodal fashion. This conclusion was verified by almost complete inhibition of hypoxemia-dependent nitrite consumption by RBCs that had been preequilibrated with CO (Fig. 2C).
Fig. 2.

RBC hemoglobin (Hb)-oxygen fractional saturation regulates nitrite consumption. A: oxygen binding curves for RBC (0.3–0.5% Hct) in Krebs-Henseleit buffer (pH 7.4) determined at 22 or 37°C in the presence of 5% CO2. Calculated 50% Hb-oxygen fractional saturation (P50 values) and Hill constants were 7.1 mmHg and 1.6, respectively, at 22°C and 32.6 mmHg and 2.9 at 37°C, respectively. B: oxygen-equilibrated RBC suspensions (5% Hct) were incubated in the presence of 100 μM nitrite at the indicated temperatures and pH 7.4. Remaining nitrite in the media was measured after 15 min. Since rates of consumption are faster at higher temperatures, data are presented as relative consumption normalized to the maximum at each respective temperature. Data are means ± SE; n = 3. Data were analyzed by one-way ANOVA and the Bonferroni post test. *P < 0.05 and **P < 0.01 vs. the highest fractional saturation at each respective temperature. C: control or carbon monoxide (CO) gas-preequilibrated RBCs (5% Hct) in Tris buffer were incubated with nitrite (100 μM) at 28-mmHg Po2, 37°C, and pH 7.4. for 15 min, and nitrite consumption was measured. Data are means ± SE; n = 3. Data were analyzed by a two-tailed Student's t-test. *P < 0.001.
Role of the RBC membrane in controlling nitrite-RBC reactions.
To study the role of the membrane in the control of the nitrite reaction with Hb, nitrite consumption was measured with intact (<0.5% hemolysis) or lysed (>70%) RBCs. Since Hb fractional saturation affects the kinetics of nitrite-heme reactions, and RBC P50 is regulated by effectors (e.g., 2,3-biphosphoglycerate), RBCs were lysed mechanically to avoid dilution and loss of allosteric effectors. P50 values of intact or hemolyzed RBCs were not different (27.6 and 25 mmHg, respectively; data not shown), ensuring that heme reaction rates were not a variable in these experiments. The results shown in Fig. 3 demonstrate that hemolysis of RBCs significantly increased nitrite consumption at half-fractional saturation. Similar results showing faster rates of nitrite consumption by lysed RBCs were observed at high oxygen fractional saturations also (Fig. 3), consistent with a previous report (64) using oxygenated RBCs. These data imply that compartmentalization by the membrane slows the overall consumption of nitrite by erythrocytic Hb and suggest that modulating how nitrite crosses the membrane will impact on nitrite metabolism by RBCs.
Fig. 3.

The RBC membrane regulates nitrite-heme reactivity. Nitrite (100 μM) was added to intact (hemolysis < 0.5%) or mechanically lysed (hemolysis > 70%) RBCs (5% Hct) at 75- or 30-mmHg Po2, 37°C, and pH 7.4, and nitrite consumption was measured after 10 min. Data are means ± SE; n = 3. Data were analyzed by an unpaired t-test versus intact cells at the respective Po2 (*P < 0.001).
Evidence for an oxygen-sensitive step in RBC nitrite transport.
We next determined the time course for nitrite consumption from the extracellular compartment and the parallel changes that occur in intracellular nitrite levels. Figure 4A shows data from one representative experiment (i.e., from one RBC donor) demonstrating that nitrite consumption occurs in a time-dependent manner that is faster at lower Hb fractional saturations. Interestingly, intracellular nitrite levels increased upon nitrite addition and immediately reached a steady state that lasted throughout the course of the observation period. Furthermore, unlike nitrite consumption, the steady state observed for intracellular nitrite was not altered by lowering the oxygen tension. The nitrite concentration inside the RBC was ∼1.5 μM, which was significantly lower than the amount of nitrite consumed from the extracellular compartment. Due to variations between RBC preparations from different donors, Fig. 4, B and C, shows normalized data from three to four independent experiments in which nitrite consumption and intracellular accumulation were followed over 30 min, further confirming the results shown in Fig. 4A. Figure 4, D and E, shows the nitrite concentration dependence on nitrite consumption from the extracellular compartment and intracellular nitrite steady-state levels as calculated by the nitrite-to-heme ratio, respectively. Nitrite consumption was higher at lower oxygen tension at all tested nitrite concentrations. However, whereas intracellular nitrite increased in proportion to the nitrite dose added, this was not altered by lowering oxygen tension. These data are consistent with intracellular nitrite levels representing a balance between nitrite entry and heme-based consumption, a result further confirmed by time-dependent increases in both metHb and HbNO (Fig. 4F). MetHb is a product of both nitrite-oxyHb and nitrite-deoxyHb reactions, whereas HbNO is an ultimate product of nitrite-deoxyHb reactions. MetHb formation was similar at both fractional saturations tested, whereas HbNO formation was significantly higher at the lower fractional saturation. These data suggest that the lack of effect of fractional saturation on intracellular nitrite levels is not due to lack of reactions with heme but instead represents a steady state comprising a balance between nitrite entry and heme-based consumption.
