

Abstract
Mechanical ventilation is a life-saving intervention in patients with respiratory failure. However, human and animal studies have demonstrated that mechanical ventilation using large tidal volumes (≥12 ml/kg) induces a potent inflammatory response and can cause acute lung injury. We hypothesized that mechanical ventilation with a “noninjurious” tidal volume of 10 ml/kg would still activate a transcriptional program that places the lung at risk for severe injury. To identify key regulators of this transcriptional response, we integrated gene expression data obtained from whole lungs of spontaneously breathing mice and mechanically ventilated mice with computational network analysis. Topological analysis of the gene product interaction network identified Jun and Fos families of proteins as potential regulatory hubs. Electrophoretic mobility gel shift assay confirmed protein binding to activator protein-1 (AP-1) consensus sequences, and supershift experiments identified JunD and FosB as components of ventilation-induced AP-1 binding. Specific recruitment of JunD to the regulatory region of the F3 gene by mechanical ventilation was confirmed by chromatin immunoprecipitation assay. In conclusion, we demonstrate a novel computational framework to systematically dissect transcriptional programs activated by mechanical ventilation in the lung, and show that noninjurious mechanical ventilation initiates a response that can prime the lung for injury from a subsequent insult.
Keywords: gene network, chromatin immunoprecipitation, activator protein-1, F3, lung injury
mechanical stretching of cells occurs under normal and abnormal physiological conditions and results in specific cellular responses collectively known as mechanotransduction (40). The lungs undergo cyclical stretching during spontaneous breathing and are well adapted to the ensuing volumetric changes (34). However, the cellular consequences of mechanical ventilation (MV), a life-saving intervention during respiratory failure, may be significantly different. Animal and human data have implicated MV as an important contributor to the development of lung injury when delivered at high volumes (e.g., 12–20 ml/kg)—a phenomenon termed ventilator-induced lung injury (VILI) or ventilator-associated lung injury (VALI) (2, 4, 15, 27, 34, 39). On the basis of these landmark studies, patients presently undergoing MV are being ventilated at volumes significantly lower (6–10 ml/kg) than those previously deemed acceptable (44).
We recently demonstrated (3) that the combination of noninjurious MV and low-dose exposure to bacterial products can cause severe lung injury in mice, implying a comodulatory role for MV in lungs that are at risk for developing injury. Using computational methods, we subsequently identified key regulatory modules activated in this “two-hit” model of acute lung injury (20). In the present work, we tested the hypothesis that noninjurious MV by itself initiates a proinflammatory transcriptional program in the lung and undertook an unbiased approach to partially decipher this complex network of pathways. We found that healthy lungs undergoing MV were “primed” and potentially vulnerable to significant injury from insults that would not have been injurious during spontaneous respiration. Importantly, we biologically validated several of our computational predictions. Our modeling methods identified regulatory nodes within activated genetic networks that, if targeted, may modify the lung's inflammatory response to MV.
MATERIALS AND METHODS
Antibodies.
Antibodies to JunD (sc-74), c-Jun (sc-45), JunB (sc-46), c-Fos (sc-52), FosB (sc-48), Fra-1 (sc-183), and Fra-2 (sc-171) were purchased from Santa Cruz Biotechnology. Antibodies to phospho-Jun (9164, binds to c-Jun phosphorylation at S73 and JunD phosphorylation at S100), phospho-JNK (9251), and phospho-ERK1/2 (4370) were purchased from Cell Signaling Technologies. Antibody to mouse actin was purchased from Abcam.
Animal experiments.
All animal experiments were approved by the University of Washington's Animal Care Committee. Experiments in which RNA was collected for microarray analysis were performed and reported as part of a previous study (3). Additional experiments were performed for quantitative RT-PCR (qRT-PCR), electrophoretic mobility gel shift assay (EMSA), and chromatin immunoprecipitation (ChIP) experiments with the same animal protocol. Briefly, 8-wk old male C57BL/6 mice (Harlan) were randomly assigned to two groups: spontaneous breathing (control) and MV. MV animals were anesthetized and mechanically ventilated with a tidal volume of 10 ml/kg, a respiratory rate of 150 min−1, a fraction of inspired O2 (FiO2) of 0.21, and 0 cmH2O end-expiratory pressure (MiniVent, Harvard Biosciences) for 4 h. The control mice breathed spontaneously during this 4-h period. At the conclusion of the intervention period, mice were euthanized by exsanguination, lungs were flushed with RNase-free PBS via the right ventricle and homogenized, and total RNA was isolated with the RNeasy Midi Kit (Qiagen). RNA integrity was confirmed with the Bioanalyzer 2100 (Agilent).
Histology.
For each group, two additional mice were killed at the end of the intervention period and their lungs were removed and fixed via intratracheal instillation of 4% formalin at a transmural pressure of 20 cmH2O. After fixation the lungs were embedded in paraffin, and 4-μm sections were stained with hematoxylin and eosin (H & E).
Oligonucleotide microarray data analysis.
Microarray experiments were performed and reported in a previous study (3). For each mouse, labeled cRNA was prepared from total RNA and hybridized to an Affymetrix GeneChip MOE430A oligonucleotide microarray with minor modifications from Affymetrix-recommended protocols. Microarrays were scanned with Affymetrix GeneChip Scanner, and image analysis was performed with Affymetrix MAS 5.0 software. Background adjustment and quantile normalization across all microarrays were performed with the Robust Multichip Average algorithm (RMAExpress) (9). Statistically significant differential gene expression during MV relative to spontaneous breathing was determined with a Bayesian t-test algorithm (5). The multiple-comparisons problem was addressed by using the false discovery rate (FDR) methodology proposed by Benjamini and Hochberg (8). Two-dimensional hierarchical clustering using the average linkage method and euclidean distance metric was applied to the normalized gene expression intensities across all samples (35). Multidimensional scaling using principal components was performed based on the covariance matrix of normalized gene expression values from all 12 animals (35). All microarray data, in compliance with Minimum Information About a Microarray Experiment (MIAME), are available at the GEO website (http://www.ncbi.nlm.nih.gov/projects/geo/; query GSE 2411).
