

Abstract
The oral pathogen Porphyromonas gingivalis, as well as its purified fimbriae, are known to activate TLR2 and induce proinflammatory and proadhesive effects. The TLR2 proinflammatory pathway induces NF-κB-dependent inflammatory cytokines, whereas the TLR2 proadhesive pathway is characterized by inside-out signaling that transactivates β2 integrin adhesive activities. In this paper, using dominant-negative or pharmacological approaches, we show that the two pathways bifurcate and proceed independently downstream of TLR2. Whereas the proinflammatory pathway is dependent on the adaptor molecules Toll/IL-1 receptor domain-containing adaptor protein (TIRAP; also known as Mal) and MyD88, the proadhesive pathway is TIRAP/MyD88-independent and proceeds through PI3K-mediated signaling. Although the Ser/Thr kinase Akt is a major downstream target of PI3K and was activated by P. gingivalis fimbriae in a TLR2- and PI3K-dependent way, Akt was shown not to play a role in the proadhesive patway. On the other hand, another PI3K downstream target, cytohesin-1, was shown to mediate P. gingivalis fimbria-induced activation of β2 integrin for ICAM-1 binding. Therefore, P. gingivalis fimbriae activate two distinct TLR2 pathways mediating proinflammatory or proadhesive effects. The delineation of these signaling pathways may provide appropriate targets for selectively inhibiting or enhancing specific activities, depending on whether they undermine or promote the host defense.
Keywords: monocytes/macrophages, cell activation, cytokines, adhesion molecules, signal transduction
Introduction
TLRs play a central role in the innate recognition of microbial pathogens and the induction of the inflammatory response aiming to control pathogen spread (1, 2). TLR2, in association with TLR1 or TLR6, recognizes a variety of pathogens (3) including Porphyromonas gingivalis, an oral pathogen associated with periodontal disease and implicated in atherosclerosis (4–7). TLR signaling is triggered upon recruitment of Toll/IL-1 receptor (TIR)3 domain-containing adaptor molecules to the cytoplasmic domains of activated TLRs (8). Initiation of TLR2 signaling, for example, involves recruitment of the TIR domain-containing adaptor protein (TIRAP; also known as Mal) and MyD88 to the TLR2 cytoplasmic domain, and downstream signaling events activate nuclear factor (NF)-κB leading to induction of proinflammatory and host-defense genes (8).
Evidence from animal models of experimental periodontitis or atherosclerosis suggests that the fimA-encoded fimbriae of P. gingivalis constitute an important virulence factor of this pathogen (9, 10). Due to their considerable length (up to 3 μm from the bacterial cell surface (11)), the fimbriae may be the first P. gingivalis molecule to interact with host structures. Although traditionally recognized as a colonization factor (11, 12), P. gingivalis fimbriae also interact with TLR2 and modulate innate immune responses (7, 13, 14). Therefore, understanding the mode whereby P. gingivalis fimbriae interact with different types of host cells (15–18) may lead to a better understanding of host-P. gingivalis interactions and their impact on disease pathogenesis.
Work from our lab has described molecular events underlying P. gingivalis-induced activation of monocytes/macrophages (6, 14, 19–21), which play an important role in the innate host response in periodontitis and other chronic infections (22–24). The binding of P. gingivalis fimbriae to CD14 greatly facilitates TLR2 activation by whole bacterial cells or purified fimbriae, thereby initiating of a core intracellular signaling cascade for NF-κB activation and induction of proinflammatory cytokines (6, 14, 16). In addition, following interaction with the CD14/TLR2 recognition complex, P. gingivalis or its purified fimbriae induce PI3K-mediated inside-out signaling for activating the ligand-binding capacity of complement receptor-3 (CR3; CD11b/CD18), (20, 21). In terms of biological significance, P. gingivalis-activated TLR2 inside-out signaling leads to enhanced CR3-dependent monocyte adhesion to endothelial ICAM-1 and transendothelial migration (20). Intriguingly, however, P. gingivalis has co-opted this TLR2/CR3 proadhesive pathway for CR3 binding and entry into macrophages (19) in a way that promotes the survival and virulence of this pathogen (25, 26).
Although the above discussed P. gingivalis-induced proinflammatory and proadhesive pathways mediate distinct biological effects, it is not currently known whether the proadhesive signaling pathway downstream of TLR2 proceeds independently of the proinflammatory pathway, i.e., by using different adaptors and/or signaling molecules. This issue was addressed in this study. The objective to characterize and delineate TLR2 downstream signaling pathways may provide appropriate targets for selectively inhibiting those signaling cascades (e.g., the TLR2 proadhesive pathway) that are exploited by P. gingivalis to enhance its survival within the host. Thus, since the innate antimicrobial response to P. gingivalis appears to depend on the TLR2/NF-κB pathway (18), it would be inappropriate to design and apply intervention strategies that inhibit TLR2 signaling altogether. The data presented in this paper show that TLR2 signaling can indeed bifurcate into two distinct pathways: The standard TLR2 proinflammatory pathway that is dependent upon the signaling adaptors TIRAP and MyD88, and a novel TLR2-mediated but TIRAP/MyD88-independent proadhesive pathway, which stimulates CR3 adhesive activity via Rac1, PI3K, and cytohesin-1.
Materials and Methods
Reagents
MAbs to human CD11b (CBRM1/5) or mouse CD11b (M1/70), and immunoglobulin isotype controls were purchased from eBioscience (San Diego, CA). PMA, wortmannin, LY294002, and LY30351 were from Sigma-Aldrich (St. Louis, MO). Recombinant human or mouse ICAM-1 was obtained from the R&D Systems (Minneapolis, MN). FimA fimbriae were chromatographically purified from P. gingivalis strain 381, as previously described (21), or from strain ATCC 33277, as described by Yoshimura et al (27). The strain 33277 fimbriae were kindly provided by Dr. F. Yoshimura, Aichi-Gakuin University, Nagoya, Japan. The final fimbrial preparations were free of any contaminating substances on silver-stained SDS-PAGE, and tested negative for endotoxin (< 0.7 ng/mg protein) according to quantitative Limulus amebocyte lysate assay (BioWhittaker, Walkersville, MD). Fimbriae from either strain (99.7% identity at the FimA amino acid level) were indistinguishable in the functional assays presented here. All reagents were used at effective concentrations determined in preliminary experiments or in previous publications (6, 20, 28).