Fig. 4.

Effects of RBC deoxygenation on intracellular nitrite concentration. A: nitrite (100 μM) was added to RBCs (5% Hct) preequilibrated at 75 mmHg (⧫, dashed lines) or 30 mmHg (○, solid lines) of oxygen at 37°C, and time-dependent losses of nitrite from the extracellular compartment and accumulation in the intracellular compartment were determined. Data show changes in absolute nitrite concentrations and are means ± SE (n = 3) from a single experiment. Data were analyzed by two-way ANOVA and the Bonferroni post test. ***P < 0.001 relative to the corresponding time at higher Po2. Intracellular nitrite concentrations were calculated by assuming a RBC volume of 100 fl and an intraerythrocytic heme concentration of 20 mM. B and C: normalized data for extracellular nitrite consumption (B) and intracellular nitrite accumulation (C) from 3 to 4 independent experiments (and RBC preparations). The absolute amounts of nitrite consumed and intracellular levels were found to vary between RBC preparations [from 52 to 139 nmol/μmol heme and from 46 to 121 pmol/μmol heme for maximum consumption and maximal intracellular nitrite levels (15 min), respectively]. Therefore, to compare results from different RBC preparations, data in B and C are plotted as relative to the maximum in each preparation. Data are means ± SE; n = 4 in B and 3 in C. Data were analyzed by two-way ANOVA and the Bonferroni post test. *P < 0.05 and **P < 0.001 vs. the corresponding time point at 75-mmHg Po2. D and E: nitrite at the indicated doses was added to RBCs (5% Hct) preequilibrated at either 73.5- or 21.4-mmHg Po2 at 37°C and pH 7.4, and both nitrite consumption from the extracellular compartment (D) and intracellular nitrite levels (E) were measured after 15 min. Data are means ± SE; n = 3. P values were determined by two-way ANOVA. The solid lines in D show the best fits as determined by linear regression. F: changes in metHb and nitrosylHb (HbNO) after the addition of 100 μM nitrite to RBCs (5% Hct) at 37°C and pH 7.4. Dashed lines show metHb at 75-mmHg (⧫) and 30-mmHg (○) Po2. Solid lines show HbNO at 75-mmHg (⧫) and 30-mmHg (○) Po2. Data were normalized to maximal metHb (99.2 nmol/μmol heme) and HbNO (1.81 nmol/μmol heme), respectively, and are means ± SE. Data were analyzed by two-way ANOVA and the Bonferroni post test. *P < 0.05 and **P < 0.001 vs. the corresponding time point at 75 mmHg for HbNO formation.
To ensure that measured intracellular nitrite levels were not affected by nitrite remaining from extracellular fractions, RBCs were washed either two, three, or four times. Intracellular nitrite levels remained the same with this procedure (not shown), suggesting that contamination from extracellular nitrite was unlikely. Other possible sources of intracellular nitrite are decomposition of HbNO (and subsequent NO autooxidation) upon sample processing and activity of RBC endothelial NO synthase (eNOS) (37, 63). We note that HbNO levels were four- to fivefold higher at lower oxygen tensions, suggesting that nitrite derived from this pool is unlikely to contribute to measured intracellular nitrite levels, which did not change with varying oxygen tensions. eNOS-dependent nitrite generation was excluded since all experiments were performed in the absence of exogenous l-arginine, and, moreover, the addition of the NO synthase inhibitor l-NAME (3 mM) did not affect either extracellular consumption rates (40.7 vs. 37.8 nmol·μmol heme−1·15 min−1 in the absence and presence of 3 mM l-NAME, respectively, P = 0.41) or intracellular nitrite levels [129.1 vs. 128.9 pmol·μmol heme−1·15 min−1 in the absence and presence of 3 mM l-NAME, respectively, P = 0.99 (not significant); oxygen tension: 30.5 mmHg, 15-min incubation, 37°C, 100 μM nitrite, significance determined by t-test].