Genetic network interactome.
A gene product interaction network was created from the transcriptional profiling experiments with Ingenuity System's database (10) and the mining of several publicly available resources: 1) Human Protein Reference Database, consisting of ∼25,000 proteins and ∼35,000 interactions (32); 2) Biomolecular Interaction Network Database, comprising ∼200,000 interactions (1); and 3) Database of Interacting Proteins, containing ∼20,000 proteins and ∼55,000 interactions (36). Gene product networks were constructed based on an iterative algorithm utilizing the connectivity of the input gene set. These subnetworks were then merged with each other to create a larger gene interaction network, or interactome. We limited the nodes of the interactome to genes differentially expressed during MV. The topology of the resulting network was studied by determining its connectivity matrix. To assess whether the interactome possessed “scale-free” properties, i.e., followed a power law distribution, the degree distribution of the nodes, P(k), was plotted versus the nodes' connectivity, k (6).
Functional enrichment analysis.
Gene annotation of all probe sets present on each GeneChip was obtained from the Gene Ontology database (19). Highly enriched biological modules activated during MV were determined with Expression Analysis Systematic Explorer (EASE) (22). Multiple hypothesis testing was addressed by performing a permutation analysis (n = 1,000) to calculate the global FDR. An FDR cutoff value of 0.1% or less was used for designating a biological module as significantly enriched. Next, a more expansive enrichment analysis was performed with the ToppGene program (http://toppgene.cchmc.org) (12). This web-based software performs simultaneous functional analysis of user-provided gene lists based on literature-derived databases covering several ontologies, disease phenotypes, biological pathways, and gene expression regulators.
Electrophoretic mobility gel shift assay.
To confirm biological activation of computationally identified transcription factors, we enriched the nuclear protein fraction of lung tissue collected from mechanically ventilated mice as previously described (7). Mice were mechanically ventilated for varying periods of time with the protocol described above. For each mouse, 10 μg of nuclear protein was incubated with 1 μl of a specific consensus binding sequence end-labeled with IRDye 700 infrared fluorescent tag (LI-COR Bioscience, Lincoln, NE) for 20 min at room temperature in a final reaction volume of 20 μl. After incubation, binding reactions were subjected to 4% nondenaturing polyacrylamide gel electrophoresis, and the gel was directly imaged on an Odyssey infrared imaging system (LI-COR Bioscience). For activator protein-1 (AP-1) EMSA, the probe sequence was sense: 5′-CGC TTG ATG ACT CAG CGG GAA-3′ and antisense: 5′-TTC CGG CTG AGT CAT CAA GCG-3′.
Optimized AP-1 binding buffer contained 10 mM Tris (pH 7.5), 50 mM KCl, 3.5 mM dithiothreitol (DTT), 1 μg of poly(dI-dC), 0.25% Tween 20, 0.05% NP-40, and 5 mM MgCl2. For cAMP response element (CRE) EMSA, the probe sequence was sense: 5′-AGA GAT TGC CTG ACG TCA GAG AGC TAG-3′ and antisense: 5′-CTA GCT CTC TGA CGT CAG GCA ATC TCT-3′.
The optimized CRE binding buffer was the same as the AP-1 buffer except without any MgCl2. For supershift EMSA, nuclear protein was preincubated in binding buffer with 2 μg of antibody at 4°C in an ultrasonic water bath (model 3510, Branson, Danbury, CT) for 30 min before incubation with the oligonucleotide probe.
Chromatin immunoprecipitation assay.
For qRT-PCR experiments, cDNA was synthesized from isolated RNA with the Superscript II kit (Invitrogen) per the manufacturer's instructions. Quantitative PCR was performed with Taqman primer/probe sets (Applied Biosystems) on an Mx3000P real-time PCR machine (Stratagene).
To isolate chromatin for subsequent ChIP, spontaneously breathing or mechanically ventilated mice were euthanized after 1 h. A combined thoracotomy and laparotomy was done, and the left renal artery was transected. The lungs were perfused free of blood via the right ventricle with 5 ml of ice-cold PBS. The left hilum was sutured and the left lung removed. The right lung was then dissected free of the chest and inflated with fresh 1% paraformaldehyde. The trachea was tied off to prevent collapse, and the lung was submerged in 2 ml of 1% paraformaldehyde. After 20 min of cross-linking at room temperature, the lung was removed from the paraformaldehyde, cleaned of trachea and any other mediastinal tissue, homogenized (Omni TH, Omni International) in 2 ml of 1% paraformaldehyde, and incubated for an additional 10 min. Lung homogenate was spun at 4,500 g for 10 min, and the supernatant was discarded. The pellet was resuspended in 1 ml of PBS containing 125 mM glycine and incubated for 5 min to inactivate any remaining paraformaldehyde. The lung homogenate was collected by centrifugation at 10,000 g for 10 min, washed once with 1 ml of PBS, and lysed in 1 ml of immunoprecipitation (IP) buffer [150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% NP-40, 50 mM Tris·HCl (pH 7.5)] containing 50 mM PMSF, 10 μg/ml leupeptin, and 1 mM DTT by stroking 10 times on the bottom of a test tube rack. The nuclear pellet was recovered by spinning at 10,000 g for 10 min at 4°C and resuspended in 1 ml of IP buffer containing PMSF, leupeptin, and DTT. Chromatin was subjected to 6 cycles of 18 pulses of ultrasonic shearing using a 250-W ultrasonic homogenizer set at a 40% power output and a 70% duty cycle (OmniRuptor 250, Omni International). This protocol was optimized to provide chromatin fragments with an average length of ∼400 bp. Chromatin was cleared by spinning at 10,000 g for 10 min at 4°C, the optical density at 260 nm (OD260) of the supernatant was determined, and the supernatant was stored in small aliquots at 80°C for later IP.