Cell culture
Human monocytic THP-1 cells stably transfected with human CD14 (THP-1/CD14) (29) were kindly provided by Dr. P. S. Tobias (The Scripps Research Institute, La Jolla, CA) and were cultured at 37°C and 5% CO2 atmosphere, in RPMI 1640 (InVitrogen-Gibco, Carlsbad, CA) supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.05 mM 2-ME (complete RPMI). Thioglycollate-elicited macrophages were isolated from the peritoneal cavity of mice deficient in TLR2 (The Jackson Laboratory, Bar Harbor, ME) or MyD88 (kindly provided by Dr. Shizuo Akira, Research Institute for Microbiological Diseases, Osaka University, Japan) or from wild-type control mice (C57BL/6; The Jackson Laboratory), as previously described (30). Mouse macrophages were cultured in complete RPMI as above. The use of animals was reviewed and approved by the institutional animal care and use committee, in compliance with established federal and state guidelines. Human or mouse cell viability was monitored using the CellTiter-Blue™ assay kit (Promega, Madison, WI). The use of fimbriae or other agonists, and cell pretreatment with dominant-negative mutants, inhibitory peptides, or S-oligos, did not affect cell viability as compared to medium-only control treatments.
Cell transfections and inhibition of intracellular signaling
Transfections of THP-1/CD14 cells were performed using the FuGene 6 transfection reagent (Roche Applied Science, Indianapolis, IN), as we previously described (20). For inhibition of intracellular signaling, THP-1/CD14 cells were transfected with plasmids expressing dominant negative versions of human TLR2 (pZERO-hTLR2tirless), MyD88 (pDeNy-hMyD88), TIRAP (pDeNy-hTIRAP), TRAM (pDeNy-hTRAM) or with empty vector controls (InVivogen, San Diego, CA). A plasmid expressing a kinase-inactive version of human Akt (K179M), which functions as a dominant negative inhibitor, as well as empty control vector pUSEamp(+), were obtained from Upstate Biotech (Lake Placid, NY). The transfected cells were used in functional assays 48h post-transfection. Alternative methods for inhibition of intracellular molecules included the following: A cell permeable peptide set, comprising a peptide that inhibits human MyD88 TIR domain dimerization and an inactive control peptide (31), was purchased from Imgenex (San Diego, CA). To block mouse TIRAP signaling, we used a cell permeable TIRAP inhibitory and control peptide set (32, 33), obtained from Calbiochem (San Diego, CA). A cell permeable peptide set, including a peptide that binds to the pleckstrin homology domain of Akt and inhibits its kinase activity, as well as an inactive control peptide (34), was purchased from Imgenex. To inhibit the expression of cytohesin-1, we followed an antisense strategy developed by Hmama et al using antisense and control sense phosphorothioate-modified oligonucleotides (S-oligos) specified in previous reports (35, 36) and synthesized by InVitrogen. Briefly, THP-1/CD14 cells (106 cells in 0.25 ml serum-free RPMI 1640 medium) were incubated at 37°C with various concentrations (0–5 μM) of S-oligos in the presence of 2.5% lipofectamine (InVitrogen). After 2h, the medium was adjusted to 1 ml and 10% fetal bovine serum was added. The cells were cultured overnight and were subsequently washed and used in functional assays.
Cellular activation assays
(a) Cytokine induction
Primary cells or transfected cell lines were stimulated with P. gingivalis fimbriae (1 μg/ml) for 16 h at 37°C and induction of release of TNF-α or IL-6 in culture supernatants was measured by ELISA, using kits from eBioscience (30). (b) Akt activation: Total and phosphorylated Akt levels were determined using a colorimetric fast-activated cell-based ELISA kit, as instructed by the manufacturer (Active Motif, Carlsbad, CA). (c) Reporter gene assays: NF-κB-dependent transcription of a luciferase reporter gene was determined as we previously described (6, 30). Briefly, THP-1/CD14 cells were cotransfected with NF-κB-dependent firefly luciferase reporter plasmid (pNF-κB-Luc; Stratagene, La Jolla, CA) and pRLnull, a Renilla luciferase transfection control (Promega, Madison, WI). Two days post-transfection, the cells were stimulated as per experimental protocol. After 6h of stimulation, the cells were lysed and the Renilla and firefly luciferase activities were measured using the Dual-Glo™ luciferase reporter assay system (Promega) and a Clarity™ luminescence microplate reader (Bio-Tek, Winooski, VT). Luciferase activity was calculated as a ratio of firefly luciferase activity to Renilla luciferase activity, to correct for transfection efficiency. The results were then be normalized to those of unstimulated controls transfected with reporter and empty vectors. (d) CR3 activation assay: The CBRM1/5 epitope induction assay was used to monitor the activation state of human CR3 (CD11b/CD18), as we have previously described (21). The assay is based on the property of the CBRM1/5 mAb to detect a conformational change on human CD11b that signifies the high-affinity binding state of CR3 (37). Activation of mouse CR3 was assessed by monitoring its binding activity for soluble ICAM-1, a ligand that binds activated but not resting CR3 (20, 38). Specifically, biotinylated sICAM-1 was allowed to bind to mouse macrophages for 30 min at 37°C. Subsequently, the cells were washed and incubated on ice with FITC-labeled streptavidin. After washing, binding was determined by measuring cell-associated fluorescence (in relative fluorescence units) on a microplate fluorescence reader (FL600, Bio-Tek) with excitation/emission wavelength settings of 485/530 nm. Background fluorescence was determined in cells treated with medium only and FITC-streptavidin.