The lack of effect of lowering fractional saturation on intracellular nitrite levels despite decreasing extracellular concentrations of nitrite at each time point provides evidence for the regulation of nitrite transport by Hb deoxygenation. This conclusion was derived by consideration of the scheme shown in Fig. 5, in which nitrite was assumed to enter the RBC in a reversible manner and independent of oxygen tension (i.e., HNO2/NO2− passive diffusion). In this model, the following criteria were applied: 1) deoxyHb reacts with nitrite faster compared with oxyHb; therefore, in this model, the only role of oxygen on nitrite consumption will be by the regulation of Hb fractional saturation; 2) nitrite transport is independent of oxygen tension; and 3) intracellular nitrite reaches steady state. These criteria were developed based on existing (36) and presented data (Fig. 4) and assuming the null hypothesis that nitrite transport was independent of oxygen tension. In this model (Fig. 5), the concentration of intracellular nitrite can be solved by applying a steady-state approximation, resulting in the following equation:
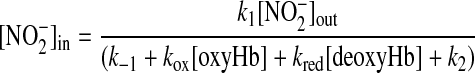 |
where [NO2−]in is the intracellular NO2− concentration, [NO2−]out is the extracellular NO2− concentration, kox is the rate constant for the reaction between nitrite and oxyHb, and kred is the rate constant for the reaction between nitrite and deoxyHb. Since [NO2−]in does not change with oxygen tension (Fig. 4C), the following four possibilities can be proposed: 1) Hb is not important for the intracellular metabolism of nitrite, i.e., k−1 and k2 >>> (kox[oxyHb] + kred[deoxyHb]) and k1, k−1, and k2 are unaffected by oxygen; 2) deoxygenation increases (kox[oxyHb] + kred[deoxyHb]) and decreases k2; 3) deoxygenation increases (kox[oxyHb] + kred[deoxyHb]) and increases nitrite import (k1); and 4) deoxygenation increases (kox[oxyHb] + kred[deoxyHb]) and decreases nitrite export (k−1). Since nitrite metabolism by RBCs was dependent on fractional saturation and inhibited by CO [suggesting Hb as the primary reaction target for nitrite, i.e., (kox[oxyHb] + kred[deoxyHb]) >>> k2], possibilities 1 and 2 can be excluded, leaving the possibility that to explain the lack of effect of deoxygenation on intracellular nitrite despite increased rates of consumption, deoxygenation also modulates nitrite transport by increasing import (k1) and/or decreasing export (k−1).
Fig. 5.

Model showing a proposed reaction scheme assuming that fractional saturation does not regulate nitrite transport. k1 and k−1 represent the rate constants for reversible nitrite transport across the membrane, and kox and kred are the rate constants for the reaction between nitrite and oxyHb or deoxyHb (dHb), respectively [where kred > kox and k2 represents other (non-Hb) sinks for nitrite consumption in the cell]. [NO2−]in is the intracellular nitrite concentration; [NO2−]out is the extracellular nitrite concentration.
AE-1 controls deoxygenation-dependent nitrite export.
We next tested the potential for AE-1 in the mediation of increased nitrite uptake by hypoxemic RBCs. The AE-1 inhibitor DIDS was added to RBCs, and hypoxia-dependent nitrite consumption and intracellular accumulation were measured. In the presence or absence of DIDS, lowering fractional saturation increased nitrite consumption to the same extent (Fig. 6A), suggesting that AE-1 is not involved in how nitrite enters the RBC. Interestingly, intracellular nitrite increased in DIDS-treated RBCs with the steady-state level being approximately three- to fourfold higher compared with control RBCs (Fig. 6B). These data suggest that AE-1 controls nitrite export from RBCs. To assess if an inhibition of an export pathway is responsible for maintaining intracellular nitrite levels in hypoxic RBCs (Fig. 4C), we hypothesized that intracellular nitrite levels would decrease when oxygen tension is lowered in DIDS-treated RBCs. In other words, if oxygen-dependent modulation of nitrite export through AE-1 is the mechanism responsible for maintaining constant intracellular nitrite levels, then in the presence of DIDS (where AE-1 is already inhibited), the only oxygen-sensitive step in RBC-nitrite interactions would be control over heme reactivity and not Hb-dependent effects on AE-1. The results shown in Fig. 6C demonstrate that in control cells, deoxygenation does not change intracellular steady-state nitrite concentrations compared with oxygenated RBCs, consistent with the data shown in Fig. 4C. However, in DIDS-treated cells, deoxygenation was associated with significant decreases in intracellular steady-state nitrite levels, suggesting that AE-1 is responsible for maintaining intracellular nitrite levels upon RBC deoxygenation. As a control, we investigated whether DIDS would affect cell-free deoxyHb reactivity with nitrite and observed no effect (not shown).
Fig. 6.

Effect of 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) on nitrite-RBC interactions. A: control and DIDS (100 μM)-treated RBCs (5% Hct) were equilibrated at either ∼75- or ∼30-mmHg Po2 at 37°C for 15 or 30 min, respectively, resulting in fractional saturations of ∼0.95 or ∼0.5, respectively. Nitrite (100 μM) was then added, and consumption from the extracellular compartment after 15 min was measured. Data are means ± SE; n = 8. *P < 0.01 by paired t-test relative to corresponding lower fractional saturation. NS, not significant by t-test. B: time-dependent changes in intracellular nitrite in control and DIDS-treated RBCs at a fractional saturation of 0.5. Data are means ± SE; n = 3. *P < 0.001 relative to the corresponding time point in control by two-way ANOVA and the Bonferroni post test. C: intracellular steady-state levels in control and DIDS-treated RBCs at oxygenated (0.95) and deoxygenated (0.5) fractional saturations. Data are means ± SE; n = 5. *P < 0.01 by paired t-test relative to the corresponding high fractional saturation condition. D: nitrite (100 μM) was added to control or DIDS-pretreated RBCs (5% Hct) at 27-mmHg Po2, 37°C, and pH 7.4, and RBC S-nitrosothiol (SNO) concentrations were measured at 60 min. Data are means ± SE; n = 3. Data were analyzed by a two-tailed Student's t-test. *P < 0.05.