ChIP assay was performed by using the method of Nelson et al. (29, 30). Briefly, the amount of chromatin used for IP from the sample with the lowest OD260 was set to 50 μl, and the input IP volume for all other samples was determined by their OD260 with additional IP buffer to bring the final volume to 50 μl. For actual IP, 2 μg of anti-JunD antibody was used. Samples were incubated with or without (mock IP) antibody for 30 min in an ultrasonic water bath at 4°C followed by centrifugation at 10,000 g for 10 min. The supernatant was transferred to fresh tubes containing 15 μl of washed protein A agarose beads (GE Healthcare, Piscataway, NJ), and the slurry was rotated at 4°C for 45 min. The beads were then washed five times with 1 ml of IP buffer. To recover DNA, 100 μl of 10% Chelex-100 (Bio-Rad Laboratories) was added to the protein A beads, and the mixture was boiled for 10 min. After cooling to room temperature, 20 μg of proteinase K was added to each sample, and the samples were incubated at 55°C while shaking for 30 min followed by boiling for 10 min to inactivate the proteinase K. The suspension was centrifuged, and the supernatant containing coprecipitated DNA was collected. One hundred twenty microliters of nuclease-free H2O was added to the beads, the slurry was vortexed and spun, and the supernatant was collected and pooled with the first collection. The DNA-containing supernatants were stored at −80°C for later quantitative PCR.
Quantitative PCR for DNA recovered by ChIP was performed with Sybr Green PCR master mix (Quantace) and the Stratagene Mx3000p real-time PCR machine. Primers (Table 1) for the 5′-flanking region and for exon 3 of F3 were designed with Primer3 software (http://primer3.sourceforge.net/) and synthesized by Operon Biotechnologies. PCR reactions contained primers at a 300 nM concentration and 5 μl of IP sample/reaction. PCR conditions were set as follows: denature at 95°C for 10 s, primer anneal at 55°C for 45 s, extension at 72°C for 45 s. Fluorescent data were collected during the anneal step for 40 cycles. Fold enrichment (R) of either the F3 5′-flanking region or exon 3 by specific IP relative to mock IP was calculated by R = 2 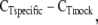 where CT is the cycle threshold for detection of fluorescence above background for either the specific IP or the mock IP. Statistical comparisons between data sets for all qRT-PCR experiments were performed by Student's t-test on log-transformed data, with a P value ≤0.05 accepted as statistically significant. Data are presented as means ± SE.
where CT is the cycle threshold for detection of fluorescence above background for either the specific IP or the mock IP. Statistical comparisons between data sets for all qRT-PCR experiments were performed by Student's t-test on log-transformed data, with a P value ≤0.05 accepted as statistically significant. Data are presented as means ± SE.
Table 1.
Primers and PCR product length for ChIP experiments
| Genomic Region | Forward/Reverse Primers | PCR Product Length, bp |
|---|---|---|
| F3 5′-flanking region | 5′-TCTCAGGCTTCATGTTGCAG-3′ | |
| (0.5 kb upstream of TSS) | 5′-CTGGTAGCAGAGGGCAGTTC-3′ | 207 |
| 5′-TGCTTCTCGACCACAGACAC-3′ | ||
| F3 exon 3 | 5′-TAAAAACTTTGGGGCGTTTG-3′ | 177 |
ChIP, chromatin immunoprecipitation; TSS, transcription start site.
Cell culture cyclical stretch model.
Mouse lung type II epithelial cells (MLE-12) were purchased from American Type Culture Collection (ATCC; CRL-2110) and grown in DMEM-F-12 medium per ATCC recommendations. Cells were seeded into a six-well collagen-coated Bioflex plate (Flexcell International, McKeesport, PA) at a density of 500,000 cells/well. Cells were incubated in a humidified atmosphere of 5% CO2-95% air at 37°C and grown until ≥80% cellular confluence. Medium was then replaced with serum-free medium, and cells were incubated for an additional 24 h.
Serum-starved cells were subjected to biaxial mechanical stretch with the FX-4000T Flexercell Tension Plus unit at 37°C. The unit applied a 15–20% stretch at 30 cycles/min in a sinusoidal pattern. Cells were harvested at time 0, 30, 60, and 120 min.
After cyclic stretch, medium was aspirated and the cells were washed twice with ice-cold 1× PBS (GIBCO) and collected with a cell scraper in lysis buffer containing (in mM) 20 Tris·HCl (pH 7.5), 150 NaCl, 1 EDTA, 1 EGTA, 2.5 sodium pyrophosphate, 1 β-glycerophosphate, and 1 sodium orthovanadate, with 1% Triton, 1 μg/ml leupeptin, 1 tablet of Complete Mini EDTA-free protease inhibitor cocktail (Roche), and 1 tablet of PhosStop phosphatase inhibitor cocktail tablet (Roche). Cell lysate was sonicated and centrifuged at 10,000 rpm for 10 min, and supernatants were collected. Protein content was measured with the bicinchoninic acid (BCA) protein assay kit by Pierce (Rockford, IL).