Statistical analysis
Data were evaluated by analysis of variance and the Dunnett multiple-comparison test using the InStat program (GraphPad Software, San Diego, CA). Where appropriate (comparison of two groups only), two-tailed t tests were also performed. P < 0.05 was taken as the level of significance. Experiments were performed using triplicate samples and were performed twice or more for verification.
Results
The TLR2 proadhesive pathway is MyD88-independent
Although P. gingivalis fimbria-activated TLR2 mediates both proinflammatory and proadhesive effects, it is unknown whether these two types of activities involve common or distinct signaling adaptors. Since MyD88 is a central signaling adaptor for TLR2 proinflammatory signaling (39), we investigated whether treatments that block MyD88 could also inhibit TLR2 inside-out signaling for CR3 activation (monitored by induction of the CBRM1/5 epitope, which signifies the high-affinity binding conformation of CR3 (37)). We found that transfection of THP-1/CD14 cells with a plasmid expressing a dominant-negative signaling mutant of human MyD88 (MyD88-DN) (40) did not affect the ability of the fimbriae to induce the CR3 activation-specific CBRM1/5 epitope, in contrast to transfection with TLR2-DN, which served as positive control (Fig. 1A). However, in the same cells, MyD88-DN significantly inhibited NF-κB activation by fimbriae, as also observed with TLR2-DN (p < 0.05; Fig. 1A). Similar results were obtained using a cell permeable peptide, which inhibits dimerization of the MyD88 TIR domains (31). This MyD88 inhibitory peptide had no effect on the ability of P. gingivalis fimbriae to induce the CBRM1/5 epitope in monocytic THP-1/CD14 cells, although it significantly suppressed fimbria-induced activation of NF-κB in the same cells (p < 0.05; Fig. 1B). Therefore, MyD88 signaling is not required for CR3 activation by P. gingivalis fimbriae, although it mediates activation of NF-κB by the same agonist.
Figure 1. P. gingivalis fimbria-induced activation of human CR3 is MyD88-independent.

Human monocytic THP-1/CD14 cells were transfected with a dominant negative mutant of MyD88 (MyD88-DN), TLR2-DN, or vector control, at the indicated μg of plasmid DNA per 2×105 cells (A), or were pretreated for 2h with MyD88 inhibitory peptide or inactive control (100 μM) (B). The cells were stimulated with P. gingivalis fimbriae (1 μg/ml) and assayed for induction of the CR3 activation-specific CBRM1/5 epitope (after staining with FITC-labeled CBRM1/5 mAb) or for NF-κB-dependent transcription of a luciferase reporter gene. CBRM1/5 induction is reported in relative fluorescent units (RFU) and NF-κB activation in relative luciferase activity (RLA). The discontinuous horizontal lines indicate CBRM1/5 epitope induction or NF-κB activation in resting cells, which were < 8% of the values of fimbria-stimulated cells. Results are presented as means ± SD (n = 3) from one of two sets of experiments yielding consistent results. The asterisks indicate statistically significant (p < 0.05) differences compared to control treatments.
These conclusions were confirmed in the mouse model using wild-type or MyD88-deficient macrophages. The cells were monitored for their adhesive activity for soluble (s)ICAM-1, which binds activated but not resting CR3 (38). We found that P. gingivalis fimbriae could activate CR3-dependent binding of sICAM-1 equally well in wild-type or MyD88-deficient macrophages, although, as expected, this activity was abrogated in TLR2-deficient macrophages (Fig. 2A). In a control experiment, the activity of a TLR4 agonist, E. coli LPS, was not influenced by TLR2 deficiency but was abrogated by MyD88 deficiency (Fig. 2A). In an additional control experiment, the ability of PMA (a cell-permeable agonist that bypasses receptor interactions (19)) to activate CR3 binding of sICAM-1 was not affected by either MyD88 or TLR2 deficiency (Fig. 2A). That the inducible binding of sICAM-1 was indeed dependent upon CR3 was confirmed by the specific blocking effect of anti-CD11b mAb (Fig. 2B). However, deficiency of either TLR2 or MyD88 abrogated proinflammatory cytokine induction by P. gingivalis fimbriae, although, as expected, not by Poly(I:C) which is a MyD88-independent TLR3 agonist (Fig. 2C). As an additional control, the cytokine-inducing ability of E. coli LPS was significantly inhibited by MyD88 deficiency but not by TLR2 deficiency (Fig. 2C). Taken together, the results from figure 1 and 2 show that human or mouse MyD88 mediates P. gingivalis fimbria-induced proinflammatory but not proadhesive signaling. In contrast, TLR2 is essential in both pathways.
Figure 2. Role of MyD88 in P. gingivalis fimbria-induced proadhesive or proinflammatory activities in mouse macrophages.

Mouse macrophages (wild-type or deficient in MyD88 or TLR2) were activated with P. gingivalis fimbriae (1 μg/ml) and assayed for CR3-dependent binding of sICAM-1 (A, B) or induction of TNF-α release (C). In B, wild-type macrophages were pretreated for 30 min with a CR3 blocking (anti-CD11b) mAb or isotype control. The agonists, E. coli LPS (0.2 μg/ml), PMA (0.1 μg/ml), and Poly(I:C) (50 μg/ml), were used as controls in A and/or C, as indicated. Data are presented as means ± SD (n = 3) from one of three (A) or two (B, C) independent experiments that yielded consistent results. Asterisks indicate statistically significant (p < 0.05) differences compared to wild-type macrophages (A, C) or compared to no-antibody treatment (B).