Finally, to demonstrate that inhibition of export results in a greater nitrite reductase activity due to higher intracellular nitrite levels, RBC SNOs were measured. These species are relatively stable byproducts of nitrite reduction by deoxyHb (3, 11, 47) and evidence the intermediate formation of reactive nitrogen species, as recently demonstrated (5). Consistent with this prediction, the results shown in Fig. 6D demonstrate that RBC SNO levels were increased approximately twofold in DIDS-treated RBCs.
DISCUSSION
How nitrite crosses biological membranes, whether this is an active or passive transport process, and whether it is subject to regulation by electrochemical and/or concentration gradients remain unclear. Nitrite transporters have been identified in plants (58), but limited data exist in mammals. Interestingly, intraperitoneal administration of nitrite results in a rapid (within 5 min) redistribution through all major organs/tissues in the rat and results in nitrite concentrations that vary significantly from tissue to tissue (8), suggesting 1) rapidity in nitrite movement and 2) either regulation of nitrite transport and/or tissue-specific rates of nitrite metabolism. A recent study (54) using a liposomal model (in the absence of protein channels) suggest that despite being predominantly (>99.9%) in the anionic form at pH 7.4, the addition of sodium nitrite to the extracellular compartment results in the acidification of the liposomal interior, consistent with the generation of nitrous acid (HNO2) that can freely diffuse across the bilayer (54).
In this study, we explored nitrite transport in RBCs and the role of oxygen as a modulator of these processes. We chose this model because 1) RBCs are endowed with multiple oxygen-sensitive ion channels/transporters and 2) RBC-nitrite interactions have been implicated in the modulation of hypoxic blood flow. We used RBCs in which sufficient time had elapsed postcollection to deplete endogenous (basal) nitrite, precluding this as a variable in our experiments. Unlike other ions, the study of nitrite transport in RBCs is complicated by the existence of a significant intracellular sink for nitrite, i.e., Hb, whose reactions with nitrite change with fractional saturation and, hence, oxygen tension. Therefore, any mechanism addressing the regulation of transport must integrate differential reactivities of nitrite with oxyHb and deoxyHb. Moreover, this reactivity indicates that measurement of nitrite consumption from the extracellular compartment alone is not sufficient if transport mechanisms are being assessed.
Consistent with increased rates of nitrite reaction with deoxyHb, nitrite loss from the extracellular compartment increased with RBC deoxygenation and was inhibited by CO, supporting an important role for intracellular reactions with deoxyHb. Moreover, nitrite consumption was faster in lysed RBCs, suggesting that compartmentalization by the membrane is a critical mechanism in controlling nitrite reactions with erythrocytic Hb. We note that a recent study (7) using sheep RBCs did not observe an effect of the RBC membrane and may reflect differences in experimental conditions and/or species-based differences in how RBCs metabolize nitrite. In the context of the latter possibility, RBCs from different species have been shown to either consume nitrite faster or at the same rate when oxygenated conditions are compared with deoxygenated conditions underlying potential species to species variations in mechanisms that regulate RBC-nitrite reactions (18, 33, 34).
Intracellular nitrite levels increased and immediately (<5 min) attained a steady state. Moreover, this effect was oxygen independent, being observed at all Hb fractional saturations tested. This suggests that nitrite can enter RBCs in a facile and presumably reversible process, consistent with a previous study (43) showing rapid equilibration of nitrite across oxygenated the RBC membrane. Whether this is mediated by a specific channel or occurs via diffusion of nitrous acid with RBCs remains to be elucidated. Surprising was the fact that intracellular steady-state nitrite levels did not change with decreasing fractional saturation despite increased rates of nitrite consumption and reactions with deoxyheme. The fact that intracellular nitrite levels remained constant supports a mechanism in which Hb fractional saturation regulates nitrite transport and, moreover, the extent to which deoxygenation affects these transport processes reflects quantitatively the extent to which it increases the rate of Hb-nitrite reactions.
Previous data have suggested a role for AE-1 in the control of peroxynitrite permeation of the RBC membrane (16). With respect to nitrite specifically, previous work using human, pig, or carp RBCs has shown no effect of DIDS on nitrite consumption by oxygenated or deoxygenated RBCs (32, 34, 43, 64), consistent with the data shown in Fig. 6A of the present study. These previous studies did not directly measure the effects of AE-1 inhibition on intracellular nitrite levels, however, which we show do increase significantly (Fig. 6C). A role for AE-1 in the mediation of nitrite export has also been suggested by Shingles et al. (55) using Hb-free RBC ghosts under oxygenated conditions. Our data extend this concept to intact RBCs and also across oxygen fractional saturations. How AE-1-mediated nitrite export occurs and is regulated are not known. Importantly, inhibition of export by DIDS increases intracellular nitrite levels, which are reflected in higher concentrations of RBC SNO, consistent with faster velocities of nitrite-deoxyHb reactions. These data also highlight the potential role of AE-1 activity and circulating nitrite levels in determining RBC SNO concentrations, an area of considerable recent interest (12, 21, 49, 57).