Cell lysate was resolved by electrophoresis in a 12% SDS-polyacrylamide gel (Invitrogen) and transferred onto a polyvinylidene difluoride (PVDF) membrane (Thermo) for detection of specific proteins by immunoblot. In one experiment, 150 μg of total protein from stretched MLE-12 cells was incubated with antibody to JunD in a 4°C ultrasonic bath for 1 h. A 50% slurry of protein A agaraose beads with cell lysis buffer was added to the cell lysate and incubated overnight at 4°C before electrophoresis and immunoblotting.
RESULTS
MV at moderate volumes does not cause lung injury.
As we previously reported (3), MV with tidal volumes of 10 ml/kg over 6 h did not independently result in significantly increased lung permeability or bronchoalveolar fluid cell count compared with spontaneously breathing mice. Figure 1 shows representative H & E stains of mouse lungs during spontaneous respiration and after 6 h of MV at a volume of 10 ml/kg. There was no histological evidence of inflammation or lung injury during MV.
Fig. 1.

Representative hematoxylin and eosin (H & E) stains (×10) of mice lungs during spontaneous ventilation (Control) and after 6 h of mechanical ventilation (MV). No histopathological evidence of lung injury is apparent in control and MV lungs.
Noninjurious MV triggers a distinct transcriptional response in the lung.
Despite the absence of overt cellular injury during MV, 710 unique genes were differentially expressed in the lungs of mechanically ventilated mice compared with spontaneously breathing animals at a FDR <5%. Hierarchical clustering of these genes demonstrates two distinct expression patterns across the samples (Fig. 2). Furthermore, principal component analysis applied to the expression values of all genes present on the microarray platform (∼14,000 unique genes) identified two separate clusters in covariance space corresponding to MV and control mice (Supplemental Fig. S1).1 These findings imply that MV, even at volumes not injurious to the lung, results in a global transcriptional signature that is distinct from spontaneous breathing.
Fig. 2.

Hierarchical clustering of gene expression profiles reveals 2 distinct patterns corresponding to spontaneously breathing (C) and MV animals.
MV activates a proinflammatory transcriptional program in uninjured lungs.
To identify putative functional interactions between the differentially expressed genes, a network-generating algorithm based on previously confirmed interactions of gene products was used to create a molecular interactome (10). A subset of 176 differentially expressed genes mapped to a single spanning interaction network as shown in Fig. 3. The topology of this interactome followed a power law distribution, P(k) ≈ k−γ (γ = 1.32, R2 = 0.91), where P(k) is the fractional degree distribution and k is the connectivity of the nodes (Fig. 4). Gene Ontology analysis of this “scale-free” interactome revealed several highly enriched modules including immune/defense response, heat shock protein activity, signal transduction pathways, and apoptotic processes (Fig. 5). Many of these functional categories were confirmed with a pathway-focused analysis with ToppGene enrichment algorithm (Table 2). These findings suggest that a topologically and biologically distinct transcriptional network is activated in the lung during noninjurious MV.
Fig. 3.

Computationally constructed gene interaction network during MV. This interactome is comprised of 176 differentially expressed genes (red: upregulated, green: downregulated). Despite complex interactions among these genes, 4 nodes (FOS, JUN, MYC, and RELA) with the highest connectivity represent potentially critical transcriptional regulators of the network. A fully labeled interactome and a table of genes with connectivity are available in the supplementary material for this article. The network was drawn with Pajek visualization software (7) based on the connectivity matrix.
Fig. 4.

A log-log plot of the topological characteristics of the MV-activated interactome shows a linear relationship between the fractional degree distribution, P(k), of the 176 nodes and their connectivity (k), consistent with a power law distribution.
Fig. 5.

Gene Ontology analysis of the gene interaction network (Fig. 3). Significantly enriched biological modules and their membership profiles are displayed for genes upregulated and downregulated during MV.
Table 2.
Computational enrichment analysis of MV-induced transcription factor binding sites
| Name | P Value | Interactome Genes with Putative cis-Binding Sites |
|---|---|---|
| HSF | 1.0×10−6 | ST13, HSPH1, DDX17, EDN1, DNAJA1, JAK1, PAFAH1B1, HSPE1, HSPA1A, HSPA1B, HSPA8 |
| DDIT3/CHOP | 4.0×10−6 | MEF2C, HERPUD1, CEBPB, S100A9, NFKBIA, SOX4, KITLG, DACH1, ATF3, ADM, NFE2L2, STMN1, LRP2 |
| ELF-1 | 4.6×10−4 | GRB2, ALOX5AP, RELA, MITF, FCER1G, RALA, CD79B, TLR4, CALCRL, SAMSN1 |
| CEBPB | 8.9×10−5 | WNT5A, GSR, CEBPB, SERPINE1, SPIB, STMN1 |
| AP-1 | 1.8×10−3 | FOS, IL6, DDX17, ROCK2, CREM, MMP9, SDCBP, GADD45A |
| CREB | 2.8×10−4 | FOS, MAFF, HERPUD1, ATF3, CREM, CXCL16, PAFAH1B1, GEM, TPM4 |
| NF-κB/RELA | 9.7×10−4 | IL6, MT2A, BCL3, NFKBIA, GEM, NR2F2, CD74 |
MV, mechanical ventilation.
Key regulators of the lung's transcriptional response to MV can be computationally identified and biologically confirmed.
To discover putative factors regulating the lung's response to MV, we focused on the connectivity of gene products within the interactome (Fig. 3, see also Supplemental Table S1). We assumed that those factors serving as central “hubs” of interaction were likely to be critical for maintaining the network's functional robustness (25, 26). Rank ordering of the network nodes based on their connectivity identified four densely connected hubs: MYC, RELA, FOS, and JUN. The fifth highly connected node was the proinflammatory cytokine IL-6. We also performed an enrichment analysis based on the interactome's putative transcription factor binding sites using ToppGene (Table 2). There was significant overlap between the two methods in identifying transcriptional regulators, including AP-1 and RELA. Furthermore, many of the highly enriched pathways identified with ToppGene were regulated by these four transcription factors (Table 3).