Role of TIRAP in P. gingivalis fimbria-induced proadhesive or proinflammatory activities
Unlike MyD88 which mediates signaling downstream of most TLRs (except for TLR3), TIRAP (or Mal) is involved only in TLR2 and TLR4 signaling (8, 41). The role of TIRAP is to facilitate the recruitment of MyD88 to the cytoplasmic domain of the activated TLR, and is thus believed to function as a sorting adaptor (42). Although TIRAP would likely be involved in P. gingivalis fimbria-induced TLR2 proinflammatory signaling, it is unknown whether it could function as an adaptor, or play any other role, in the TLR2 proadhesive pathway. To examine this possibility, we first determined the ability of THP-1/CD14 cells to induce the CR3 activation-specific CBRM1/5 epitope in response to P. gingivalis fimbriae, upon transfection with a TIRAP mutant (P125H) which acts as dominant negative inhibitor (TIRAP-DN) (43). Transfection with TLR2-DN served as a positive control, whereas transfections with empty vector or a dominant negative version (C117H) of the TRIF-related adaptor molecule (TRAM), which is exclusively involved in TLR4 signaling (44, 45), served as negative controls. We found that TIRAP-DN failed to suppress CBRM1/5 epitope induction by P. gingivalis fimbriae (as seen with the negative controls), although TLR2-DN readily inhibited the same activity (p < 0.05; Fig. 3). However, as expected, both TLR2-DN and TIRAP-DN (but not TRAM-DN) inhibited fimbria-induced NF-κB activation (Fig. 3).
Figure 3. TIRAP-dependent and –independent proinflammatory and proadhesive activities of P. gingivalis fimbriae.

Human THP-1/CD14 cells were transfected with dominant negative (DN) point mutants of human TIRAP (TIRAP-DN) or TRAM (TRAM-DN; negative control), or a TIR domain-deficient mutant of TLR2 that acts as a dominant negative inhibitor (TLR2-DN; positive control), at the indicated μg amounts of plasmid DNA per 2×105 cells. The cells were stimulated with P. gingivalis fimbriae (1 μg/ml) and assayed for induction of the CR3 activation-specific CBRM1/5 epitope (after staining with FITC-labeled CBRM1/5 mAb) or for NF-κB-dependent transcription of a luciferase reporter gene. CBRM1/5 induction is reported in relative fluorescent units (RFU) and NF-κB activation in relative luciferase activity (RLA). The discontinuous horizontal lines indicate CBRM1/5 epitope induction or NF-κB activation in resting cells, which were < 12 % of the values of fimbria-stimulated cells. Data are means ± SD (n = 3) from one of two experiments that yielded consistent results. The asterisks indicate statistically significant (p < 0.05) differences compared to empty vector control treatments.
The notion that TIRAP is involved in the TLR2 proinflammatory but not the proadhesive pathway was confirmed using mouse macrophages. Indeed, a TIRAP inhibitory peptide, which blocks TIRAP-dependent signaling (32, 33), significantly (p < 0.05) diminished the ability of P. gingivalis fimbriae to induce IL-6 production, although it had no effect on the ability of fimbria to stimulate CR3 adhesive activity, as measured by sICAM-1 binding (Fig. 4). Treatments with a control peptide had no effect on either assay (Fig. 4). Therefore, TIRAP signaling has no effect on P. gingivalis fimbria-induced proadhesive activities, although it readily inhibits P. gingivalis fimbria-induced proinflammatory activities. These findings, taken together with those from figures 1 and 2, indicate that the proadhesive pathway is TIRAP- and MyD88-independent.
Figure 4. Role of TIRAP in P. gingivalis fimbria-induced proadhesive or proinflammatory activities in mouse macrophages.
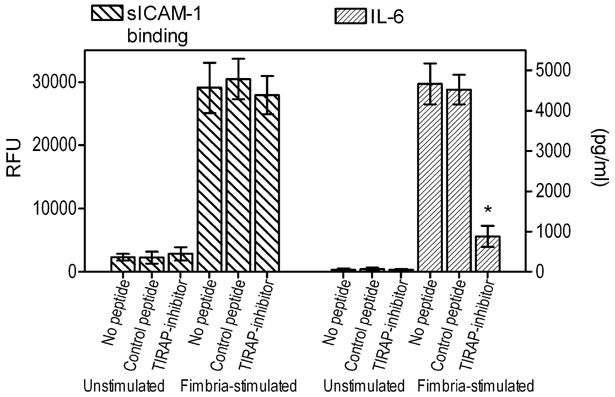
Mouse macrophages were pretreated for 1h with 10 μM of a cell-permeable TIRAP inhibitory peptide or inactive control. The cells were subsequently left unstimulated or were stimulated with P. gingivalis fimbriae (1 μg/ml) and assayed for sICAM-1 binding or induction of IL-6 release. Data are means ± SD (n = 3) from one of two independent experiments that yielded consistent findings. Asterisks indicate statistically significant (p < 0.05) differences compared to medium-treated (no peptide) controls.
Akt is activated in a TLR2/PI3K-dependent way but is not involved in the proadhesive pathway
PI3K plays an essential role in the P. gingivalis fimbria-induced TLR2 proadhesive signaling pathway (20, 21). Since the Ser/Thr kinase Akt was previously implicated in inside-out proadhesive signaling (46) and is one of the most likely downstream targets of PI3K for a variety of cellular functions (47), we examined possible involvement of Akt in the fimbria-activated proadhesive pathway.
We first examined if Akt is activated by P. gingivalis fimbriae in a TLR2- and PI3K-dependent way. Indeed, stimulation of THP-1/CD14 cells by fimbriae led to activation (phosphorylation) of Akt, although this activity was significantly (p < 0.05) suppressed in cells transfected with TLR2-DN or pretreated with the PI3K inhibitor, LY294002 (but not with its inactive analog, LY303511) (Fig. 5A). None of the treatments influenced total Akt levels (Fig. 5A). We then determined whether transfection of THP-1/CD14 cells with a dominant negative inhibitor of Akt signaling (Akt-DN) could inhibit induction of the CR3 activation-specific CBRM1/5 epitope by P. gingivalis fimbriae. Empty vector and TLR2-DN were used as negative and positive controls, respectively. Akt-DN failed to inhibit CBRM1/5 induction, in contrast to TLR2-DN which displayed dose-dependent inhibitory action (Fig. 5B). We observed a similar lack of inhibitory effects on CBRM1/5 epitope induction, using a range of concentrations (25–125 μM) of a cell permeable peptide that blocks Akt kinase activity (34) or a pharmacological inhibitor of Akt (5–10 μM; 1L-6-Hydroxymethyl-chiro-inositol2-[(R)-2-O-methyl-3-O-octadecylcarbonate]) (data not shown). Therefore, a different PI3K downstream effector may link TLR2 to CR3 activation, and we set out to identify it.