We propose that maintaining intracellular nitrite levels during deoxygenation is achieved by deoxyHb-dependent inhibition of nitrite export via AE-1. Precedence for such a mechanism is provided first by studies (9, 45) showing that deoxyHb (T state conformation) has a higher affinity for binding to the NH2-terminus of AE-1 compared with oxyHb (R state conformation) and that this phenomenon is responsible for the displacement and activation of AE-1-bound glycolytic enzymes in RBCs under low fractional saturations. Moreover, deoxyHb binding to AE-1 has been shown to inhibit AE-1 activity in the context of sulfate transport (19). To investigate a nitrite export-based mechanism experimentally, we employed the AE-1 inhibitor DIDS. We reasoned that in the presence of DIDS, AE-1 will be already inhibited and therefore no longer subject to deoxyHb-mediated inhibition. In this scenario, we predicted that intracellular levels of nitrite would decrease upon deoxygenation due to faster reactions with heme. This result was observed experimentally, supporting this model. Taken together with intracellular nitrite levels increasing in the presence of DIDS, we propose the following scheme (Fig. 7) in which nitrite moves into the RBC down an electrochemical/concentration gradient through either a channel and/or as nitrous acid (Fig. 7, step 1). Under oxygenated conditions, the nitrite concentration gradient is regulated by intracellular reactions with oxyHb, resulting in nitrite oxidation (Fig. 7, step 2) and export via AE-1 (Fig. 7, step 3). As RBCs desaturate, nitrite consumption is accelerated due to deoxyHb reactions, resulting in a species [NOx, recently proposed to be N2O3 (5)] that can ultimately produce NO outside the RBC (Fig. 7, step 4) reaching maximal rates at the Hb P50. Concomitantly, deoxyHb binds to and inhibits AE-1, thereby preventing export and maintaining intracellular nitrite levels (Fig. 7, step 5). We propose that such a mechanism allows more nitrite to react with deoxyheme relative to being exported as RBCs transit from the arterial to venous circulation with the concomitant decrease in fractional saturation and thereby allowing for a more efficient coupling between hypoxia and vasodilation. Put another way, if nitrite transport (modulation of export) was not an oxygen-sensitive process, increased Hb reactivity at lower fractional saturations could result in intracellular nitrite levels being lowered to levels that may preclude significant rates of NO formation. Moreover, a recent study (53) has indicated that deoxyHb bound to AE-1 is a faster nitrite reductase compared with Hb in bulk solution. Taken together, this suggests a central role for Hb-AE-1 interactions in the regulation of nitrite reduction to NO by RBCs.
Fig. 7.

Scheme showing the proposed mechanism by which Hb deoxygenation regulates nitrite metabolism by RBCs. In step 1, nitrite moves into the RBC down an electrochemical/concentration gradient through either a channel and/or as nitrous acid. Under oxygenated conditions, the nitrite concentration gradient is regulated by intracellular reactions with oxyHb, resulting in nitrite oxidation (step 2) and export via anion exchanger (AE-1) (step 3). As RBCs desaturate, nitrite consumption is accelerated due to deoxyHb reactions, resulting in a species (NOx) that can ultimately produce NO outside the RBC (step 4) reaching maximal rates at the Hb P50. Concomitantly, deoxyHb binds to and inhibits AE-1, thereby preventing export and maintaining intracellular nitrite levels (step 5).
Limitations.
We note that our proposed model is based on results obtained by inhibiting AE-1 using DIDS, which, despite being a relatively specific inhibitor for AE-1 in isolated RBCs (30, 40) and being widely used to demonstrate a role for AE-1 in the control of transport of species that also react with Hb (e.g., peroxynitrite and NO) (16, 25), does not provide molecular-based evidence for a role of AE-1-deoxyHb interactions in the regulation of nitrite metabolism. We did test the effect DIDS on the ability of RBCs to modulate nitrite-dependent vasodilation at different oxygen tensions, as shown in Fig. 1; however, AE-1 inhibition also decreased NO scavenging (not shown), consistent with a previous report (25). This observation precludes the use of aortic ring vasodilation as a functional readout to assess the role of nitrite transport regulation on RBC nitrite reductase activity. Moreover, recent studies (9, 10) have shown that the incorporation of an antibody into permeabilized RBCs that specifically binds to the cytoplasmic NH2-terminal domain of AE-1 prevents deoxyHb binding to AE-1. We attempted to use this approach but observed significant lysis of resealed RBCs at low oxygen tensions (not shown), precluding this approach to assess nitrite transport mechanisms by RBCs. Further studies using molecular targeted approaches to modulate Hb-AE-1 interactions are required to definitely demonstrate a role for deoxyHb-AE-1 binding in the inhibition of nitrite export.
Conclusions.
In summary, the data presented here extend the paradigm of allosteric regulation of RBC-dependent nitrite reduction to include the inhibition of nitrite export. We propose that inhibition of nitrite export during hypoxia is crucial in ensuring increased rates of NO production from intraerythrocytic nitrite as the oxygen tension is lowered and speculate that this may be a general biological mechanism in the regulation of nitrite transport. Finally, these data suggest that inhibition of nitrite export is integrated with diverse RBC functions that include the allosteric regulation of glycolytic flux, ion homeostasis, and oxygen/NO delivery.