Table 3.
Computational enrichment analysis of MV-induced pathways
| Biological Pathways | P Value | Interactome Genes in Pathway |
|---|---|---|
| Apoptosis signaling | 1×10−10 | FOS, ATF4, JDP2, ATF3, MCL1, EIF2S1, JUN, RELA, NFKBIA, HSPA1A, HSPA1B, HSPA8 |
| IL-6 signaling | 1×10−10 | FOS, IL6, CEBPB, GRB2, JUN, JAK1, SRF |
| Oxidative stress | 1×10−6 | FOS, MAFF, ATF4, HMOX1, JUN, NFE2L2 |
| Toll-like receptor | 4×10−6 | FOS, IL6, CEBPB, GRB2, JUN, SRF |
| Glycogen synthase kinase | 7×10−6 | MYD88, LY96, RELA, TLR4, APC, DVL1 |
| Mitogen-activated protein kinase | 3.4×10−5 | MEF2C, IL1R2, GRB2, HSPA1A, HSPA1B, SRF, FOS, ATF4, RPS6KA2, ARRB1, JUN, GADD45G, STMN1, GADD45A, MYC, HSPA8 |
| Platelet-derived growth factor | 9.7×10−4 | FOS, GRB2, JUN, JAK1, SRF |
Densely connected transcription factors are indicated in bold.
We then tested whether these computationally identified transcription regulators were, in fact, activated by MV by subjecting mice to MV as described above for 0, 1, 2, or 6 h (n = 2 for each time point), followed by isolation of lung nuclear proteins and EMSA with probes containing the AP-1 consensus sequence. Protein binding to the AP-1 sequence was progressively increased by MV at 1, 2, and 6 h (Fig. 6), implying temporal activation of this key transcriptional regulator of the immune system (17). Because AP-1 is a heterodimer made up of various combinations of Jun and Fos family proteins, we performed a supershift experiment using the nuclear proteins isolated from a lung subjected to 6 h of MV and antibodies to c-Jun, JunB, JunD, c-Fos, FosB, Fra-1, and Fra-2 (Santa Cruz Biotechnology). The strongest supershifts observed were for lanes with JunD and FosB (Fig. 6B).
Fig. 6.

Electrophoretic mobility shift assays (EMSAs) at multiple time points during MV. A: progressive increase in activator protein-1 (AP-1) binding during MV during early time points. B: persistence of gel shift at 6 h and supershift assay reveal that the AP-1 heterodimer is primarily comprised of JUN-D and FOS-B molecules.
The activation of JunD has not been previously associated with mechanical stretch. To confirm that MV recruits JunD to the regulatory region of a gene induced by MV, we first identified a candidate gene by qRT-PCR that was induced by MV (F3) and then used ChIP to evaluate for JunD interaction with the gene's promoter region. Whole lung RNA was isolated from three mechanically ventilated (4 h) mice and three spontaneously breathing control mice. qRT-PCR confirmed that F3, the gene encoding for tissue factor, was significantly induced by mechanical ventilation (3.8 ± 1.3-fold increased expression, P = 0.03; Fig. 7A). F3 was chosen as a candidate gene because 1) F3 is a member of the gene interactome (Fig. 3), 2) F3 contains an AP-1 binding site in its regulatory region (28), 3) F3 is implicated in the pathogenesis of lung injury (11, 18, 41), and 4) lung epithelial cells express F3 (16, 21). Next, chromatin was isolated from six mice mechanically ventilated for 1 h and from six control mice. Compared with mock IP, IP using antibodies to JunD resulted in significant enrichment of DNA from the JunD promoter region (Fig. 7B). JunD recruitment to the F3 5′-flanking region was significantly increased by MV (4.9 ± 0.87 vs. 2.5 ± 0.88, P = 0.05; Fig. 7B). Importantly, JunD was not recruited to exon 3 of F3, indicating specificity for the regulatory region of the gene (Fig. 7B).
Fig. 7.

Chromatin immunoprecipitation (ChIP) experiments. A: confirmation by quantitative RT-PCR (qRT-PCR) that MV induces expression of F3 (tissue factor) (n = 3/group). B: relative enrichment of DNA from the 5′-flanking region and from exon 3 of F3 by JunD immunoprecipitation compared with mock immunoprecipitation. n.s., Not significant.
Since the above results were obtained by mechanically ventilating whole lungs, many cell types could have contributed to our findings. We hypothesized that alveolar epithelial cells were responsive to cyclical stretch and tested whether cyclical stretch in cell culture could reproduce the in vivo response to MV. We found that MLE-12 cells, derived from an alveolar epithelial cell line (42), expressed F3 in response to cyclical stretch (Fig. 8A). Importantly, this finding was associated with phosphorylation of JunD at serine 100 (Fig. 8, B–D), reflecting its transcriptional activation. Furthermore, two kinases known to phosphorylate JunD, i.e., JNK and ERK1/2 MAP, were in turn activated by cyclical stretch (Fig. 8, B and D). These results recapitulate our in vivo findings and imply that alveolar epithelial cells are key effectors of the lung's transcriptional response to noninjurious MV.
Fig. 8.

In vitro cyclical stretch experiments. A: qRT-PCR measurement of F3 in cyclically stretched epithelial cells (summary of 2 experiments, 3 replicates/experiment). B: immunoblot of phospho (p)-Jun, phospho-JNK, and phospho-ERK1/2. The phospho-Jun antibody detects both c-Jun phosphorylated at S73 (top band) and phospho-JunD (bottom band). C: densitometry of phospho-JunD, phospho-JNK, and phospho-ERK1/2 normalized to actin from immunoblots in B. D: immunoprecipitation (IP) using either JunD antibody or FLAG antibody (control) followed by phospho-Jun immunoblot.