Figure 5. Akt is activated by P. gingivalis fimbriae in a TLR2/PI3K-dependent way but is not involved in CR3 activation.

THP-1/CD14 cells were transfected or not with dominant negative inhibitors of TLR2 (TLR2-DN) [A and B] or Akt (Akt-DN) [B] at the indicated μg of plasmid DNA per 2×105 cells. In A, untransfected THP-1/CD14 cells were pretreated with medium only, LY294002 (PI3K inhibitor) or the inactive analog LY30351 (both at 20 μM). Cells were then activated with P. gingivalis fimbriae and assayed for total and phosphorylated Akt [A], or for induction of the CBRM1/5 epitope, measured in relative fluorescence units (RFU) [B]. The discontinuous horizontal line in B indicates CBRM1/5 epitope induction in resting cells which was < 10% of the activity of fimbria-stimulated cells. Data are means ± SD (n = 3), from one of three (A) or two (B) independent experiments yielding consistent results. Asterisks indicate statistically significant (p < 0.05) differences compared to corresponding control treatments.
Role of cytohesin-1 in P. gingivalis fimbria-induced CR3 activation
In addition to Akt, cytohesin-1 is another pleckstrin homology domain-containing cytoplasmic protein that binds to phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) generated by PI3K (48). Moreover, cytohesin-1 is also implicated in induction of integrin adhesive capacity (36, 48). We have thus investigated whether cytohesin-1 participates in the P. gingivalis fimbria-induced pathway for CR3 activation, using an established antisense strategy for inhibiting cytohesin-1 expression (35). Treatment of THP-1/CD14 cells with antisense S-oligos to cytohesin-1 mRNA (but not with control sense S-oligos) significantly inhibited the ability of fimbria-stimulated cells to induce the CBRM1/5 epitope and bind sICAM-1 (p < 0.05; Fig. 6). However, the antisense S-oligo treatments had no significant effect on NF-κB activation by P. gingivalis fimbriae (Fig. 6). Therefore, cytohesin-1 appears to mediate P. gingivalis fimbria-induced activation of the proadhesive but not of the proinflammatory pathway.
Figure 6. Cytohesin-1 is involved in P. gingivalis fimbria-induced proadhesive but not proinflammatory activities.

THP-1/CD14 cells were treated with antisense S-oligos (AS) to cytohesin-1 mRNA or with control sense S-oligos (S), at the indicated concentrations (μM). Following stimulation with P. gingivalis fimbriae (1 μg/ml), the cells were assayed for induction of the CBRM1/5 epitope, binding of sICAM-1, or for NF-κB-dependent transcription of a luciferase reporter gene. CBRM1/5 induction and sICAM-1 binding are reported in relative fluorescent units (RFU) and NF-κB activation in relative luciferase activity (RLA). Unstimulated cells displayed < 9 % of the activities of fimbria-stimulated cells in all three assays. Results are means ± SD (n = 3) from one of two independent experiments that yielded consistent findings. Asterisks indicate statistically significant (p < 0.05) inhibition of activities compared to cells that were not treated with anti-sense S-oligos.
Discussion
As more information becomes increasingly available on the intricacies of TLR signaling, it is appreciated that the control of adverse host responses in infection-driven inflammatory diseases would require highly selective and precisely targeted intervention. This in turn requires precise delineation of TLR signaling pathways in response to a given pathogen, such as P. gingivalis. It is generally thought that TLR4 signaling is more complex than TLR2 signaling, since TLR4 activation initiates two divergent signaling cascades: The TIRAP/MyD88 pathway which activates NF-κB, and the TRAM/TRIF (TIR-domain-containing adapter-inducing interferon-β) pathway, which additionally triggers activation of the interferon regulatory factor-3 (8). In this paper, we have presented evidence that TLR2 activation also initiates two divergent signaling pathways.
Specifically, TLR2 activation by P. gingivalis fimbriae induces a TIRAP/MyD88-dependent proinflammatory pathway and a TIRAP/MyD88-independent but PI3K-dependent proadhesive pathway, as illustrated in the Fig. 7 model. The former pathway induces production of NF-κB-dependent proinflammatory cytokines and the antimicrobial molecule nitric oxide, within hours upon cell stimulation with P. gingivalis (6, 18). The proadhesive pathway is characterized by inside-out signaling which, within minutes, transactivates the adhesive capacity of CR3 (CD11b/CD18) (21), a β2 integrin which can binds both host and microbial molecules (49). More recently, other agonists were also shown to activate the TLR2 proadhesive pathway (50). In particular, peptidoglycan and the Pam3CysSerLys4 lipopeptide were demonstrated to activate TLR2- and CD11b/CD18-dependent monocyte adhesion to ICAM-1, although intermediate signaling molecules were not examined (50).
Figure 7. P. gingivalis-activated TLR2 induces distinct proadhesive and proinflammatory signaling pathways.

The model is based on the current findings and our previously published studies (6, 19–21). P. gingivalis fimbriae interact with the CD14/TLR2 complex leading to TLR2 activation. Activated TLR2 induces TIRAP/MyD88-dependent signaling for NF-κB activation and induction of proinflammatory cytokines (proinflammatory pathway), and TIRAP/MyD88-independent signaling for transactivation of the CR3 adhesive activity (proadhesive pathway). The latter pathway proceeds via Rac1, PI3K and cytohesin-1 (Cyt-1). Cytohesin-1 is likely to function immediately downstream of PI3K, since it was previously shown to use PI3K-generated PIP3 for membrane docking and interaction with the CD18 cytoplasmic tail (48).
