

Abstract
Tri-n-octylphosphine oxide (TOPO) is the most commonly used solvent for the synthesis of colloidal nanocrystals. Here we show that the use of different batches of commercially obtained TOPO solvent introduces significant variability into the outcomes of CdSe quantum-wire syntheses. This irreproducibility is attributed to varying amounts of phosphorus-containing impurities in the different TOPO batches. We employ 31P NMR to identify 10 of the common TOPO impurities. Their beneficial, harmful, or negligible effects on quantum-wire growth are determined. The impurity di-n-octylphosphinic acid (DOPA) is found to be the important beneficial TOPO impurity for the reproducible growth of high-quality CdSe quantum wires. DOPA is shown to beneficially modify precursor reactivity through ligand substitution. The other significant TOPO impurities are ranked according to their abilities to similarly influence precursor reactivity. The results are likely of general relevance to most nanocrystal syntheses conducted in TOPO.
Introduction
We report identification of 10 phosphorus-containing impurities in commercially obtained tri-n-octylphosphine oxide (n-octyl3PO, TOPO) through spectroscopic analyses. The effects of individual impurities on the solution-liquid-solid (SLS)1,2 growth of CdSe quantum wires (QWs) are studied, and their roles in the synthesis are defined. The results allow optimization of the structural quality of the CdSe QWs, for straightness, defect densities, uniformity, diameter distributions, and length. Moreover, the study emphasizes the significant influences that trace TOPO-solvent impurities may exert over colloidal-nanocrystal growth.
TOPO has long been used for acetic-acid recovery3,4 and metal-ion extraction.3,5 It is currently available under the trade name CYANEX 921 for these purposes.3 More recently, TOPO has emerged as one of the most commonly used solvents for colloidal-nanocrystal synthesis, including semiconductor quantum dots (QDs),6–15 rods (QRs),16–27 and tetrapods.17,21,26–30 The origin of this usage is interesting, and is briefly recounted here.
The synthesis of semiconductor nanoparticles in reverse micelles was introduced in the 1980s.31–38 During that early synthetic era, the Steigerwald group discovered that poorly crystalline CdSe nanoparticles prepared in reverse micelles could be ripened and recrystallized in n-Bu3P/n-Bu3PO mixtures.39,40 The phosphine-oxide component was found necessary to the process.41 Shortly thereafter, Murray, Norris, and Bawendi introduced an organometallic synthesis of CdE nanocrystals (E = S, Se, and Te) using n-octyl3P (TOP)/TOPO solvent mixtures, which constituted a breakthrough in the preparation of semiconductor nanocrystals.6 Subsequently, TOPO and TOPO mixtures have been employed as solvents in a wide range of nanocrystal syntheses.7–30,42–53 More recently, “greener” syntheses of QDs and QRs employing non-coordinating solvents have emerged.54–58 However, TOPO remains an important option for the synthesis of semiconductor nanocrystals.6–30,59–70
The likely participation of adventitious TOPO impurities in nanocrystal growth has been recognized for several years.16 TOPO is available from research-chemical suppliers in nominal 90%-purity (technical) and 99%-purity grades (hereafter referred to as 90% TOPO and 99% TOPO, respectively). The 90% TOPO is known to contain a variety of phosphorus-containing impurities (Scheme 1): n-octylphosphonic acid (OPA), mono-n-octylphosphinic acid (MOPA), di-n-octylphosphine oxide (DOPO), di-n-octylphosphinic acid (DOPA), and n-octyl di-n-octylphosphinate (ODOP).71,72 The 99% TOPO contains some of the same impurities, in smaller amounts.67,72 Although such impurities were believed to influence the quality, morphology, and growth kinetics of QDs and QRs (see below),14,16,73 none of the active impurities had been specifically identified until our recent preliminary communication of this work.72
Scheme 1.
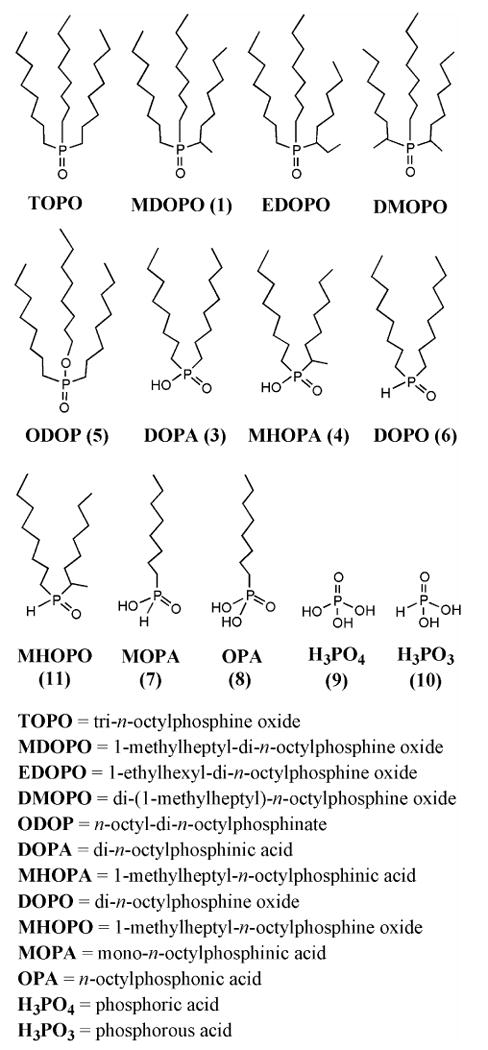
Molecular structures of TOPO and its suspected impurities. All are found in commercially obtained TOPO specimens except EDOPO and DMOPO.
In 2000, Alivisatos and coworkers reported that CdSe QRs grew at a slower rate and with better shape control in 90% TOPO than in 99% TOPO, apparently as a result of impurities present in the 90% solvent.16 The active impurities were presumed to be alkylphosphonic and/or alkylphosphinic acids. Purposeful addition of n-hexylphosphonic acid to 99% TOPO allowed the synthesis of CdSe QRs in a more controlled and reproducible manner,16 initiating extensive subsequent research activities and interest in semiconductor QRs. A separate study by Alivisatos and coworkers found that the growth kinetics and morphologies of hyperbranched nanocrystals varied when using 99% TOPO from different batches and/or suppliers.74 A related synthetic reproducibility problem was also reported for the growth of CdSe QDs, but attempts to analyze the influential TOPO impurities were unsuccessful.73 Similarly, we experienced significant irreproducibility problems in the growth of CdSe QWs2,59 using various batches of 99% TOPO.72
Such TOPO-induced irreproducibility could have at least three possible origins. (1) Essential, beneficial impurities may be present in some TOPO batches, in which high-quality QWs may be prepared, but absent in other batches. (2) Harmful impurities may be present in some TOPO batches, but absent in others. (3) Batch-to-batch variations in the concentrations of either beneficial or harmful impurities may be responsible for the synthetic irreproducibility. We note that if beneficial impurities are important (as we will show), then successful, reproducible syntheses cannot be achieved merely by using purified TOPO. Therefore, determining the effects of individual impurities present in TOPO on the SLS growth of CdSe QWs was the major goal of this work.
In this paper, we first analyze the impurities present in commercially obtained TOPO by 31P NMR spectroscopy, four of which were identified in our preliminary study.72 The spectroscopic behavior of individual impurities is concentration dependent, and influenced by TOPO and other species present in a TOPO specimen. Here we describe such dependences in detail, and identify six additional impurities that were not previously characterized.72 Second, we establish the influences of individual TOPO impurities on CdSe QW growth. As we previously reported,72 DOPA is the essential impurity for the synthesis of high-quality CdSe QWs.
The major new result presented here is an elucidation of the mechanism by which DOPA and other TOPO impurities influence nanocrystal syntheses. We provide experimental evidence that the beneficial role of DOPA is a modification of precursor reactivity through ligand substitution. Variations in precursor reactivity through impurity-based ligand substitutions influence the rate of CdSe generation, which in turn influences the nucleation and growth of nanocrystals, in either a harmful or beneficial manner. Here we rank the relative abilities of the significant TOPO impurities to produce precursors of greater or lesser reactivity toward CdSe formation. This proposed impurity (or additive) ranking should be generally useful for tuning precursor reactivity in nanocrystal syntheses conducted in TOPO.
Finally, we discuss how the spectrum of typical impurities (which includes all but two of the Scheme-1 compounds) arises from the TOPO-manufacturing process.
Experimental Section
Materials
n-Octylphosphonic acid (OPA, 99%, Lancaster), tri-n-octylphosphine (TOP, 90%, Aldrich), oleic acid (OA, 90%, Aldrich), selenium (Se, 99.99%, Aldrich), cadmium oxide (CdO, 99.99%, Aldrich), and 1-octadecene (ODE, 90%, Aldrich) were used as received. n-Hexadecylamine (HDA, 90%, Aldrich) was vacuum distilled at 200 °C (∼0.1 torr). Tri-n-octylphosphine oxide (TOPO, 90% and 99%) specimens were purchased from Aldrich, if not otherwise specified. The batch numbers for the 99% TOPO were 00529CD (TOPO A, for growth of high-quality CdSe QWs), 03008CE (TOPO F, resulting in poor-quality CdSe QWs), 08716AH (TOPO C, resulting in average-quality CdSe QWs). The batch numbers for the 90% TOPO were 04028DJ, 04220MB, 04301BA, 06520ED, 06820CH, 08506DD, B4751105 (Strem), and B1026024 (Strem). A CYANEX 921 sample (batch no. WE7060753B) was provided by Cytec Canada Inc. A 9.0-nm-diameter Bi-nanoparticle stock solution (containing 0.04 mmol Bi atoms/g solution) was prepared as previously described.75 The TOPSe stock solution was prepared by dissolving Se (4 g, 0.051 mol) into TOP (22 g, 0.059 mol) at ∼100 °C under dry, O2-free N2 (g). A second portion of Se (1 g, 0.013 mol) was added into the mixture to ensure all TOP had been converted to TOPSe. The liquid fraction of the mixture was subsequently used.