GRANTS
This work was supported by American Heart Association Southeast Affiliate Grant 0655312B (to R. P. Patel), Predoctoral Fellowship 0815248E (to D. A. Vitturi), and National Institutes of Health Grants HL-71189 and HL-074391 (to J. R. Lancaster, Jr.), U01-ES-015676 and RO1-HL-075540 (to S. Matalon), and U54-ES-017218 (to S. Matalon and R. P. Patel).
Acknowledgments
Present address of J. C. Toledo: Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André CEP 09210-170, Brazil.
REFERENCES
- 1.Adragna NC, Lauf PK. Role of nitrite, a nitric oxide derivative, in K-Cl cotransport activation of low-potassium sheep red blood cells. J Membr Biol 166: 157–167, 1998. [DOI] [PubMed] [Google Scholar]
- 2.Alzawahra WF, Talukder MA, Liu X, Samouilov A, Zweier JL. Heme proteins mediate the conversion of nitrite to nitric oxide in the vascular wall. Am J Physiol Heart Circ Physiol 295: H499–H508, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proc Natl Acad Sci USA 103: 8366–8371, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barvitenko NN, Adragna NC, Weber RE. Erythrocyte signal transduction pathways, their oxygenation dependence and functional significance. Cell Physiol Biochem 15: 1–18, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Basu S, Grubina R, Huang J, Conradie J, Huang Z, Jeffers A, Jiang A, He X, Azarov I, Seibert R, Mehta A, Patel R, King SB, Hogg N, Ghosh A, Gladwin MT, Kim-Shapiro DB. Catalytic generation of N2O3 by the concerted nitrite reductase and anhydrase activity of hemoglobin. Nat Chem Biol 3: 785–794, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Bertuglia S, Giusti A. Role of nitric oxide in capillary perfusion and oxygen delivery regulation during systemic hypoxia. Am J Physiol Heart Circ Physiol 288: H525–H531, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Blood AB, Power GG. In vitro and in vivo kinetic handling of nitrite in blood: effects of varying hemoglobin oxygen saturation. Am J Physiol Heart Circ Physiol 293: H1508–H1517, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Bryan NS, Fernandez BO, Bauer SM, Garcia-Saura MF, Milsom AB, Rassaf T, Maloney RE, Bharti A, Rodriguez J, Feelisch M. Nitrite is a signaling molecule and regulator of gene expression in mammalian tissues. Nat Chem Biol 1: 290–297, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci USA 102: 2402–2407, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campanella ME, Chu H, Wandersee NJ, Peters LL, Mohandas N, Gilligan DM, Low PS. Characterization of glycolytic enzyme interactions with murine erythrocyte membranes in wild-type and membrane protein knockout mice. Blood 112: 3900–3906, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, Yang BK, Waclawiw MA, Zalos G, Xu X, Huang KT, Shields H, Kim-Shapiro DB, Schechter AN, Cannon RO, 3rd, Gladwin MT. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med 9: 1498–1505, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Crawford JH, Chacko BK, Pruitt HM, Piknova B, Hogg N, Patel RP. Transduction of NO-bioactivity by the red blood cell in sepsis: novel mechanisms of vasodilation during acute inflammatory disease. Blood 104: 1375–1382, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, Kerby JD, Lang JD Jr, Kraus D, Ho C, Gladwin MT, Patel RP. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood 107: 566–574, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dalsgaard T, Simonsen U, Fago A. Nitrite-dependent vasodilation is facilitated by hypoxia and is independent of known NO-generating nitrite reductase activities. Am J Physiol Heart Circ Physiol 292: H3072–H3078, 2007. [DOI] [PubMed] [Google Scholar]
- 15.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood 106: 734–739, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Denicola A, Souza JM, Radi R. Diffusion of peroxynitrite across erythrocyte membranes. Proc Natl Acad Sci USA 95: 3566–3571, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drabkin DL, Austin JH. Spectrophotometric studies. II. Preparations from washed blood cells: nitric oxide, hemoglobin, and sulfhemoglobin. J Biol Chem 112: 51–65, 1935. [Google Scholar]
- 18.Feelisch M, Fernandez BO, Bryan NS, Garcia-Saura MF, Bauer S, Whitlock DR, Ford PC, Janero DR, Rodriguez J, Ashrafian H. Tissue processing of nitrite in hypoxia: an intricate interplay of nitric oxide-generating and -scavenging systems. J Biol Chem 283: 33927–33934, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galtieri A, Tellone E, Romano L, Misiti F, Bellocco E, Ficarra S, Russo A, Di Rosa D, Castagnola M, Giardina B, Messana I. Band-3 protein function in human erythrocytes: effect of oxygenation-deoxygenation. Biochim Biophys Acta 1564: 214–218, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Gibson JS, Cossins AR, Ellory JC. Oxygen-sensitive membrane transporters in vertebrate red cells. J Exp Biol 203: 1395–1407, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Gladwin MT, Lancaster JR Jr, Freeman BA, Schechter AN. Nitric oxide's reactions with hemoglobin: a view through the SNO-storm. Nat Med 9: 496–500, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Gladwin MT, Raat NJ, Shiva S, Dezfulian C, Hogg N, Kim-Shapiro DB, Patel RP. Nitrite as a vascular endocrine nitric oxide reservoir that contributes to hypoxic signaling, cytoprotection, and vasodilation. Am J Physiol Heart Circ Physiol 291: H2026–H2035, 2006. [DOI] [PubMed] [Google Scholar]