Our demonstration that the TLR2/PI3K proadhesive pathway proceeds independently of MyD88 and TIRAP is consistent with earlier findings by another group that the cytoplasmic domain of TLR2 contains two PI3K binding motifs (YXXM/W) that could serve as docking sites for PI3K (51). Taken together, these findings suggest that PI3K may be recruited to P. gingivalis fimbria-activated TLR2 in a MyD88- and TIRAP-independent way. MyD88, however, was required for LPS-induced binding of soluble ICAM-1 by CR3, in line with a previous report that activation of the αMβ2 integrin (CR3) by LPS involves MyD88-mediated signaling (52). Therefore, P. gingivalis fimbriae acting through TLR2 may stimulate a different inside-out signaling pathway from that activated by LPS acting through TLR4. This difference may be attributable to the inability of TLR4 to directly recruit PI3K to its cytoplasmic tail which lacks PI3K binding motifs (51). Our previous work has also shown that the small GTPase Rac1 acts upstream of PI3K and plays a positive regulatory role in the TLR2 proadhesive pathway (20) (Fig. 7), presumably through its ability to bind PI3K and augment its activity (53). Despite these earlier developments, the downstream effector of PI3K in the P. gingivalis fimbria-activated proadhesive pathway had remained elusive.
Here, we identified cytohesin-1, rather than Akt, as the downstream PI3K effector in the TLR2 proadhesive pathway. Interestingly, although PMA induces β2 integrin activation through a distinct pathway involving protein kinase C, this pathway also uses cytohesin-1 as an effector (54, 55). Moreover, the ability of LPS to activate the ligand-binding capacity of leukocyte function-associated antigen-1 (CD11a/CD18) is similarly dependent on cytohesin-1 activity (35). Therefore, cytohesin-1 appears to functionally integrate distinct signaling pathways for β2 integrin activation. Earlier studies have established that the inside-out signaling regulation of integrin binding affinity involves targeting of intracellular proteins to the cytoplasmic tails of the integrin, thereby causing high-affinity conformational changes on its ligand-binding domain (reviewed in ref. (56). Since cytohesin-1 can utilize PI3K-generated PIP3 phospholipids as docking sites to interact with the CD18 cytoplasmic tail (48), it can be suggested that cytohesin-1 forms a link between PI3K and CR3 (CD11b/CD18) in the P. gingivalis fimbria-induced TLR2 proadhesive pathway (Fig. 7). Although a role for Akt in this pathway is not supported by our data, Akt was previously implicated as an essential PI3K downstream effector for glycoprotein Ib-IX induced inside-out signaling for αIIbβ3 integrin activation in platelets (46).
Monocyte transmigration is mediated by interacting sets of cell adhesion molecules, including the CR3 (CD11b/CD18) ICAM-1 pair (57). In this regard, activation of TLR2 inside-out signaling by P. gingivalis (or purified fimbriae) leads to CR3-dependent monocyte adhesion to endothelial ICAM-1 and transmigration across endothelial cell monolayers (20). Although this is a potentially protective mechanism that may contribute to the recruitment of monocytes to sites of P. gingivalis infection, adhesion of monocytes to the arterial endothelium and their subsequent migration into the subendothelial area is a hallmark of early atherogenesis (57). Whether this may form a mechanistic basis linking this pathogen to inflammatory atherosclerotic processes, in which it has been implicated (4), is uncertain at the moment. However, there is sufficient evidence that P. gingivalis has co-opted the TLR2/CR3 proadhesive pathway for CR3 binding and entry into macrophages in a way that promotes its virulence (19, 26). Strikingly, the same pathway is exploited by mycobacteria for promoting their uptake by monocytes/macrophages through activated CR3 (36) leading to their survival and intracellular persistence (58), possibly because this receptor is not linked to vigorous microbicidal mechanisms (reviewed in ref. (59). It is intriguing to speculate that pathogen-induced TLR2 inside-out signaling for CR3 activation may be a universal pathway exploited by a number of pathogens and, therefore, ways to block this pathway could be beneficial in certain infections.
In addition to a better understanding of the signaling pathways activated by P. gingivalis or its purified fimbriae, our current findings may be significant from a translational viewpoint. In this regard, delineation of signaling pathways may provide potential therapeutic targets for selectively enhancing or inhibiting pathway activities, depending on whether they promote or undermine host defense. If TLR2/NF-κB-mediated immunity helps control P. gingivalis infection, whereas exploitation of TLR2-transactivated CR3 favors P. gingivalis persistence, as supported by our recent studies (18, 25, 26, 59), it would make sense to inhibit TLR2 signaling in a way that would inhibit specifically the proadhesive pathway. Indeed, inhibition of the TLR2 proadhesive pathway may prevent CR3-mediated uptake of P. gingivalis, an internalization route that suppresses the ability of macrophages to clear P. gingivalis infection (25, 26). So far, we have identified three targets in the proadhesive pathway downstream of TLR2; Rac1, PI3K, and cytohesin-1. Considering that Rac1 and PI3K are involved in a great number of essential physiological functions (60, 61), it appears that cytohesin-1 may be a more feasible target for intervention. More studies are warranted to determine the usefulness of a cytohesin-based approach as a strategy to counteract immune evasion by P. gingivalis.
Footnotes
This work was supported by U.S. Public Health Service Grants DE015254 and DE018292 from the National Institutes of Health/NIDCR (to G.H.).