Finally, we proceeded to assess whether the transcription factors computationally predicted to bind to members of the interactome based on ToppGene's enrichment analysis were indeed activated by MV (Table 2). We performed EMSA using probes containing cAMP-responsive element binding protein (CREB) consensus sequence under experimental conditions identical to those of the AP-1 experiments. We observed a progressive increase in CREB binding at 1, 2, and 6 h of mechanical ventilation (Supplemental Fig. S3).
DISCUSSION
In this work, we investigated the transcriptional response of intact lungs to noninjurious MV. We initially showed that MV at a volume of 10 ml/kg, while causing no overt injury, resulted in a distinct transcriptional signature enriched in proinflammatory programs in the lung. Next, we developed a computational framework to identify MV-induced pathways in the context of their transcriptional regulators and genetic networks. We validated our approach by biologically confirming the activation of several of these factors, including the identification of a previously unknown role for JunD signaling in mechanical stretch.
To our knowledge, this is the first global assessment of gene expression in lung tissue undergoing noninjurious MV in vivo. Previous investigators have demonstrated that high-volume MV in animals and humans results in lung injury and is associated with elevated levels of inflammatory mediators in the alveolar space and plasma (4, 13, 31, 33, 38, 43). Using a rat model of high-tidal volume MV, Copland et al. (13) demonstrated the early transcription of ∼20 genes before the histological appearance of lung injury. Although we performed our studies in a different species and during a later time point, eight of their genes (or close family members) were also differentially expressed in our analysis, including heat shock protein 70 (HSPA1B), whose expression was confirmed by Copland et al. to be localized to airway and alveolar epithelial cells. This observation suggested that although the transcriptional response to mechanical stretch is a temporally dynamic process, there is significant overlap among different models in activation of an early inflammatory program.
We decided to further explore our hypothesis that a core set of common pathways are activated in lungs because of mechanical stretch, regardless of the volume used. Recently, Ma et al. (27) utilized transcriptional profiling and bioinformatics approaches to identify differential gene expression in mouse and rat models of VILI. Despite differences in the volumes used during MV in our noninjurious model and the VILI model of Ma et al. (10 vs. 17–35 ml/kg), we proceeded to compare our microarray results. We utilized the same image processing, normalization, and statistical procedures on Ma's VILI murine data as we had performed on our microarray experiments. Of 75 differentially expressed genes in the VILI model (at FDR < 0.05), 36 were also differentially expressed in our noninjurious MV strategy (P < 5 × 10−24 for occurring by chance), strongly implying a significant overlap between the lung's transcriptional response during mild and severe stretch. Interestingly, both F3 and MYC were significantly upregulated during VILI (P = 1.3 × 10−7, FDR = 4.0 × 10−4 and P = 4.8 × 10−5, FDR = 1.5 × 10−2, respectively), and JUN approached statistical significance (P = 4.8 × 10−4, FDR = 6.5 × 10−2) in the model of Ma et al.
We investigated the mechanisms controlling the transcriptional response to mechanical stretch by identifying a gene interaction network comprised of 176 differentially expressed genes during MV (Fig. 3). To better understand the complex relationships within this interactome, we functionally categorized its members with Gene Ontology and pathway analyses (Fig. 5, Table 3). These functional analyses revealed the enrichment of several proinflammatory modules such as defense/immune response, apoptosis, heat shock protein activity, and cytokine biosynthesis. Members of these modules included well-known components of cellular inflammatory response such as lymphocyte antigen 96 (LY96) chemokine receptor 1 (CCR1), interleukin-6 (IL-6, one of the top 5 densely connected network nodes), plasminogen activator (PLAUR), and several heat shock proteins (HSPA1A, HSPA1B, HSPA8, HSPCA, HSPE1, HSPH1). Other investigators have demonstrated elevated cytokine levels and heat shock protein activity in rodent models of VILI (13, 37). However, our findings extend these results to a ventilation strategy that is not injurious to the lung and implies that a proinflammatory program is activated in vivo in response to even mild cyclical stretching.
Topological analysis of the MV-activated interactome confirmed that it is approximately scale-free, a common characteristic of complex biological networks (23). There is increasing evidence that the functional stability of such networks is dependent on hubs of high connectivity (26). The cornerstones of this interactome were four highly connected transcription factors: MYC, RELA, JUN, and FOS. These hubs were ubiquitous members of the enriched pathways mapped to this interactome (Table 3). JUN and FOS can dimerize to form the AP-1 transcription factor, a key regulator of proinflammatory genes especially in response to RELA (p65 NF-κB) (17). We confirmed the temporal activation of AP-1 due to MV with EMSA and determined the individual components of this heterodimeric molecule with supershift analysis (Fig. 6). Our results are consistent with recent studies reporting increased binding activity of AP-1 in isolated, perfused, mechanically ventilated rabbit lungs (24) and the activation of FOS in both isolated rat lungs during VILI (37) and pulmonary epithelial cell lines undergoing mechanical stretch (45). We have previously demonstrated (2) increased AP-1 activity in rabbit lungs ventilated at mildly injurious volumes of 15 ml/kg. Our present results extend these findings with an in vivo murine model of noninjurious MV, which is more representative of ventilation strategies currently employed in clinical practice.