- 23.Gladwin MT, Schechter AN, Kim-Shapiro DB, Patel RP, Hogg N, Shiva S, Cannon RO 3rd, Kelm M, Wink DA, Espey MG, Oldfield EH, Pluta RM, Freeman BA, Lancaster JR Jr, Feelisch M, Lundberg JO. The emerging biology of the nitrite anion. Nat Chem Biol 1: 308–314, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Grubina R, Huang Z, Shiva S, Joshi MS, Azarov I, Basu S, Ringwood LA, Jiang A, Hogg N, Kim-Shapiro DB, Gladwin MT. Concerted nitric oxide formation and release from the simultaneous reactions of nitrite with deoxy- and oxyhemoglobin. J Biol Chem 282: 12916–12927, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Han TH, Pelling A, Jeon TJ, Gimzewski JK, Liao JC. Erythrocyte nitric oxide transport reduced by a submembrane cytoskeletal barrier. Biochim Biophys Acta 1723: 135–142, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Hendgen-Cotta UB, Merx MW, Shiva S, Schmitz J, Becher S, Klare JP, Steinhoff HJ, Goedecke A, Schrader J, Gladwin MT, Kelm M, Rassaf T. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc Natl Acad Sci USA 105: 10256–10261, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB, Hogg N. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem 280: 31126–31131, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115: 2099–2107, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isbell TS, Gladwin MT, Patel RP. Hemoglobin oxygen fractional saturation regulates nitrite-dependent vasodilation of aortic ring bioassays. Am J Physiol Heart Circ Physiol 293: H2565–H2572, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Jay D, Cantley L. Structural aspects of the red cell anion exchange protein. Annu Rev Biochem 55: 511–538, 1986. [DOI] [PubMed] [Google Scholar]
- 31.Jensen FB Nitric oxide formation from the reaction of nitrite with carp and rabbit hemoglobin at intermediate oxygen saturations. FEBS J 275: 3375–3387, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Jensen FB Nitrite and red cell function in carp: control factors for nitrite entry, membrane potassium ion permeation, oxygen affinity and methemoglobin formation. J Exp Biol 152: 149–166, 1990. [Google Scholar]
- 33.Jensen FB Nitrite disrupts multiple physiological functions in aquatic animals. Comp Biochem Physiol A Mol Integr Physiol 135: 9–24, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Jensen FB Nitrite transport into pig erythrocytes and its potential biological role. Acta Physiol Scand 184: 243–251, 2005. [DOI] [PubMed] [Google Scholar]
- 35.Kevil CG, Patel RP. Preserving vessel function during ischemic disease: new possibilities of inorganic nitrite therapy. Expert Rev Cardiovasc Ther 6: 1175–1179, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim-Shapiro DB, Gladwin MT, Patel RP, Hogg N. The reaction between nitrite and hemoglobin: the role of nitrite in hemoglobin-mediated hypoxic vasodilation. J Inorg Biochem 99: 237–246, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, Kumara I, Gharini P, Kabanova S, Ozuyaman B, Schnurch HG, Godecke A, Weber AA, Robenek M, Robenek H, Bloch W, Rosen P, Kelm M. Red blood cells express a functional endothelial nitric oxide synthase. Blood 107: 2943–2951, 2006. [DOI] [PubMed] [Google Scholar]
- 38.Kozlov AV, Costantino G, Sobhian B, Szalay L, Umar F, Nohl H, Bahrami S, Redl H. Mechanisms of vasodilatation induced by nitrite instillation in intestinal lumen: possible role of hemoglobin. Antioxid Redox Signal 7: 515–521, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Lang JD, Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, Liu Y, Jhala N, Crowe DR, Smith AB, Cross RC, Frenette L, Kelley EE, Wilhite DW, Hall CR, Page GP, Fallon MB, Bynon JS, Eckhoff DE, Patel RP. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J Clin Invest 117: 2583–2591, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lepke S, Fasold H, Pring M, Passow H. A study of the relationship between inhibition of anion exchange and binding to the red blood cell membrane of 4,4′-diisothiocyano stilbene-2,2′-disulfonic acid (DIDS) and its dihydro derivative (H2DIDS). J Membr Biol 29: 147–177, 1976. [DOI] [PubMed] [Google Scholar]
- 41.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7: 156–167, 2008. [DOI] [PubMed] [Google Scholar]
- 42.Maher AR, Milsom AB, Gunaruwan P, Abozguia K, Ahmed I, Weaver RA, Thomas P, Ashrafian H, Born GV, James PE, Frenneaux MP. Hypoxic modulation of exogenous nitrite-induced vasodilation in humans. Circulation 117: 670–677, 2008. [DOI] [PubMed] [Google Scholar]