References
- 1.Medzhitov R. Toll-like receptors and innate immunity. Nat Rev Immunol. 2001;1:135–145. doi: 10.1038/35100529. [DOI] [PubMed] [Google Scholar]
- 2.Beutler B, Jiang Z, Georgel P, Crozat K, Croker B, Rutschmann S, Du X, Hoebe K. Genetic analysis of host resistance: Toll-like receptor signaling and immunity at large. Annu Rev Immunol. 2006;24:353–389. doi: 10.1146/annurev.immunol.24.021605.090552. [DOI] [PubMed] [Google Scholar]
- 3.Carpenter S, O’Neill LA. How important are Toll-like receptors for antimicrobial responses? Cell Microbiol. 2007;9:1891–1901. doi: 10.1111/j.1462-5822.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 4.Gibson FC, 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–121. doi: 10.1177/154405910608500202. [DOI] [PubMed] [Google Scholar]
- 5.Burns E, Bachrach G, Shapira L, Nussbaum G. Cutting Edge: TLR2 is required for the innate response to Porphyromonas gingivalis: Activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- 6.Hajishengallis G, Tapping RI, Harokopakis E, Nishiyama SI, Ratti P, Schifferle RE, Lyle EA, Triantafilou M, Triantafilou K, Yoshimura F. Differential interactions of fimbriae and lipopolysaccharide from Porphyromonas gingivalis with the Toll-like receptor 2-centred pattern recognition apparatus. Cell Microbiol. 2006;8:1557–1570. doi: 10.1111/j.1462-5822.2006.00730.x. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun. 2005;73:935–943. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 9.Malek R, Fisher JG, Caleca A, Stinson M, van Oss CJ, Lee JY, Cho MI, Genco RJ, Evans RT, Dyer DW. Inactivation of Porphyromonas gingivalis fimA gene blocks periodontal damage in gnotobiotic rats. J Bacteriol. 1994;176:1052–1059. doi: 10.1128/jb.176.4.1052-1059.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson FC, 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, Wong J, Genco CA. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 11.Lamont RJ, Jenkinson HF. Life below the gum line: Pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amano A. Molecular interaction of Porphyromonas gingivalis with host cells: implication for the microbial pathogenesis of periodontal disease. J Periodontol. 2003;74:90–96. doi: 10.1902/jop.2003.74.1.90. [DOI] [PubMed] [Google Scholar]
- 13.Asai Y, Ohyama Y, Gen K, Ogawa T. Bacterial fimbriae and their peptides activate human gingival epithelial cells through Toll-like receptor 2. Infect Immun. 2001;69:7387–7395. doi: 10.1128/IAI.69.12.7387-7395.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hajishengallis G, Ratti P, Harokopakis E. Peptide mapping of bacterial fimbrial epitopes interacting with pattern recognition receptors. J Biol Chem. 2005;280:38902–38913. doi: 10.1074/jbc.M507326200. [DOI] [PubMed] [Google Scholar]
- 15.Jotwani R, Cutler CW. Fimbriated Porphyromonas gingivalis is more effcient than fimbria-deficient P. gingivalis in entering human dendritic cells in vitro and induces an inflammatory Th1 effector response. Infect Immun. 2004;72:1725–1732. doi: 10.1128/IAI.72.3.1725-1732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eskan MA, Hajishengallis G, Kinane DF. Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect Immun. 2007;75:892–898. doi: 10.1128/IAI.01604-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davey M, Liu X, Ukai T, Jain V, Gudino C, Gibson FC, 3rd, Golenbock D, Visintin A, Genco CA. Bacterial fimbriae stimulate proinflammatory activation in the endothelium through distinct TLRs. J Immunol. 2008;180:2187–2195. doi: 10.4049/jimmunol.180.4.2187. [DOI] [PubMed] [Google Scholar]
- 18.Hajishengallis G, Wang M, Liang S, Triantafilou M, Triantafilou K. Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc Natl Acad Sci U S A. 2008;105:13532–13537. doi: 10.1073/pnas.0803852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas gingivalis fimbriae proactively modulate β2 integrin adhesive activity and promote binding to and internalization by macrophages. Infect Immun. 2006;74:5658–5666. doi: 10.1128/IAI.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 transmodulates monocyte adhesion and transmigration via Rac1- and PI3K-mediated inside-out signaling in response to Porphyromonas gingivalis fimbriae. J Immunol. 2006;176:7645–7656. doi: 10.4049/jimmunol.176.12.7645. [DOI] [PubMed] [Google Scholar]
- 21.Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35:1201–1210. doi: 10.1002/eji.200425883. [DOI] [PubMed] [Google Scholar]
- 22.Muthukuru M, Jotwani R, Cutler CW. Oral mucosal endotoxin tolerance induction in chronic periodontitis. Infect Immun. 2005;73:687–694. doi: 10.1128/IAI.73.2.687-694.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren L, Leung WK, Darveau RP, Jin L. The expression profile of lipopolysaccharide-binding protein, membrane-bound CD14, and toll-like receptors 2 and 4 in chronic periodontitis. J Periodontol. 2005;76:1950–1959. doi: 10.1902/jop.2005.76.11.1950. [DOI] [PubMed] [Google Scholar]
- 24.Linton MF, Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obes Relat Metab Disord. 2003;27(Suppl 3):S35–40. doi: 10.1038/sj.ijo.0802498. [DOI] [PubMed] [Google Scholar]
- 25.Hajishengallis G, Shakhatreh MAK, Wang M, Liang S. Complement receptor 3 blockade Promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J Immunol. 2007;179:2359–2367. doi: 10.4049/jimmunol.179.4.2359. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Shakhatreh M-AK, James D, Liang S, Nishiyama S-i, Yoshimura F, Demuth DR, Hajishengallis G. Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J Immunol. 2007;179:2349–2358. doi: 10.4049/jimmunol.179.4.2349. [DOI] [PubMed] [Google Scholar]
- 27.Yoshimura F, Takahashi K, Nodasaka Y, Suzuki T. Purification and characterization of a novel type of fimbriae from the oral anaerobe Bacteroides gingivalis. J Bacteriol. 1984;160:949–957. doi: 10.1128/jb.160.3.949-957.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang S, Wang M, Tapping RI, Stepensky V, Nawar HF, Triantafilou M, Triantafilou K, Connell TD, Hajishengallis G. Ganglioside GD1a is an essential coreceptor for toll-like receptor 2 signaling in response to the B subunit of Type IIB enterotoxin. J Biol Chem. 2007;282:7532–7542. doi: 10.1074/jbc.M611722200. [DOI] [PubMed] [Google Scholar]