Purification of TOPO
Three TOPO samples were fractionally distilled under vacuum to decrease the concentrations of impurities (see Table S1 in the Supporting Information for impurity concentrations). TOPO F was purified by distillation, retaining the fraction transferred between 190–230 °C at 0.35 torr. 90% TOPO (04220MB) was purified by distillation, retaining the fraction transferred between 200–260 °C at 0.5 torr. The Strem 90% TOPO (B4751105) was purified by distillation, retaining the fraction transferred between 180–240 °C at 0.25 torr.
Purified TOPO (∼80 g) was obtained via recrystallization of TOPO C (100 g) in acetonitrile (200 mL).72 In a similar manner, purified TOPO could be obtained from 90% TOPO by double recrystallization. In a typical procedure, the 90% TOPO (04028DJ, 500 g) was dissolved in acetonitrile (2 L) at ∼80 °C. The solution was allowed to cool to room temperature to recrystallize the TOPO solute (1–2 days). The white solid isolated by suction filtration was redissolved in acetonitrile (1 L) at ∼80 °C. The solution was allowed to cool to room temperature overnight to recrystallize the TOPO solute. Suction filtration yielded shiny, free flowing, needle-like crystals (264 g, 57% overall yield based on the starting ∼465 g (500 g × 93%) of TOPO. All yields for TOPO reported below followed this calculation. If the 90% TOPO appeared to be sticky and oily (i.e., Strem B4751105), a fractional distillation was performed, followed by a double recrystallization (with ∼50% overall yield).
Syntheses of TOPO impurities
Syntheses of DOPA76, MOPA77, DOPO78, and ODOP79 were adapted from literature methods as described previously72 and in the Supporting Information. Syntheses of MDOPO, EDOPO, DMOPO, MHOPA, and MHOPO are described in the Supporting Information.
Synthesis of CdSe QWs
All synthetic procedures were conducted under dry, O2-free N2 (g), but the isolation and purification steps were conducted in the ambient atmosphere. In a typical preparation of high-quality CdSe QWs, CdO (6 mg, 0.047 mmol), OA (53 mg, 0.19 mmol), HDA (50 mg, 0.21 mmol), DOPA (10 mg, 0.034 mmol), and 99% or purified TOPO (5 g, 12.9 mmol) were loaded into a 50-mL Schlenk reaction tube. A Bi-nanoparticle stock solution (23 mg, 0.00092 mmol Bi atoms), TOPSe (500 mg, 1.1 mmol), and TOP (100 mg, 0.27 mmol) were combined in a separate vial, which was septum capped. The mixture of Bi nanoparticles and TOPSe was then loaded into a 3-mL syringe. The reaction mixture in the Schlenk tube was degassed under vacuum (0.01–0.1 torr) at 100 °C for 15 min, back-filled with N2(g), and then inserted into a 320 °C salt bath (NaNO3/KNO3, 46:54 by weight) to achieve a clear solution. (The time required to dissolve CdO in 90% and 99% TOPO and the optical clarity of the resulting solutions are listed in Table S2 in the Supporting Information). The Schlenk tube was switched to a 250 °C salt bath, and the mixture of Bi nanoparticles and TOPSe was quickly injected into the tube. A reddish to brown color resulted in about 5 s. The tube was withdrawn from the bath after 5 min and allowed to cool to room temperature.
The QWs were isolated as a brown precipitate from a 1-mL aliquot of the reaction mixture (before its solidification, or upon gentle warming to melt the TOPO) by adding toluene (ca. 2 mL) and methanol (ca. 2 mL), followed by centrifugation (benchtop centrifuge) and decanting of the supernatant. The precipitate was redispersed in toluene to form a uniform and optically clear light-brown dispersion for spectroscopic analysis. For TEM analysis, the QW precipitate was subjected to an additional dispersion-centrifugation cycle in a mixture of toluene (ca. 2 mL) and methanol (ca. 6 mL). The QWs ultimately were redispersed in toluene. Carbon-coated copper grids were dipped in the toluene solution of QWs and then immediately taken out to evaporate the solvent.
Synthesis of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n
This procedure was adapted from Peng and coworkers.57 Briefly, CdO (128 mg, 1.0 mmol), OA (593 mg, 2.1 mmol), DOPA (290 mg, 1.0 mmol), and ODE (3 g) were loaded into a 50-mL Schlenk reaction tube and degassed under vacuum (0.01–0.1 torr) at 100 °C for 15 min, back-filled with N2(g), then inserted into a 310 °C salt bath (NaNO3/KNO3, 46:54 by weight) to achieve a clear colorless solution (∼ 3min). The tube was withdrawn from the bath and allowed to cool to room temperature. Acetone was added to cause a white cloudy suspension. A translucent gel was separated by centrifugation, which was washed three additional times by acetone, and finally dried under dynamic vacuum for 3 h to yield a clear gel-like solid (650 mg, 0.94 mmol, 94%). 31P{1H} NMR (δ, ppm, d8-toluene, 55 °C): 54.2 (Figure 6a). 113Cd NMR (δ, ppm, d8-toluene, 25 °C): −10.5 (Figure 6b). IR (KBr, cm-1, 25 °C): 3005 (sh), 2955 (vs), 2923 (vs), 2853 (vs), 1556 (s), 1466 (s), 1411 (s), 1305 (w), 1238 (w), 1197 (w), 1200 (s), 1012 (s), 909 (w), 814 (w), 722 (m), 496 (m) (Figure S30). Anal. Calcd for CdC34.32H64.32P0.84O4: C, 60.40; H, 9.80; P, 3.83. Found: C, 63.48; H, 10.42; P, 3.83 (by Galbraith Laboratories Inc.).
Figure 6.
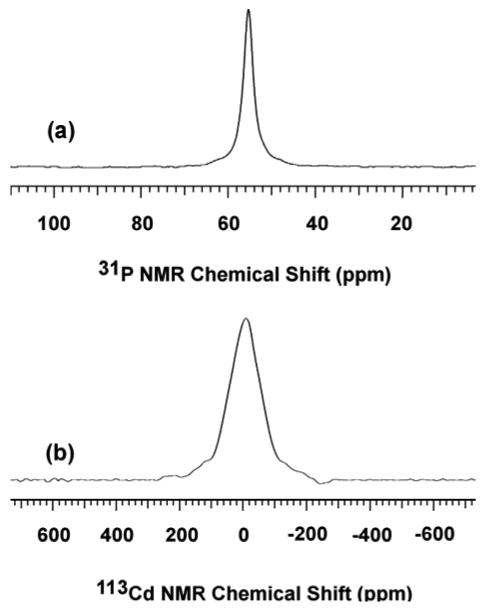
31P{1H} (a) and 113Cd (b) NMR spectra of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n collected in d8-toluene at 55 and 25 °C, respectively.
NMR Methods
Variable-temperature 31P{1H} NMR spectra of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n were acquired at 121.4 MHz in d8-toluene on a Varian INOVA-300 spectrometer, and the chemical shifts reported (in ppm) relative to external 85% phosphoric acid (δ = 0 ppm). The 113Cd NMR spectrum of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n was acquired at 133.0 MHz in d8-toluene on a Varian Unity Inova-600 spectrometer and the chemical shift (in ppm) was referenced to external 0.1 M aqueous cadmium perchlorate (Cd(ClO4)2, δ = 0 ppm).
1H, 13C{1H}, and 31P NMR spectra of TOPO and TOPO impurities were acquired on a Varian Unity Plus-300 spectrometer, if not otherwise specified, with CDCl3 or d6-acetone as solvent. 1H and 13C{1H} NMR spectra were recorded at 300 MHz and 75 MHz, respectively, and the chemical shifts (in ppm) were referenced to CDCl3 (δH= 7.24 ppm, δC= 77.2 ppm). 31P NMR spectra were recorded in d6-acetone (instead of the CDCl3 previously used72) at 121.4 MHz and the chemical shifts were referenced to external 85% phosphoric acid (δ = 0 ppm). The FID data were transformed with a 1-Hz line broadening, allowing the TOPO 13C satellites and the impurity-2 resonance to be resolved. (The spectroscopic behavior of individual impurities is less concentration dependent in d6-acetone than in CDCl3.) A mixture of TOPO and an independently synthesized or obtained impurity in d6-acetone was heated at ∼50 °C for 2 min in a sealed NMR tube with frequent shaking to ensure thorough mixing, and cooled to room temperature before 31P NMR analysis.
The molar concentrations of TOPO and impurities were estimated from peak integrals in the 31P{1H} NMR spectra. To ensure all the 31P NMR signals were fully relaxed, a series of relaxation delays (2, 5, 10, 15, 30, and 50 s) were tested using a standard mixture of recrystallized TOPO, DOPA, DOPO, OPA, and H3PO3 with known molar ratio. The measured ratio from the peak-area integration obtained at relaxation delay of ≥ 5 s was consistent with the known ratio. To further ensure that 5 s was appropriate for estimating the concentrations of other phosphorous-containing impurities, relaxation delays of 5, 10, 15, and 30 s were tested on a 90% TOPO (08506DD) containing the greatest number of impurities. The measured concentrations of impurities from the four tested delays were essentially the same, thus the 5-s relaxation delay was applied for all the 31P{1H} NMR experiments.
The impurity-2 resonance generally overlapped with the TOPO resonance (see the text and Figures 2 and S13 for details). To extrapolate a more accurate impurity-2 concentration, DOPA was added to TOPO so that the impurity-2 peak shifted away from the TOPO peak (see Figure 3), although it some times still overlapped with one of the TOPO 13C satellites. The integrated peak area of impurity 2, from which the contribution from the TOPO 13C satellite was deducted, was finally subtracted from the peak area of TOPO. The measurement errors for TOPO and impurity 2 were approximately ± 0.5–1%. The errors for other impurities were approximately ± 0.05–0.2%.