Additionally, we found that JunD was the primary Jun family protein activated by MV. By adapting the ChIP assay to whole lung, we confirmed recruitment of JunD to the regulatory region of F3 by MV, supporting the relevance of JunD activation to the transcriptional response to mechanical stretch (Fig. 7). To our knowledge, JunD activation by mechanical stretch/ventilation has not been previously reported. Furthermore, utilizing a well-established in vitro cyclical stretch model, we demonstrated that at least part of these findings can be attributed to the transcriptional response of alveolar epithelial cells. It is important to note that our findings were the direct result of an unbiased and systematic assessment of the global transcriptional response to mechanical stretch, thereby minimizing the “pick and choose” option often utilized for analysis of complex biological processes.
There are several limitations in our analysis. Because transcriptional profiling was performed on whole lungs, we cannot assess the contribution of different cell types to overall gene expression values. However, we proceeded to confirm several of our findings with an in vitro mechanical stretch model on pulmonary epithelial cells. A general shortcoming of our computational approach is the inherent bias and limitation in the current state of knowledge of gene product function and interactions. Therefore, by definition, our analysis is incomplete and future iterations of the same data will yield (slightly) different results.
Although the findings from this study are not directly applicable to humans, the insights gained from it may have several important clinical implications. We demonstrate that even at currently accepted “safe” volumes, MV activates a broad repertoire of proinflammatory and immune-mediated pathways in the lung. Our results suggest that noninjurious MV can “prime” the lung to a state in which a secondary insult, such as infection or ischemia-reperfusion, could precipitate an exaggerated inflammatory response and result in acute injury (3, 14). Targeting the critical regulators of these stretch-induced programs before the impact of a “second hit” may attenuate the inflammatory consequences that lead to lung injury.
In conclusion, we present a novel computational framework to systematically dissect MV-activated transcriptional programs in the lung. Using this approach, we identify and biologically verify several key transcriptional regulators of MV-induced pathways. These factors may serve as critical targets for modulating VALI.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-074223, HL-071020, HL-073996, and HL-072370. W. C. Liles is supported by a Canada Research Chair in Infectious Disease and Inflammation from the Canadian Institutes of Health Research.
Acknowledgments
The authors thank Oleg Denisenko and Joel D. Nelson for their assistance in developing the tissue ChIP protocol and Dowon An for assisting with the mouse mechanical ventilation experiments.
Address for reprint requests and other correspondence: S. A. Gharib, Box 358052, 815 Mercer St., Seattle, WA 98109 (e-mail:sagharib@u.washington.edu).
Footnotes
The online version of this article contains supplemental material.
REFERENCES
- 1.Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E, Buzadzija K, Cavero R, D'Abreo C, Donaldson I, Dorairajoo D, Dumontier MJ, Dumontier MR, Earles V, Farrall R, Feldman H, Garderman E, Gong Y, Gonzaga R, Grytsan V, Gryz E, Gu V, Haldorsen E, Halupa A, Haw R, Hrvojic A, Hurrell L, Isserlin R, Jack F, Juma F, Khan A, Kon T, Konopinsky S, Le V, Lee E, Ling S, Magidin M, Moniakis J, Montojo J, Moore S, Muskat B, Ng I, Paraiso JP, Parker B, Pintilie G, Pirone R, Salama JJ, Sgro S, Shan T, Shu Y, Siew J, Skinner D, Snyder K, Stasiuk R, Strumpf D, Tuekam B, Tao S, Wang Z, White M, Willis R, Wolting C, Wong S, Wrong A, Xin C, Yao R, Yates B, Zhang S, Zheng K, Pawson T, Ouellette BF, Hogue CW. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res 33: D418–D424, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altemeier WA, Matute-Bello G, Frevert CW, Kawata Y, Kajikawa O, Martin TR, Glenny RW. Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am J Physiol Lung Cell Mol Physiol 287: L533–L542, 2004. [DOI] [PubMed] [Google Scholar]
- 3.Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, Liles WC. Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol 175: 3369–3376, 2005. [DOI] [PubMed] [Google Scholar]
- 4.ARDSNet. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 342: 1301–1308, 2000. [DOI] [PubMed] [Google Scholar]
- 5.Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17: 509–519, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Barabasi AL, Albert R. Emergence of scaling in random networks. Science 286: 509–512, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Batagelj V, Mrvr A. Pajek-analysis and visualization of large networks. In: Graph Drawing Software, edited by Jèunger M, Mutzel P. Berlin: Springer, 2004, p. xii.