- 43.May JM, Qu ZC, Xia L, Cobb CE. Nitrite uptake and metabolism and oxidant stress in human erythrocytes. Am J Physiol Cell Physiol 279: C1946–C1954, 2000. [DOI] [PubMed] [Google Scholar]
- 44.McNulty PH, Scott S, Kehoe V, Kozak M, Sinoway LI, Li J. Nitrite consumption in ischemic rat heart catalyzed by distinct blood-borne and tissue factors. Am J Physiol Heart Circ Physiol 295: H2143–H2148, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Messana I, Orlando M, Cassiano L, Pennacchietti L, Zuppi C, Castagnola M, Giardina B. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett 390: 25–28, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J Biol Chem 278: 46349–46356, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Nagababu E, Ramasamy S, Rifkind JM. S-nitrosohemoglobin: a mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide 15: 20–29, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Petersen MG, Dewilde S, Fago A. Reactions of ferrous neuroglobin and cytoglobin with nitrite under anaerobic conditions. J Inorg Biochem 102: 1777–1782, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Rassaf T, Bryan NS, Maloney RE, Specian V, Kelm M, Kalyanaraman B, Rodriguez J, Feelisch M. NO adducts in mammalian red blood cells: too much or too little? Nat Med 9: 481–483, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Rassaf T, Heiss C, Hendgen-Cotta U, Balzer J, Matern S, Kleinbongard P, Lee A, Lauer T, Kelm M. Plasma nitrite reserve and endothelial function in the human forearm circulation. Free Radic Biol Med 41: 295–301, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Rifkind JM, Nagababu E, Barbiro-Michaely E, Ramasamy S, Pluta RM, Mayevsky A. Nitrite infusion increases cerebral blood flow and decreases mean arterial blood pressure in rats: a role for red cell NO. Nitric Oxide 16: 448–456, 2007. [DOI] [PubMed] [Google Scholar]
- 52.Rogers SC, Khalatbari A, Datta BN, Ellery S, Paul V, Frenneaux MP, James PE. NO metabolite flux across the human coronary circulation. Cardiovasc Res 75: 434–441, 2007. [DOI] [PubMed] [Google Scholar]
- 53.Salhany JM Kinetics of reaction of nitrite with deoxy hemoglobin after rapid deoxygenation or predeoxygenation by dithionite measured in solution and bound to the cytoplasmic domain of band 3 (SLC4A1). Biochemistry 47: 6059–6072, 2008. [DOI] [PubMed] [Google Scholar]
- 54.Samouilov A, Woldman YY, Zweier JL, Khramtsov VV. Magnetic resonance study of the transmembrane nitrite diffusion. Nitric Oxide 16: 362–370, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shingles R, Roh MH, McCarty RE. Direct measurement of nitrite transport across erythrocyte membrane vesicles using the fluorescent probe, 6-methoxy-N-(3-sulfopropyl) quinolinium. J Bioenerg Biomembr 29: 611–616, 1997. [DOI] [PubMed] [Google Scholar]
- 56.Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley-Usmar VM, Gladwin MT. Deoxymyoglobin is a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res 100: 654–661, 2007. [DOI] [PubMed] [Google Scholar]
- 57.Stamler JS S-nitrosothiols in the blood: roles, amounts, and methods of analysis. Circ Res 94: 414–417, 2004. [DOI] [PubMed] [Google Scholar]
- 58.Sugiura M, Georgescu MN, Takahashi M. A nitrite transporter associated with nitrite uptake by higher plant chloroplasts. Plant Cell Physiol 48: 1022–1035, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Tsai AG, Johnson PC, Intaglietta M. Oxygen gradients in the microcirculation. Physiol Rev 83: 933–963, 2003. [DOI] [PubMed] [Google Scholar]
- 60.Tsujita K, Shiraishi T, Kakinuma K. Microspectrophotometry of nitric oxide-dependent changes in hemoglobin in single red blood cells incubated with stimulated macrophages. J Biochem 122: 264–270, 1997. [DOI] [PubMed] [Google Scholar]
- 61.Walder JA, Chatterjee R, Steck TL, Low PS, Musso GF, Kaiser ET, Rogers PH, Arnone A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. J Biol Chem 259: 10238–10246, 1984. [PubMed] [Google Scholar]
- 62.Webb A, Bond R, McLean P, Uppal R, Benjamin N, Ahluwalia A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc Natl Acad Sci USA 101: 13683–13688, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Webb AJ, Milsom AB, Rathod KS, Chu WL, Qureshi S, Lovell MJ, Lecomte FM, Perrett D, Raimondo C, Khoshbin E, Ahmed Z, Uppal R, Benjamin N, Hobbs AJ, Ahluwalia A. Mechanisms underlying erythrocyte and endothelial nitrite reduction to nitric oxide in hypoxia: role for xanthine oxidoreductase and endothelial nitric oxide synthase. Circ Res 103: 957–964, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zavodnik IB, Lapshina EA, Rekawiecka K, Zavodnik LB, Bartosz G, Bryszewska M. Membrane effects of nitrite-induced oxidation of human red blood cells. Biochim Biophys Acta 1421: 306–316, 1999. [DOI] [PubMed] [Google Scholar]
- 65.Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1: 804–809, 1995. [DOI] [PubMed] [Google Scholar]