Figure 2.
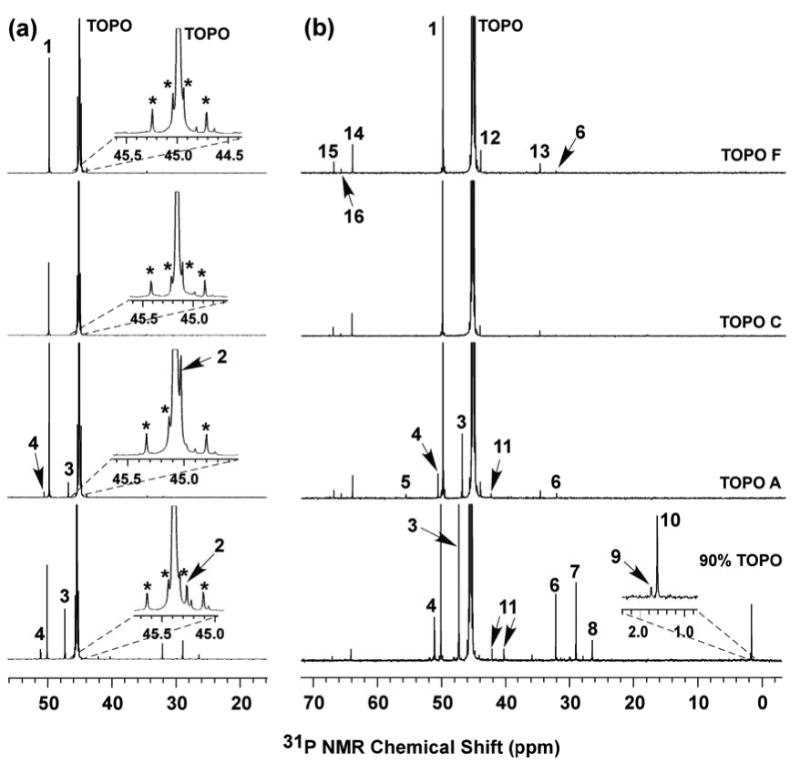
31P{1H} NMR spectra of TOPO F (03008CE), TOPO C (08716AH), TOPO A (00529CD), and 90% TOPO (06520ED) collected in d6-acetone. The peak intensities for TOPO (∼45 ppm) were normalized and the impurities were arbitrarily labeled with compound numbers. (a) The TOPO peak intensities were decreased to a lower value to show impurity 1. Insets: magnified view of the TOPO peaks to show impurity 2 and the 13C satellites (*). (b) The TOPO peak intensities were increased to a higher value to show all the impurity peaks. Inset: magnified view of impurities 9 and 10 in the 90% TOPO (06520ED).
Figure 3.
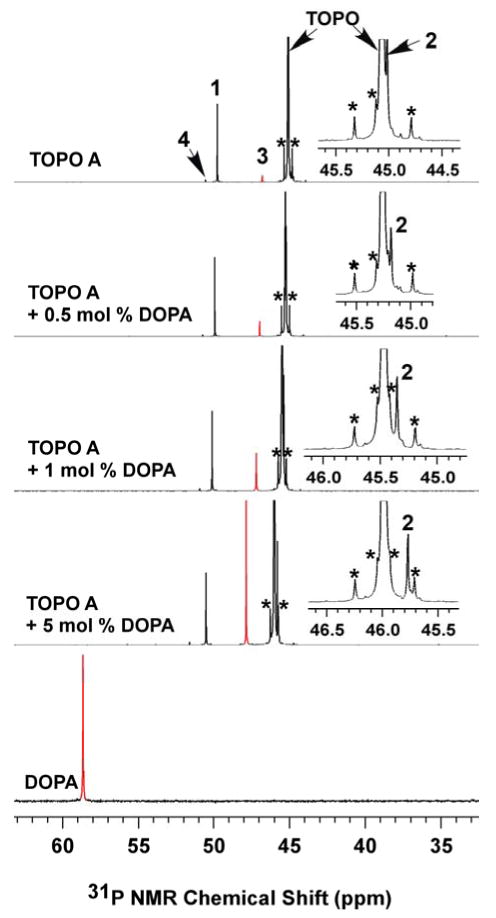
31P{1H} NMR spectra for identifying DOPA in TOPO A (00529CD). The spectra were collected in d6-acetone. The peak intensities for TOPO were normalized and the DOPA peaks labeled in red. Note the DOPA resonance in the mixtures shifted upfield from its pure form, whereas the resonance of TOPO and other impurities shifted downfield with increasing DOPA concentration. Insets: magnified view of the TOPO peaks to show the shifting of impurity 2. The 13C satellites (*) are labeled.
Results
Synthesis of CdSe QWs in various batches of TOPO
In this study, we compared the synthetic utility of three batches of 99% and several batches of 90% TOPO. The three 99% specimens are labeled with the performance letter grades TOPO A, TOPO C, and TOPO F, corresponding to the excellent, average, and failed synthetic results achieved in each of these specimens, respectively (see below). The Aldrich/Strem batch numbers for the 99% and 90% specimens employed are listed in the Experimental Section.
The synthetic procedure for the SLS growth of CdSe QWs (eq 1) was adapted from a reported method,59 with the optimization of some reaction parameters as previously reported.2 In the present syntheses (eq 1), the previously used selenium reagent tri-n-butylphosphine selenide (TBPSe) was replaced with tri-n-octylphosphine selenide (TOPSe).
| (1) |
As mentioned above, high-quality CdSe QWs were not reproducibly synthesized in the various 99% TOPO batches. The QWs grown in TOPO A were long (>5 μm) and straight, with uniform diameters along their lengths and narrow diameter distributions (Figure 1a,b). In contrast, the QWs grown in TOPO F were short (<1 μm), and exhibited kinks, branches, diameter fluctuations along their lengths, and wide diameter distributions (Figure 1c,d). The quality of the QWs grown in TOPO C (Figure 1e,f) was clearly improved over those from TOPO F; however, it was lower than that of the QWs grown in TOPO A. The varying qualities of these QW specimens were also evident in the absorption spectra (Figure S10). The first four excitonic features were sharper and better resolved for QWs grown in TOPO A as compared to those grown in TOPO C or TOPO F.
Figure 1.

Representative TEM images of CdSe QWs grown in TOPO A (a, b), TOPO F (c, d), and TOPO C (e, f) having mean diameters of 8.4 nm ± 11%, 7.6 nm ± 17%, and 7.9 nm ± 18%, respectively (± one standard deviation in the diameter distribution, expressed as a percentage of the mean diameter). The right panels show lower-magnification images of the left panels.
The quality of the CdSe QWs grown in 90% TOPO samples was even lower than that of QWs grown in 99% TOPO C or F (Figure S12). The poorly formed wires, other irregularly shaped nanocrystals, and occasional presence of dots indicated poor control over wire growth, which we attributed to the presence of impurities in the TOPO samples. Notably, the quality of the QWs was little improved after recrystallization or distillation of 99% TOPO C, TOPO F, or 90% TOPO (Figures S11 and S12). We will show below that a single recrystallization or distillation of 90% TOPO is insufficient to completely remove all harmful impurities, and that recrystallized 99% TOPO is lacking a beneficial impurity (DOPA).
31P NMR spectroscopic analysis of TOPO impurities
The 31P{1H} NMR spectra for TOPO specimens used in this study are shown in Figures 2 and S13. The TOPO resonance appeared at δ ≈ 44.5–46.5 ppm, along with many small resonances arising from impurities. 7–12 impurity peaks were found in 99% TOPO (see also Table S1), whereas over 16 impurity peaks were found in 90% TOPO (see Figure S13 for the 31P NMR spectra of other 90% TOPO samples). The purities of the various TOPO specimens were determined by calibrated integration of the 31P NMR spectra (see Experimental), and were found to vary from 93 to 98 mol % for 99% TOPO, and from 87 to 95 mol % for 90% TOPO (see Table S1).
The impurity species in Figure 2 were arbitrarily assigned the compound labels 1–16. TOPO A and TOPO F differed by the presence of impurities 2, 3, 4, 5, and 11 and a larger amount of impurity 1 in TOPO A (Figure 2 and Table S1). The remaining impurities in TOPO A and F were present in similar amounts, and thus were unlikely to be significantly beneficial or harmful impurities. TOPO C and TOPO F differed by the presence of impurity 6 and larger amounts of impurities 1 and 12 in TOPO F. Comparisons of the amount of impurity 1 present in these three TOPO specimens (3.1 mol % in A, 2.1 mol % in F, and 1.7 mol % in C) suggested that impurity 1 was unlikely a beneficial or harmful impurity. Additionally, the QWs synthesized in TOPO C before and after recrystallization were of similar quality. We will show below that recrystallization of 99% TOPO reduced all impurity levels, including 1 and 12, below our 31P NMR detection limit, indicating that impurities 1 and 12–16, which were present in TOPO C, were not significantly influential to the wire growth.
The above reasoning suggested that impurity 6 may have been a harmful impurity in TOPO F, and that impurities 2, 3, 4, 5, and 11 may have been beneficial impurities in TOPO A. Our next task was thus to identify impurities 2, 3, 4, 5, 6, and 11 among the Scheme-1 substances. Therefore, each of the Scheme-1 compounds was either obtained commercially or independently synthesized by adapting literature procedures (as detailed in the Supporting Information). These independent specimens were used to identify compounds 3, 4, 5, 6, and 11 by 31P NMR, as described below. Compound 2 has not been positively identified, but is not necessary to the growth of high-quality CdSe QWs (see below).