- 8.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57: 289–300, 1995. [Google Scholar]
- 9.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, Miller-Graziano C, Moldawer LL, Mindrinos MN, Davis RW, Tompkins RG, Lowry SF. A network-based analysis of systemic inflammation in humans. Nature 437: 1032–1037, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Carraway MS, Welty-Wolf KE, Miller DL, Ortel TL, Idell S, Ghio AJ, Petersen LC, Piantadosi CA. Blockade of tissue factor: treatment for organ injury in established sepsis. Am J Respir Crit Care Med 167: 1200–1209, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Xu H, Aronow BJ, Jegga AG. Improved human disease candidate gene prioritization using mouse phenotype. BMC Bioinformatics 8: 392, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Copland IB, Kavanagh BP, Engelberts D, McKerlie C, Belik J, Post M. Early changes in lung gene expression due to high tidal volume. Am J Respir Crit Care Med 168: 1051–1059, 2003. [DOI] [PubMed] [Google Scholar]
- 14.Dhanireddy S, Altemeier WA, Matute-Bello G, O'Mahony DS, Glenny RW, Martin TR, Liles WC. Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab Invest 86: 790–799, 2006. [DOI] [PubMed] [Google Scholar]
- 15.dos Santos CC, Slutsky AS. The contribution of biophysical lung injury to the development of biotrauma. Annu Rev Physiol 68: 585–618, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Drake TA, Cheng J, Chang A, Taylor FB Jr. Expression of tissue factor, thrombomodulin, and E-selectin in baboons with lethal Escherichia coli sepsis. Am J Pathol 142: 1458–1470, 1993. [PMC free article] [PubMed] [Google Scholar]
- 17.Foletta VC, Segal DH, Cohen DR. Transcriptional regulation in the immune system: all roads lead to AP-1. J Leukoc Biol 63: 139–152, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Gando S, Kameue T, Matsuda N, Hayakawa M, Morimoto Y, Ishitani T, Kemmotsu O. Imbalances between the levels of tissue factor and tissue factor pathway inhibitor in ARDS patients. Thromb Res 109: 119–124, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Gene Ontology Consortium. Creating the gene ontology resource: design and implementation. Genome Res 11: 1425–1433, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gharib SA, Liles WC, Matute-Bello G, Glenny RW, Martin TR, Altemeier WA. Computational identification of key biological modules and transcription factors in acute lung injury. Am J Respir Crit Care Med 173: 653–658, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Gross TJ, Simon RH, Sitrin RG. Tissue factor procoagulant expression by rat alveolar epithelial cells. Am J Respir Cell Mol Biol 6: 397–403, 1992. [DOI] [PubMed] [Google Scholar]
- 22.Hosack DA, Dennis G Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol 4: R70, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature 407: 651–654, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Kirchner EA, Mols G, Hermle G, Muehlschlegel JD, Geiger KK, Guttmann J, Pahl HL. Reduced activation of immunomodulatory transcription factors during positive end-expiratory pressure adjustment based on volume-dependent compliance in isolated perfused rabbit lungs. Br J Anaesth 94: 530–535, 2005. [DOI] [PubMed] [Google Scholar]
- 25.Lovegrove FE, Gharib SA, Patel SN, Hawkes CA, Kain KC, Liles WC. Expression microarray analysis implicates apoptosis and interferon-responsive mechanisms in susceptibility to experimental cerebral malaria. Am J Pathol 171: 1894–1903, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luscombe NM, Babu MM, Yu H, Snyder M, Teichmann SA, Gerstein M. Genomic analysis of regulatory network dynamics reveals large topological changes. Nature 431: 308–312, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Ma SF, Grigoryev DN, Taylor AD, Nonas S, Sammani S, Ye SQ, Garcia JG. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol 289: L468–L477, 2005. [DOI] [PubMed] [Google Scholar]
- 28.Mackman N Regulation of the tissue factor gene. FASEB J 9: 883–889, 1995. [DOI] [PubMed] [Google Scholar]
- 29.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Nelson JD, Denisenko O, Sova P, Bomsztyk K. Fast chromatin immunoprecipitation assay. Nucleic Acids Res 34: e2, 2006. [DOI] [PMC free article] [PubMed]
- 31.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 33: 1–6, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Peri S, Navarro JD, Amanchy R, Kristiansen TZ, Jonnalagadda CK, Surendranath V, Niranjan V, Muthusamy B, Gandhi TK, Gronborg M, Ibarrola N, Deshpande N, Shanker K, Shivashankar HN, Rashmi BP, Ramya MA, Zhao Z, Chandrika KN, Padma N, Harsha HC, Yatish AJ, Kavitha MP, Menezes M, Choudhury DR, Suresh S, Ghosh N, Saravana R, Chandran S, Krishna S, Joy M, Anand SK, Madavan V, Joseph A, Wong GW, Schiemann WP, Constantinescu SN, Huang L, Khosravi-Far R, Steen H, Tewari M, Ghaffari S, Blobe GC, Dang CV, Garcia JG, Pevsner J, Jensen ON, Roepstorff P, Deshpande KS, Chinnaiyan AM, Hamosh A, Chakravarti A, Pandey A. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res 13: 2363–2371, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ranieri VM, Suter PM, Tortorella C, De Tullio R, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 282: 54–61, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Rodarte JR, Hubmayr RD, Stamenovic D, Walters BJ. Regional lung strain in dogs during deflation from total lung capacity. J Appl Physiol 58: 164–172, 1985. [DOI] [PubMed] [Google Scholar]
- 35.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34: 374–378, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32: D449–D451, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 99: 944–952, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tremblay LN, Miatto D, Hamid Q, Govindarajan A, Slutsky AS. Injurious ventilation induces widespread pulmonary epithelial expression of tumor necrosis factor-alpha and interleukin-6 messenger RNA. Crit Care Med 30: 1693–1700, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med 171: 1328–1342, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol 7: 265–275, 2006. [DOI] [PubMed] [Google Scholar]
- 41.Welty-Wolf KE, Carraway MS, Idell S, Ortel TL, Ezban M, Piantadosi CA. Tissue factor in experimental acute lung injury. Semin Hematol 38: 35–38, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Wikenheiser KA, Vorbroker DK, Rice WR, Clark JC, Bachurski CJ, Oie HK, Whitsett JA. Production of immortalized distal respiratory epithelial cell lines from surfactant protein C/simian virus 40 large tumor antigen transgenic mice. Proc Natl Acad Sci USA 90: 11029–11033, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson MR, Choudhury S, Goddard ME, O'Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol 95: 1385–1393, 2003. [DOI] [PubMed] [Google Scholar]
- 44.Wongsurakiat P, Pierson DJ, Rubenfeld GD. Changing pattern of ventilator settings in patients without acute lung injury: changes over 11 years in a single institution. Chest 126: 1281–1291, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Ying B, Fan H, Wen F, Xu D, Liu D, Yang D, Chen G, Dou L, Jiang F. Mechanical strain-induced c-fos expression in pulmonary epithelial cell line A549. Biochem Biophys Res Commun 347: 369–372, 2006. [DOI] [PubMed] [Google Scholar]