- 29.Pugin J, V, Kravchenko V, Lee JD, Kline L, Ulevitch RJ, Tobias PS. Cell activation mediated by glycosylphosphatidylinositol-anchored or transmembrane forms of CD14. Infect Immun. 1998;66:1174–1180. doi: 10.1128/iai.66.3.1174-1180.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajishengallis G, Tapping RI, Martin MH, Nawar H, Lyle EA, Russell MW, Connell TD. Toll-like receptor 2 mediates cellular activation by the B subunits of type II heat-labile enterotoxins. Infect Immun. 2005;73:1343–1349. doi: 10.1128/IAI.73.3.1343-1349.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loiarro M, Sette C, Gallo G, Ciacci A, Fanto N, Mastroianni D, Carminati P, Ruggiero V. Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J Biol Chem. 2005;280:15809–15814. doi: 10.1074/jbc.C400613200. [DOI] [PubMed] [Google Scholar]
- 32.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-β-induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 33.Horng T, Barton GM, Medzhitov R. TIRAP: an adapter molecule in the Toll signaling pathway. Nat Immunol. 2001;2:835–841. doi: 10.1038/ni0901-835. [DOI] [PubMed] [Google Scholar]
- 34.Hiromura M, Okada F, Obata T, Auguin D, Shibata T, Roumestand C, Noguchi M. Inhibition of Akt Kinase Activity by a Peptide Spanning the βA Strand of the Proto-oncogene TCL1. J Biol Chem. 2004;279:53407–53418. doi: 10.1074/jbc.M403775200. [DOI] [PubMed] [Google Scholar]
- 35.Hmama Z, Knutson KL, Herrera-Velit P, Nandan D, Reiner NE. Monocyte adherence induced by lipopolysaccharide involves CD14, LFA-1, and Cytohesin-1. Regulation by Rho and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:1050–1057. doi: 10.1074/jbc.274.2.1050. [DOI] [PubMed] [Google Scholar]
- 36.Sendide K, Reiner NE, Lee JS, Bourgoin S, Talal A, Hmama Z. Crosstalk between CD14 and complement receptor 3 promotes phagocytosis of mycobacteria: regulation by phosphatidylinositol 3-kinase and cytohesin-1. J Immunol. 2005;174:4210–4219. doi: 10.4049/jimmunol.174.7.4210. [DOI] [PubMed] [Google Scholar]
- 37.Diamond MS, Springer TA. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol. 1993;120:545–556. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. J Cell Biol. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 40.Burns K, Martinon F, Esslinger C, Pahl H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein involved in interleukin-1 signaling. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- 41.Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, Hoshino K, Takeuchi O, Kobayashi M, Fujita T, Takeda K, Akira S. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature. 2002;420:324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 42.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 43.Fitzgerald KA, Palsson-McDermott EM, Bowie AG, Jefferies CA, Mansell AS, Brady G, Brint E, Dunne A, Gray P, Harte MT, McMurray D, Smith DE, Sims JE, Bird TA, O’Neill LA. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 44.Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T. TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that induces interferon-beta. J Biol Chem. 2003;278:49751–49762. doi: 10.1074/jbc.M305820200. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto M, Sato S, Hemmi H, Uematsu S, Hoshino K, Kaisho T, Takeuchi O, Takeda K, Akira S. TRAM is specifically involved in the Toll-like receptor 4-mediated MyD88-independent signaling pathway. Nat Immunol. 2003;4:1144–1150. doi: 10.1038/ni986. [DOI] [PubMed] [Google Scholar]
- 46.Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111:658–665. doi: 10.1182/blood-2007-04-085514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 48.Nagel W, Zeitlmann L, Schilcher P, Geiger C, Kolanus J, Kolanus W. Phosphoinositide 3-OH kinase activates theβ2 integrin adhesion pathway and induces membrane recruitment of cytohesin-1. J Biol Chem. 1998;273:14853–14861. doi: 10.1074/jbc.273.24.14853. [DOI] [PubMed] [Google Scholar]
- 49.Ehlers MRW. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–294. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 50.Nijhuis MM, Pasterkamp G, Sluis NI, de Kleijn DP, Laman JD, Ulfman LH. Peptidoglycan increases firm adhesion of monocytes under flow conditions and primes monocyte chemotaxis. J Vasc Res. 2007;44:214–222. doi: 10.1159/000100420. [DOI] [PubMed] [Google Scholar]
- 51.Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-κB activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- 52.Schmidt A, Caron E, Hall A. Lipopolysaccharide-Induced Activation of {beta}2-Integrin Function in Macrophages Requires Irak Kinase Activity, p38 Mitogen-Activated Protein Kinase, and the Rap1 GTPase. Mol Cell Biol. 2001;21:438–448. doi: 10.1128/MCB.21.2.438-448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bokoch GM, Vlahos CJ, Wang Y, Knaus UG, Traynor-Kaplan AE. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem J. 1996;315(Pt 3):775–779. doi: 10.1042/bj3150775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dierks H, Kolanus J, Kolanus W. Actin cytoskeletal association of cytohesin-1 is regulated by specific phosphorylation of its carboxyl-terminal polybasic domain. J Biol Chem. 2001;276:37472–37481. doi: 10.1074/jbc.M101502200. [DOI] [PubMed] [Google Scholar]
- 55.Mayer G, Blind M, Nagel W, Bohm T, Knorr T, Jackson CL, Kolanus W, Famulok M. Controlling small guanine-nucleotide-exchange factor function through cytoplasmic RNA intramers. Proc Natl Acad Sci U S A. 2001;98:4961–4965. doi: 10.1073/pnas.091100698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annu Rev Biophys Biomol Struct. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- 57.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 58.Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun. 1998;66:1277–1281. doi: 10.1128/iai.66.4.1277-1281.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hajishengallis G, Wang M, Liang S, Shakhatreh MA, James D, Nishiyama S, Yoshimura F, Demuth DR. Subversion of innate immunity by periodontopathic bacteria via exploitation of complement receptor-3. Adv Exp Med Biol. 2008;632:203–219. doi: 10.1007/978-0-387-78952-1_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol. 2003;15:590–597. doi: 10.1016/s0955-0674(03)00097-8. [DOI] [PubMed] [Google Scholar]
- 61.Koyasu S. The role of PI3K in immune cells. Nat Immunol. 2003;4:313–319. doi: 10.1038/ni0403-313. [DOI] [PubMed] [Google Scholar]