Impurity 3 was identified as DOPA by measuring the 31P NMR spectrum of DOPA (d6-acetone) in the presence and absence of TOPO. Like many Scheme-1 compounds, DOPA exhibited a concentration-dependent chemical shift in mixtures with TOPO, due to hydrogen bonding. DOPA exhibited a chemical shift of 58.6 ppm in the absence of TOPO (Figure 3). However, when a small concentration of DOPA (0.5 mol %) was added to TOPO A, the resonance for impurity 3 was enhanced and shifted to 47.0 ppm (Figure 3), slightly downfield of its position in TOPO A and significantly upfield of the chemical shift of “pure” DOPA (in d6-acetone). As the amount of DOPA added to TOPO A was increased, the impurity-3 resonance was further enhanced and progressively shifted downfield, towards the position of “pure” DOPA. Consequently, DOPA and 3 are the same substance, with a concentration of 0.44 mol % in TOPO A. DOPA was similarly identified as 3 in 90% TOPO (Table S1), in which its concentration varied from 0.3–3 mol %.
In the course of the DOPA identification, we observed that the impurity-4 resonance remained a constant 3.8 ppm downfield of the DOPA resonance in TOPO (Figure 3), irrespective of the concentration of DOPA and therefore the absolute chemical shift of DOPA (Table S1, Figures 2, 3 and S13). Because of its similar spectroscopic behavior, we surmised that impurity 4 was possibly a DOPA isomer with a branched octyl (i.e., 1-methylheptyl) chain. This was confirmed by addition of the DOPA isomer MHOPA (Scheme 1) into 90% TOPO, for which the MHOPA resonance merged with that of 4 (see Figure S14).
Figure 4 describes the identification of impurity 6 as DOPO. The resonance for DOPO added to a 90%-TOPO sample superposed with that of impurity 6, at 32.1 ppm. The proton-coupled 31P NMR spectrum of DOPO matched that of impurity 6 in 90% TOPO, giving the expected doublet with a coupling constant of 1JH-P = 446 Hz. DOPO (6) was also found in TOPO A, TOPO F, and some of the other 90% TOPO samples (Table S1), in which its concentrations varied from 0.01–1.3 mol %. The branched-chain MHOPO isomers (Scheme 1) were similarly identified as impurity 11 (consisting of two diastereomers) in some TOPO specimens (see Table S1 and Figure S15).
Figure 4.
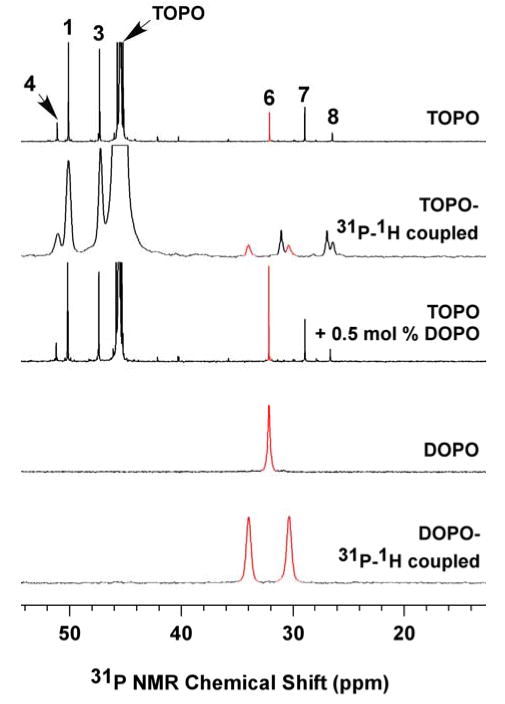
31P NMR spectra (in d6-acetone) for identifying DOPO in 90% TOPO (06520ED). The 31P{1H} and 31P–1H coupled NMR spectra are indicated. The peak intensities for TOPO in the 31P{1H} NMR spectra were normalized. The DOPO peaks are labeled in red.
Through related 31P NMR experiments, six additional TOPO impurities were identified as indicated by the compound numbers (labels) in Scheme 1. The details of these experiments and the spectroscopic results are given in the Supporting Information. The most-prominent impurity in nearly all of the TOPO specimens, compound 1, was confirmed to be the branched-chain TOPO isomer MDOPO. It is neither a harmful or beneficial impurity. Other significant impurities in the 90%-TOPO specimens (but not in the 99% specimens) were MOPA (7), OPA (8), phosphoric acid (H3PO4, 9) and phosphorous acid (aka phosphonic acid, H3PO3, 10). The synthetic influences of these impurities are discussed later.
As noted above, another prominent impurity, compound 2, was not positively identified. The 31P NMR chemical shift of 2 was very close to that of TOPO, such that it was always observed on the high-field shoulder of the TOPO resonance (Figure 2a). Because the chemical shift of 2 (relative to TOPO) was unchanged after addition of NH4OH (aq, Figure S23), it contained no acidic proton and therefore no P-OH group. We concluded that 2 was likely a trialkylphosphine oxide, like TOPO. The two likely branched-chain TOPO isomers EDOPO and DMOPO (Scheme 1) were synthesized, but their chemical shifts were significantly downfield from that of TOPO (Figure S19), excluding them as significant impurities. We speculate that 2 is a trialkylphosphine oxide with two n-octyl groups and one longer-chain, unbranched substituent (see the Discussion). As such its 31P NMR chemical shift and chemical properties would be very similar to those of TOPO.
Purification of 90% and 99% TOPO
A single recrystallization of 99% TOPO in acetonitrile was sufficient to reduce the levels of all P-containing impurities below the 31P NMR detection limit (see Figure S21); the TOPO was recovered in 80% yield. However, a single recrystallization of 90% TOPO proceeded in lower yield (72–78%), and provided material still containing appreciable amounts of impurities 1, 3, 4, 7, 8, and 9 (Figure S21). Fractional distillation (in vacuo) of 90% TOPO proceeded in higher yield (89%), but the impurities noted above were retained, along with 5, 11, and others (Figure S22). Singly recrystallized or singly distilled 90% TOPO gave very poor results for the synthesis of CdSe QWs (Figures S11e, f, and S12f).
We established the following protocol for purifying 90% TOPO. Some batches of 90% TOPO were dry, white, opaque, polycrystalline powders. Such batches contained comparatively lower initial impurity levels, and could be purified to leave no 31P-NMR-detectable impurities with two consecutive recrystallizations in acetonitrile (55–60% overall yield). Other batches of 90% TOPO contained higher initial impurity levels, and appeared as sticky, glistening (“wet” or oily), nearly translucent, chunky solids. These batches were first vacuum distilled, and then twice recrystallized in acetonitrile to afford purified TOPO with no 31P-NMR-detectable impurities (50% overall yield). The purified TOPO was a free-flowing white powder consisting of small, needle-shaped crystals.
Purified TOPO from 99% and 90% purity grades was equally suitable for synthetic use (see below). Of course, purification of 90% TOPO required more steps and proceeded in lower overall yield than did the purification of 99% TOPO. Even so, 99% TOPO (from the typical research-chemical suppliers) is approximately ten times as costly as 90% TOPO. Therefore, researchers adept with standard preparative techniques will likely find the purification and use of 90% TOPO to be time and cost effective.
Interestingly, we found that the 31P NMR spectra of the commodity chemical CYANEX 921 (provided by Cytec Canada Inc.) and 90% TOPO were essentially identical (Figure S24), indicating that the materials were most likely manufactured by the same process, or may even be the same material relabeled as 90% TOPO for small-quantity distribution. CYANEX 921 must be purchased on a scale too large for most research laboratories (200 kg minimum). However, the purification method we used could be translated to a standard chemical manufacturing practice, which should not severely impact the price of a purified commodity-scale TOPO for commercial production of SLS-grown CdSe QWs. Although commercial applications of semiconductor nanowires do not appear to be imminent, and while commercialization of such materials might eventually be impeded by application and environmental factors, the availability and affordability of raw materials to manufacture commercial quantities of CdSe QWs should not be a barrier. The same may be true for other colloidal nanocrystals synthesized in TOPO.
Effects of impurities on the growth of CdSe QWs
The results above suggested that DOPA (3), MHOPA (4), ODOP (5), and/or MHOPO (11) could be beneficial to the growth of CdSe QWs in TOPO, because they were present in TOPO A, but absent from TOPO C. Furthermore, H3PO4 (9), H3PO3 (10), DOPO (6), MOPA (7), and OPA (8) were identified as potentially harmful impurities, because they were present in singly distilled or singly recrystallized 90% TOPO. Consequently, the compounds DOPA, ODOP, DOPO, OPA, MOPA, H3PO4, and H3PO3 were tested individually to determine their effects on the growth of CdSe QWs. Syntheses of QWs were conducted in purified TOPO (recrystallized TOPO C) with the addition of measured amounts of the independently synthesized or obtained impurity species listed above.
Figure 5 shows representative TEM images of CdSe QWs synthesized in the presence of DOPA. The QWs grown with DOPA (5 mg, 0.13 mol%, Figure 5a,b) were straighter, much longer (>10 μm), exhibited narrower diameter distributions, and had more-uniform diameters along their lengths than did those grown in purified TOPO without added DOPA (Figure S11c, d). The quality of the QWs synthesized with added DOPA was comparable to that of the QWs synthesized in TOPO A (Figures 1 and S10). As the amount of added DOPA was increased, the QWs became slightly shorter, but the straightness, uniformity, and narrow diameter distributions were retained (Figure 5c–f). That TOPO A contains a comparable concentration of DOPA (0.44 mol %) as those tested here (0.13–0.53 mol %) established DOPA as the primary beneficial impurity in TOPO A. However, the addition of large amounts of DOPA (i.e., 100 mg or 2.65 mol %) resulted in low-solubility QWs having broad diameter distributions (Figure S25).
Figure 5.

Representative TEM images of CdSe QWs grown in recrystallized TOPO C with addition of (a, b) 5, (c, d) 10, and (e, f) 20 mg of DOPA having mean diameters of 8.2 nm ± 11%, 8.2 nm ± 11%, and 8.8 nm ± 11%, respectively (± one standard deviation in the diameter distribution, expressed as a percentage of the mean diameter). The right panels show lower-magnification images of the left panels.
The additives MOPA and OPA were similarly found to have beneficial effects on QW growth, but less than those achieved with DOPA. Addition of MOPA (10 mg, 0.4 mol %) improved the straightness and diameter distribution of the QWs (Figure S26b) relative to those obtained in purified TOPO (Figure S11c, d), or in the presence of a lower concentration of MOPA (5 mg, 0.2 mol %, Figure S26a). However, larger concentrations of MOPA (20 mg, 0.6 mol %) produced shorter (<500 nm) and tapered wires (Figure S26c). Straight QWs having lengths of <2 μm and uniform diameters along their lengths were grown in the presence of 5 mg of OPA (0.2 mol%, Figure S26d), whereas larger amounts of OPA induced short (<1 μm), branched, and slightly tapered wires (Figure S26e, f). Neither MOPA or OPA could reproduce the optimal results obtained with DOPA.
Other additives were found to have deleterious effects on QW growth. DOPO, even in a small amount (i.e., 5 mg, 0.1 mol %), induced the growth of CdSe QDs (Figure S27a–c), whereas wires grown in the presence of ODOP (20 mg, 0.4 mol %) were short (<1 μm), with kinks and branching (Figure S27f). The deleterious effects of ODOP were much less noticeable at lower ODOP concentrations (Figure S27d).72 The acids H3PO3 (4 mg, 0.4 mol %) and H3PO4 (5 mg, 0.4 mol %) generated insoluble white suspensions when combined with the precursor mixture containing CdO, oleic acid (OA), HDA, and TOPO. No QWs or other CdSe nanostructures were subsequently produced from the corresponding reaction mixtures.
Finally, we demonstrated the growth of high-quality CdSe QWs from the initially unsuccessful 99% TOPO specimens (TOPO C and TOPO F), by adding to them the beneficial impurities DOPA, MOPA, or OPA. Figure S28 shows representative TEM images of QWs grown in the presence of DOPA (10 mg, 0.26 mol%), MOPA (10 mg, 0.40 mol%), or OPA (10 mg, 0.40 mol%). The straightness and diameter uniformity of the QWs were dramatically improved over those achieved in TOPO C or TOPO F. The best results were obtained with DOPA (10 mg, 0.26 mol %), as expected, for which very narrow diameter distributions were obtained (Figure S28a, b). Therefore, 99% TOPO specimens are sufficiently free of deleterious impurities that only an appropriate concentration of DOPA is required to produce excellent synthetic results. That is, 99% TOPO can probably be used without further purification.
The results above established that small impurity concentrations (0.1–0.5 mol %) dramatically influenced the synthesis of CdSe QWs. MOPA, OPA, and especially DOPA were beneficial at low concentrations, whereas larger amounts were detrimental to the quality of the wires. DOPO, H3PO3, and H3PO4 were deleterious at all concentrations studied. We speculate that small concentrations of MHOPA (4, the branched-chain DOPA isomer), as was DOPA, would be beneficial to the wire growth, whereas MHOPO (11, the branched-chain DOPO isomers), as was DOPO, would be deleterious. The observant will note that the best 99% TOPO specimen, TOPO A, contained both DOPA and DOPO, indicating that the beneficial effects of DOPA counteracted the harmful effects of the small amount of DOPO present. We return to this point in the Discussion.
Although the various beneficial and harmful impurities were influential at comparatively low concentrations in TOPO, we note that they were present in molar quantities comparable to those of precursor species such as Cd(oleate)2. For example, a typical, optimized QW synthesis employed 0.047 mmol of CdO and 0.034 mmol of DOPA. Therefore, the impurity levels in the commercially obtained 90% and 99% TOPO specimens, and the optimal levels of beneficial additives to purified TOPO, were stoichiometrically significant.
We also considered if impurities in the reagents employed might influence the synthetic results. For example, two prior studies of CdSe-nanocrystal synthesis found that the purity of n-octadecylphosphonic-acid reagents significantly influenced the resulting nanocrystal morphologies.30,68 The TOP and OA reagents we used for CdSe QW synthesis were of technical-grade purity (nominally 90%). Although these reagents were used in much lower quantities than the TOPO solvent, a small probability allowed that the impurities in the technical-grade reagents might achieve stoichiometrically significant levels. However, the CdSe QW syntheses that we conducted using 97% TOP and/or 99% OA in 99% TOPO gave no discernible improvement in the quality of the QWs. We therefore concluded that the important active impurities were contained in the TOPO solvent.
Precursor modification by DOPA
We next sought to determine the mechanism by which DOPA beneficially influenced the growth of CdSe QWs. A possible mechanism was imagined to involve incorporation of DOPA into the Cd precursor, which was nominally [Cd(oleate)2]n. The reaction chemistry employed for the synthesis of CdSe QWs (eq 1) was adapted from Peng and coworkers.13 Following this method, CdO, OA, and TOPO were initially combined at high temperature to generate [Cd(oleate)2]n in situ and prior to the addition of the Se precursor. [Cd(oleate)2]n is a soluble oligomer or polymer under the reaction conditions.67 We considered that, under the reaction conditions, the DOPA impurity in TOPO may exchange into [Cd(oleate)2]n, generating a mixed-ligand precursor containing di-n-octylphosphinate, (n-octyl)2PO2–, ligands. If all of the available DOPA exchanged into [Cd(oleate)2]n, then under our optimal conditions (see the Experimental section) the Cd precursor would become {Cd[O2P(n-octyl)2]0.72[oleate]1.28}n.
To examine this possibility, CdO, OA, and DOPA were combined in the hydrocarbon solvent 1-octadecene (ODE) at 310 °C in the molar ratio 1:2.1:1, respectively, generating a homogeneous solution. The solution was cooled and acetone was added to precipitate a clear, colorless, gel-like solid. This solid product was characterized by spectroscopic and elemental analyses. (A corresponding experiment conducted in TOPO solvent also resulted in a homogeneous solution at high temperature, but ultimately afforded an insoluble product that was not easily amenable to further spectroscopic characterization.)
The characterization results identified the product as the mixed-ligand complex {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n. The 31P and 113Cd spectra of the product are shown in Figure 6. Both contained a single, broad resonance, consistent with a fluxional, oligomeric structure. We note that the broad 31P NMR resonance (at 54.2 ppm) corresponding to the (n-octyl)2PO2– ligands was shifted from that of free, neutral DOPA (58.6 ppm, see Figure 3). The 1H NMR spectrum was similarly broad. The IR spectrum contained characteristic bands for the di-n-octylphosphinate and oleate ligands (see Figure S30). Finally, elemental analyses confirmed the empirical formula given above. The results established that under the reaction conditions employed for the synthesis of CdSe QWs, the DOPA “impurity” in TOPO rapidly exchanged into the Cd-precursor complex, affording a mixed-ligand precursor of the general formula {Cd[O2P(n-octyl)2]x[oleate]y}n. This mixed-ligand precursor exhibited much greater thermal stability than did the oleate precursor [Cd(oleate)2]n, which quickly decomposed to a gray insoluble material at 310 °C in ODE, previously shown to be nanocrystalline cadmium.80 Therefore, we argue below that the incorporation of (n-octyl)2PO2– ligands beneficially modified the reactivity of the precursor, aiding the formation of high-quality QWs.
Discussion
Origins of TOPO impurities
TOPO is manufactured by the sequential radical addition of 1-octene to PH3, followed by oxidation of the P(III) center (see the central, black branch of Scheme 2).81,82 The varying numbers of alkyl chains, oxygen contents, and structures of the chains in the typical TOPO impurities (Scheme 1) are consistent with this manufacturing process. The Scheme-1 impurities have 0-3 alkyl substituents corresponding to the number of 1-octene addition steps that occur prior to oxidation. Thus, phosphoric acid (9) and phosphorous acid (10) are derived from oxidation of residual PH3, MOPA (7) and OPA (8) from (n-octyl)PH2, and DOPO (6) and DOPA (3) from (n-octyl)2PH (blue branches of Scheme 2).
Scheme 2.
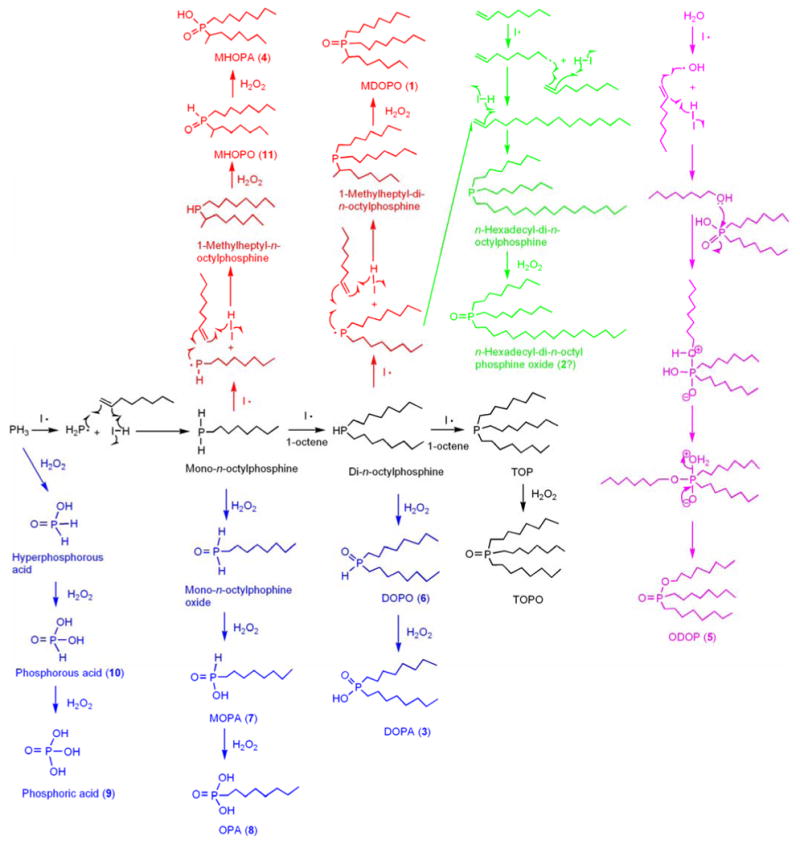
Synthetic pathway for the manufacture of TOPO, and proposed pathways for the formation of the common TOPO impurities. The symbol I· represents a radical initiator.
Occasionally, the radical addition occurs with reversed regiochemistry, resulting in a bond between P and C2 (rather than C1) of 1-octene. Such reversed additions are responsible for the branched-chain isomers MHOPO (11), MHOPA (4), and MDOPO (1), the predominant TOPO impurity (red branches of Scheme 2). We propose that ODOP (5) results from small amounts of 1-octanol formed in a side reaction (magenta branch of Scheme 2). Finally, we speculate that the unknown impurity 2 may be a TOPO analog having one n-hexadecyl chain replacing an n-octyl chain (green branch of Scheme 2). This compound may form by radical dimerization of 1-octene to 1-hexadecene, followed by its addition to (n-octyl)2PH, and subsequent oxidation of the resulting tertiary phosphine. Although most of these impurities are typically present in sub-percentage quantities, the amounts are stoichiometrically significant for nanocrystal syntheses conducted in TOPO, as noted above.
Roles of TOPO impurities in syntheses of colloidal nanocrystals
At least three synthetic influences have been suggested for TOPO impurities and the related additives that have been used to simulate their effects. First, some impurities and additives selectively passivate specific facets of growing nanocrystals, inhibiting growth on those facets, and producing anisotropic nanocrystal morphologies such as QRs and tetrapods.17,83–85 Thus, the growth of CdSe QRs16,17 and CdTe tetrapods30 with optimized diameters and lengths was achieved by the addition of n-hexyl- and methylphosphonic acid, respectively. The anisotropic growth of QRs and tetrapods along the [001] direction was attributed to the increased growth rate of the (001) facet relative to other facets as a consequence of the weaker binding of alkylphosphonic acids on the (001) facet.17,83–85
Second, some TOPO impurities or related additives may significantly influence rates of homogeneous nucleation. Peng and coworkers have argued that alkylphosphonate ligands bind more strongly to Cd in precursor complexes than do alkylcarboxylate ligands and thus decrease the reactivities of the precursors.21,55,57 We note that alkylphosphonate ligands may also be less labile (see below), and therefore Cd-alkylphosphonate complexes may be both thermodynamically more stable and kinetically less reactive than Cd-alkylcarboxylate complexes. Consequently, Peng and coworkers proposed that the use of alkylphosphonate additives dramatically decreased the numbers of nuclei formed in CdSe QR syntheses, allowing high monomer concentrations to be attained, favoring anisotropic growth.18,21,57 Similarly, we previously found that the SLS synthesis of InP QWs was accompanied by the formation of significant quantities of InP QRs, which resulted from a competitive homogeneous-nucleation process.62,63 Fortunately, small amounts of a strongly binding alkylphosphonic-acid additive (OPA) quenched the homogeneous nucleation of the QRs, such that only SLS-derived InP QWs were grown.62,63 Therefore, certain impurities or additives may modify precursor reactivity through ligand substitution to inhibit nanocrystal nucleation.
Third, as a related effect, such precursor modifications by impurities or additives may strongly influence nanocrystal growth rates. Insights into the chemical pathway followed in the eq-1 synthesis of CdSe QWs were provided by the elucidations of the reaction mechanisms for the analogous syntheses of II–VI67 and IV–VI86 QDs. The syntheses were shown to follow the pathway outlined in Scheme 3, in which CdSe formation is presented as a specific example. The first step is the Lewis-acid activation of R3PSe by the Cd(OX)2 precursor (XO– = alkylcarboxylate or alkylphosphonate). Alivisatos and coworkers argued that Cd-alkylphosphonate complexes have lower Lewis acidities than do Cd-alkylcarboxylate complexes because of the polydentate coordination and bridging tendencies of alkylphosphonate ligands, which enhances coordinative saturation.67 Consequently, Scheme 3 is kinetically slower for Cd-alkylphosphonate precursors than for Cd-alkylcarboxylate precursors. We note that the XO– ligands in Scheme 3 function as both leaving groups and nucleophiles, and so both the stability and lability of the Cd precursors may also influence Scheme-3 kinetics (depending on which step is rate determining).
Scheme 3.
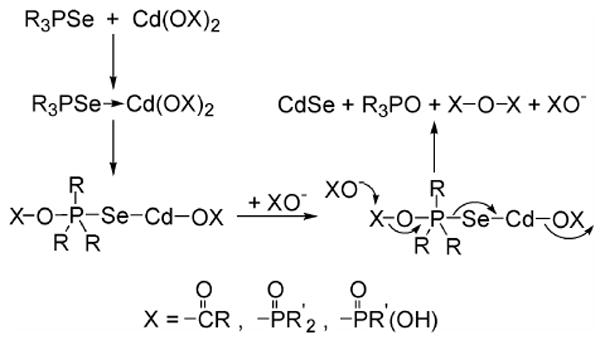
Because Scheme 3 provides CdSe to both nucleation and growth processes, in principle its kinetics should influence both the homogeneous-nucleation and growth rates of nanocrystals. However, homogeneous nucleation does not participate in the SLS growth of QWs. (The nucleation of QWs is heterogeneous upon the catalyst droplet, after the droplet has become supersaturated.2,87) Consequently, the Scheme-3 kinetics should primarily influence the QW growth rates. We propose that the QWs of highest quality result from an optimal growth rate. If the growth rate is too fast, the QWs may acquire higher defect densities and diameter fluctuations, as we observed of QWs synthesized in TOPO C or purified TOPO (see Figures 1c, d, and S11c, d). If the growth rate is too slow, the QWs may be short, tapered, and branched, as we observed of QWs synthesized in purified TOPO with added MOPA or OPA (see Figure S26c, e, f).
Therefore, we propose that the SLS growth rates of CdSe QWs are significantly influenced by the stabilities, labilities, and Lewis acidities of the Cd(OX)2 precursors employed. In general, growth rates should decrease as the precursor stabilities increase, and the labilities and Lewis acidities decrease. Consequently, we propose the reactivity scale in Scheme 4, which attempts a ranking of the important impurities, reagents, and additives investigated in this study in terms of the composite stabilities, labilities, and Lewis acidities of the corresponding Cd precursors.
Scheme 4.
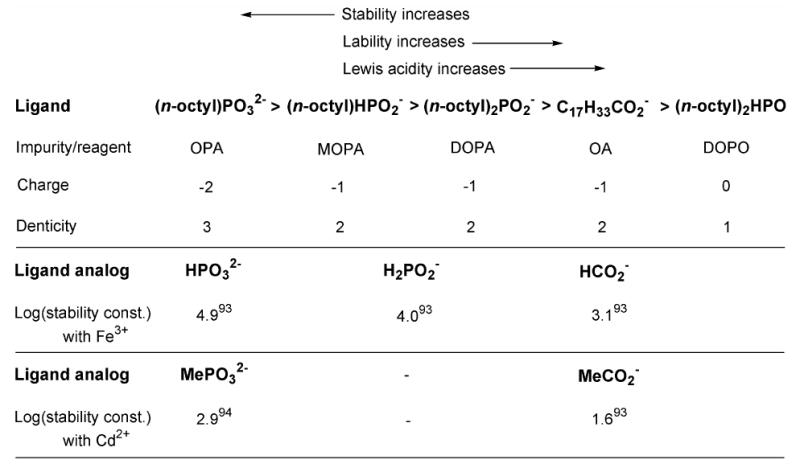
Proposed precursor lability, stability, and Lewis acidity on the basis of the impurity- or additive-derived ligands present. Stability constants93,94 (aqueous solution, 25 °C) of ligand analogs with Fe3+ and Cd2+ are provided for comparison.
We estimate the relative stabilities first by the maximum ligand charge (more negative = more stable), and second by the maximum ligand denticity (higher denticity = higher stability). We also take the bridging tendencies of the ligands into account. Thus, the doubly negative (n-octyl)PO32– ligand (derived from OPA) should provide the most-stable Cd precursors, whereas the uncharged DOPO ligand should provide the least stable. The ligand denticities follow the same trend. This ranking system does not distinguish the relative placement of the DOPA-derived (n-octyl)2PO2– and C7H33CO2– (oleate) ligands; we assign higher stability to (n-octyl)2PO2–-containing precursors in accord with the greater bridging tendencies of phosphonate and phosphinate vs. carboxylate ligands.67,88-92 This ranking is consistent with the greater thermal stability of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n relative to [Cd(oleate)2]n described above.
To our knowledge, no quantitative data, measure, parameter, or scale exists to confirm the stability order we have proposed in Scheme 4, because the ligands are uncommon and the set is diverse. Consequently, we refer to aqueous stability constants of ligand analogs, recognizing that the values of these constants will differ in TOPO solvent, but in the hopes that the relative ranking of the constants will not. We could not find a complete set of stability constants for ligand analogs with Cd2+,93,94 so we also provide values for Fe3+ (Scheme 4).93 Notably, the available analog data provided in Scheme 4 fully support our proposed stability order.
By reference to the Hammond postulate,95 we propose that precursor labilities should follow the reverse of the stability ordering. Similarly, Lewis acidities should increase with decreasing denticities and bridging tendencies, as described by Alivisatos and coworkers.67 Thus, Scheme-3 kinetics and QW growth rates should increase from left to right in the Scheme-4 ligand series. Interestingly, DOPA appears in the middle of the series, providing a precursor of moderate reactivity. Apparently, this moderate reactivity is the key to the synthesis of high-quality CdSe QWs.
DOPO appears at the right end of the Scheme-4 sequence, and thus its presence should promote rapid Scheme-3 kinetics. As previously described, syntheses employing purified TOPO with DOPO as the sole additive enhanced the homogeneous nucleation of QDs, which competed with the SLS growth of QWs (Figure S27a–c). However, when the stronger-binding ligand DOPA was added in conjunction with DOPO, the reactivity of the Cd precursor was significantly decreased, and the formation of QDs was eliminated (Figure S31).
Finally, we note that the amount of stronger-binding ligand added will influence reactivity by varying the composition of the resulting {Cd[ligand]x[oleate]y}n precursor. Thus, the syntheses employing higher concentrations of DOPA, OPA, or MOPA resulted in the growth of lower-quality CdSe QWs (Figures S25, and S26c, e, f) due to the decreased reactivity of the cadmium precursor.
Conclusion
The solvent TOPO has been critically important to the development of colloidal nanoscience. In TOPO the first modern synthesis of semiconductor quantum dots was achieved.6 The adventitious impurities in TOPO were responsible for the discovery of semiconductor quantum rods.16 These impurities significantly influence the rates of nanocrystal formation, the nanocrystal morphologies, and the reproducibilities of nanocrystal syntheses in TOPO. In this study, we have identified the spectrum of typical TOPO impurities, and have elucidated their roles in the reaction chemistry that supports the nucleation and growth of colloidal semiconductor nanocrystals. The results reported here will free nanocrystal researchers and their synthetic results from reliance on particular “special” batches of commercially obtained TOPO. Now the use of purified TOPO with controlled amounts of specific additives, such as those included in the reactivity ranking of Scheme 4, will allow rational and reproducible nanocrystal syntheses.
Supplementary Material
Acknowledgments
We thank Dr. Haitao Liu (Columbia U.), Dr. Michael L. Steigerwald (Columbia U.), Prof. Evamarie Hey-Hawkins (Universität Leipzig), Prof. Guy Bertrand (U. of California, Riverside), and Prof. John A. Gladysz (Texas A&M U.) for helpful discussions. We also thank Cytec Canada Inc for providing the CYANEX 921 sample and Nippon Chemical Industrial for providing manufacturing information. We are grateful to the National Science Foundation for funding this work under grant no. CHE-0518427. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, an NIH Research Resource (Grant No. P41RR0954).
Footnotes
Supporting Information Available: Syntheses and characterizations of DOPA, MOPA, DOPO, ODOP, MDOPO, EDOPO, DMOPO, MHOPA, and MHOPO, methods for TEM and absorption spectroscopy, a table listing the time required to dissolve CdO in various TOPO samples, the optical clarity of resulting solutions, and the final products in the wire synthesis, a table listing the concentrations of impurities present in various TOPO samples, 31P{1H} NMR spectra of 90% TOPO specimens, vacuum-distilled/recrystallized TOPO, and ammonia-treated TOPO A, 31P NMR spectra for identifying ODOP, MDOPO, MHOPA, MHOPO, phosphoric acid, and phosphorous acid, IR and various-temperature 31P{1H} NMR spectra of {Cd[O2P(n-octyl)2]0.84[oleate]1.16}n, and additional TEM images. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Trentler TJ, Hichman KM, Goel SC, Viano AM, Gibbons PC, Buhro WE. Science. 1995;270:1791–1794. [Google Scholar]
- 2.Wang F, Dong A, Sun J, Tang R, Yu H, Buhro WE. Inorg Chem. 2006;45:7511–7521. doi: 10.1021/ic060498r. [DOI] [PubMed] [Google Scholar]
- 3.www.cytec.com/specialty-chemicals/AlkylphosphineDervivatives/CYANEX921.pdf
- 4.Golob J, Grilc V, Zadnik B. Ind Eng Chem Process Des Dev. 1981;20:433–435. [Google Scholar]
- 5.Flett DS. J Organomet Chem. 2005;690:2426–2438. [Google Scholar]
- 6.Murray CB, Norris DJ, Bawendi MG. J Am Chem Soc. 1993;115:8706–8715. [Google Scholar]
- 7.Katari JEB, Colvin VL, Alivisatos AP. J Phys Chem. 1994;98:4109–4117. [Google Scholar]
- 8.Mićić OI, Sprague J, Curtis CJ, Jones KM, Machol JL, Nozik AJ, Giessen H, Fluegel B, Mohs G, Peyghambarian N. J Phys Chem. 1995;99:7754–7759. [Google Scholar]
- 9.Guzelian AA, Katari JEB, Kadavanich AV, Banin U, Hamad K, Juban E, Alivisatos AP, Wolters RH, Arnold CC, Heath JR. J Phys Chem. 1996;100:7212–7219. [Google Scholar]
- 10.Peng X, Wickham J, Alivisatos AP. J Am Chem Soc. 1998;120:5343–5344. [Google Scholar]
- 11.Peng ZA, Peng X. J Am Chem Soc. 2001;123:183–184. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]
- 12.Qu L, Peng ZA, Peng X. Nano Lett. 2001;1:333–337. [Google Scholar]
- 13.Qu L, Peng X. J Am Chem Soc. 2002;124:2049–2055. doi: 10.1021/ja017002j. [DOI] [PubMed] [Google Scholar]
- 14.Mekis I, Talapin DT, Kornowski A, Haase M, Weller H. J Phys Chem B. 2003;107:7454–7462. [Google Scholar]
- 15.Qu L, Yu WW, Peng X. Nano Lett. 2004;4:465–469. [Google Scholar]
- 16.Peng X, Manna L, Yang W, Wickham J, Scher E, Kadavanich A, Alivisatos AP. Nature. 2000;404:59–61. doi: 10.1038/35003535. [DOI] [PubMed] [Google Scholar]
- 17.Manna L, Scher EC, Alivisatos AP. J Am Chem Soc. 2000;122:12700–12706. [Google Scholar]
- 18.Peng ZA, Peng X. J Am Chem Soc. 2001;123:1389–1395. doi: 10.1021/ja003633m. [DOI] [PubMed] [Google Scholar]
- 19.Li LS, Hu J, Yang W, Alivisatos AP. Nano Lett. 2001;1:349–351. [Google Scholar]
- 20.Hu J, Li LS, Yang W, Manna L, Wang LW, Alivisatos AP. Science. 2001;292:2060–2063. doi: 10.1126/science.1060810. [DOI] [PubMed] [Google Scholar]
- 21.Peng ZA, Peng X. J Am Chem Soc. 2002;124:3343–3353. doi: 10.1021/ja0173167. [DOI] [PubMed] [Google Scholar]
- 22.Manna L, Scher EC, Li LS, Alivisatos AP. J Am Chem Soc. 2002;124:7136–7145. doi: 10.1021/ja025946i. [DOI] [PubMed] [Google Scholar]
- 23.Mokari T, Banin U. Chem Mater. 2003;15:3955–3960. [Google Scholar]
- 24.Kan S, Mokari T, Rothenberg E, Banin U. Nat Mater. 2003;2:155–158. doi: 10.1038/nmat830. [DOI] [PubMed] [Google Scholar]
- 25.Kan S, Aharoni A, Mokari T, Banin U. Faraday Discuss. 2004;125:23–38. doi: 10.1039/b302898d. [DOI] [PubMed] [Google Scholar]
- 26.Talapin DV, Nelson JH, Shevchenko EV, Aloni S, Sadtler B, Alivisatos AP. Nano Lett. 2007;7:2951–2959. doi: 10.1021/nl072003g. [DOI] [PubMed] [Google Scholar]
- 27.Carbone L, et al. Nano Lett. 2007;7:2942–2950. doi: 10.1021/nl0717661. [DOI] [PubMed] [Google Scholar]
- 28.Manna L, Milliron DJ, Meisel A, Scher EC, Alivisatos AP. Nat Mater. 2003;2:382–385. doi: 10.1038/nmat902. [DOI] [PubMed] [Google Scholar]
- 29.Milliron DL, Hughes SM, Cui Y, Manna L, Li J, Wang LW, Alivisatos AP. Nature. 2004;430:190–195. doi: 10.1038/nature02695. [DOI] [PubMed] [Google Scholar]
- 30.Carbone L, Kudera S, Carlino E, Parak WJ, Giannini C, Cingolani R, Manna L. J Am Chem Soc. 2006;128:748–755. doi: 10.1021/ja054893c. [DOI] [PubMed] [Google Scholar]
- 31.Meyer M, Wallberg C, Kurihara K, Fendler JH. J Chem Soc Chem Commun. 1984:90–91. [Google Scholar]
- 32.Lianos P, Thomas JK. Chem Phys Lett. 1986;125:299–302. [Google Scholar]
- 33.Fendler JH. Chem Rev. 1987;87:877–899. [Google Scholar]
- 34.Dannhauser T, O'Neil M, Johansson K, Whitten D, McLendon G. J Phys Chem. 1986;90:6074–6076. [Google Scholar]
- 35.Petit C, Pileni MP. J Phys Chem. 1988;92:2282–2286. [Google Scholar]
- 36.Steigerwald ML, Alivisatos AP, Gibson JM, Harris TD, Kortan R, Muller AJ, Thayer AM, Duncan TM, Douglass DC, Brus LE. J Am Chem Soc. 1988;110:3046–3050. [Google Scholar]
- 37.Kortan AR, Hull R, Opila RL, Bawendi MG, Steigerwald ML, Carroll PJ, Brus LE. J Am Chem Soc. 1990;112:1327–1332. [Google Scholar]
- 38.Steigerwald ML, Brus LE. Acc Chem Res. 1990;23:183–188. [Google Scholar]
- 39.Bawendi MG, Wilson WL, Rothberg L, Carroll PJ, Jedju TM, Steigerwald ML, Brus LE. Phys Rev Lett. 1990;65:1623–1626. doi: 10.1103/PhysRevLett.65.1623. [DOI] [PubMed] [Google Scholar]
- 40.Bawendi MG, Carroll PJ, Wilson WL, Brus LE. J Chem Phys. 1992;96:946–954. [Google Scholar]
- 41.Steigerwald, M. L., personal communication.
- 42.Trentler TJ, Denler TE, Bertone JF, Agrawal A, Colvin VL. J Am Chem Soc. 1999;121:1613–1614. [Google Scholar]
- 43.Green M, O'Brien P. Chem Commun. 2000:183–184. [Google Scholar]
- 44.Park SJ, Kim S, Lee S, Khim ZG, Char K, Hyeon T. J Am Chem Soc. 2000;122:8581–8582. [Google Scholar]
- 45.Puntes VF, Krishnan KM, Alivisatos AP. Science. 2001;291:2115–2117. doi: 10.1126/science.1057553. [DOI] [PubMed] [Google Scholar]
- 46.Puntes VF, Zanchet D, Erdonmez CK, Alivisatos AP. J Am Chem Soc. 2002;124:12874–12880. doi: 10.1021/ja027262g. [DOI] [PubMed] [Google Scholar]
- 47.Joo J, Yu T, Kim TW, Park HM, Wu F, Zhang JZ, Hyeon T. J Am Chem Soc. 2003;125:6553–6557. doi: 10.1021/ja034258b. [DOI] [PubMed] [Google Scholar]
- 48.Perera SC, Tsoi G, Wenger LE, Brock SL. J Am Chem Soc. 2003;125:13960–13961. doi: 10.1021/ja038037h. [DOI] [PubMed] [Google Scholar]
- 49.Jun YW, Casula MF, Sim JH, Kim SY, Cheon J, Alivisatos AP. J Am Chem Soc. 2003;125:15981–15985. doi: 10.1021/ja0369515. [DOI] [PubMed] [Google Scholar]
- 50.Qian C, Kim F, Ma L, Tsui F, Yang P, Liu J. J Am Chem Soc. 2004;126:1195–1198. doi: 10.1021/ja038401c. [DOI] [PubMed] [Google Scholar]
- 51.Park J, Koo B, Yoon KY, Hwang Y, Kang M, Park JG, Hyeon T. J Am Chem Soc. 2005;127:8433–8440. doi: 10.1021/ja0427496. [DOI] [PubMed] [Google Scholar]
- 52.Li Y, Malik MA, O'Brien P. J Am Chem Soc. 2005;127:16020–16021. doi: 10.1021/ja055963i. [DOI] [PubMed] [Google Scholar]
- 53.Samia ACS, Schlueter JA, Jiang JS, Bader SD, Qin CJ, Lin XM. Chem Mater. 2006;18:5203–5212. [Google Scholar]
- 54.Yu WW, Peng X. Angew Chem, Int Ed. 2002;41:2368–2371. doi: 10.1002/1521-3773(20020703)41:13<2368::AID-ANIE2368>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 55.Battaglia D, Peng X. Nano Lett. 2002;2:1027–1030. [Google Scholar]
- 56.Li JJ, Wang YA, Guo W, Keay JC, Mishima TD, Johnson MB, Peng X. J Am Chem Soc. 2003;125:12567–12575. doi: 10.1021/ja0363563. [DOI] [PubMed] [Google Scholar]
- 57.Yu WW, Wang YA, Peng X. Chem Mater. 2003;15:4300–4308. [Google Scholar]
- 58.Xie R, Battaglia D, Peng X. J Am Chem Soc. 2007;129:15432–15433. doi: 10.1021/ja076363h. [DOI] [PubMed] [Google Scholar]
- 59.Yu H, Li J, Loomis RA, Gibbons PC, Wang LW, Buhro WE. J Am Chem Soc. 2003;125:16168–16169. doi: 10.1021/ja037971+. [DOI] [PubMed] [Google Scholar]
- 60.Grebinski JW, Hull KL, Zhang J, Kosel TH, Kuno M. Chem Mater. 2004;16:5260–5272. [Google Scholar]
- 61.Dong A, Yu H, Wang F, Buhro WE. Nano Lett. 2007;7:1308–1313. doi: 10.1021/nl070293v. [DOI] [PubMed] [Google Scholar]
- 62.Wang F, Yu H, Li J, Hang Q, Zemlyanov D, Gibbons PC, Wang LW, Janes DB, Buhro WE. J Am Chem Soc. 2007;129:14327–14335. doi: 10.1021/ja074049h. [DOI] [PubMed] [Google Scholar]
- 63.Wang F, Buhro WE. J Am Chem Soc. 2007;129:14381–14387. doi: 10.1021/ja075015c. [DOI] [PubMed] [Google Scholar]
- 64.Sun J, Buhro WE. Angew Chem, Int Ed. 2008;47:3215–3218. doi: 10.1002/anie.200705142. [DOI] [PubMed] [Google Scholar]
- 65.Dong A, Yu H, Wang F, Buhro WE. J Am Chem Soc. 2008;130:5954–5961. doi: 10.1021/ja711408t. [DOI] [PubMed] [Google Scholar]
- 66.Sun J, Wang LW, Buhro WE. J Am Chem Soc. 2008;130:7997–8005. doi: 10.1021/ja800837v. [DOI] [PubMed] [Google Scholar]
- 67.Liu H, Owen JS, Alivisatos AP. J Am Chem Soc. 2007;129:305–312. doi: 10.1021/ja0656696. [DOI] [PubMed] [Google Scholar]
- 68.Wang W, Banerjee S, Jia S, Steigerwald ML, Herman IP. Chem Mater. 2007;19:2573–2580. [Google Scholar]
- 69.Kopping JT, Patten TE. J Am Chem Soc. 2008;130:56895–5698. doi: 10.1021/ja077414d. [DOI] [PubMed] [Google Scholar]
- 70.Owen JS, Park J, Trudeau PE, Alivisatos AP. J Am Chem Soc. 2008;130:12279–12281. doi: 10.1021/ja804414f. [DOI] [PubMed] [Google Scholar]
- 71.Kolosky M, Vialle J. J Chromatogr. 1984;299:436–444. [Google Scholar]
- 72.Wang F, Tang R, Buhro WE. Nano Lett. 2008;8:3521–3524. doi: 10.1021/nl801692g. [DOI] [PubMed] [Google Scholar]
- 73.Kawa M, Morii H, Ioku A, Saita S, Okuyama K. J Nanopart Res. 2003;5:81–85. [Google Scholar]
- 74.Kanaras AG, Sonnichsen C, Liu H, Alivisatos AP. Nano Lett. 2005;5:2164–2167. doi: 10.1021/nl0518728. [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Tang R, Yu H, Gibbons PC, Buhro WE. Chem Mater. 2008;20:3656–3662. [Google Scholar]
- 76.Peppard DF, Mason GW, Lewey S. J Inorg Nucl Chem. 1965;27:2065–2073. [Google Scholar]
- 77.Agustin CH, Martinez MM. J Dispersion Sci Tech. 1988;9:209–221. [Google Scholar]
- 78.Williams RH, Hamilton LA. J Am Chem Soc. 1952;74:5418–5420. [Google Scholar]
- 79.Kosolapoff GM, Watson RM. J Am Chem Soc. 1951;73:4101–4102. [Google Scholar]
- 80.Kloper V, Osovsky R, Kolny-Olesiak J, Sashchiuk A, Lifshitz E. J Phys Chem C. 2007;111:10336–10341. [Google Scholar]
- 81.Hoye PAT, Ellis JW, Park W. U S Patent. 1994:5, 284, 555.
- 82.http://cytec.com/specialty-chemicals/technology1.htm
- 83.Puzder A, Williamson AJ, Zaitseva N, Galli G, Manna L, Alivisatos AP. Nano Lett. 2004;4:2361–2365. [Google Scholar]
- 84.Rempel JY, Trout BL, Bawendi MG, Jensen KF. J Phys Chem B. 2006;110:18007–18016. doi: 10.1021/jp064051f. [DOI] [PubMed] [Google Scholar]
- 85.Manna L, Wang LW, Cingolani R, Alivisatos AP. J Phys Chem B. 2005;109:6183–6192. doi: 10.1021/jp0445573. [DOI] [PubMed] [Google Scholar]
- 86.Steckel JS, Yen BKH, Oertel DC, Bawendi MG. J Am Chem Soc. 2006;128:13032–13033. doi: 10.1021/ja062626g. [DOI] [PubMed] [Google Scholar]
- 87.Trentler TJ, Goel SC, Hickman KM, Viano AM, Chiang MY, Beatty AM, Gibbons PC, Buhro WE. J Am Chem Soc. 1997;119:2172–2181. [Google Scholar]
- 88.Harrison W, Trotter J. J Chem Soc, Dalton Trans. 1972:956–960. [Google Scholar]
- 89.Cao G, Lynch VM, Yacullo LN. Chem Mater. 1993;5:1000–1006. [Google Scholar]
- 90.Rose SH, Block BP. J Am Chem Soc. 1965;87:2076–2077. [Google Scholar]
- 91.Gillman HD. Inorg Chem. 1974;13:1921–1924. [Google Scholar]
- 92.Gillman HD, Eichelberger JL. Inorg Chem. 1976;15:840–843. [Google Scholar]
- 93.Martel AE, Smith RM. Critical Stability Constants. 3, 4 Plenum; New York: 1977 1976. [Google Scholar]
- 94.Popov K, Ronkkomaki H, Lajunen LHJ. Pure Appl Chem. 2001;73:1641–1677. [Google Scholar]
- 95.Hammond GS. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.