

Abstract
The reaction center (RC) from Rhodobacter sphaeroides converts light into chemical energy through the light induced two-electron, two-proton reduction of a bound quinone molecule QB (the secondary quinone acceptor). A unique pathway for proton transfer to the QB site had so far not been determined. To study the molecular basis for proton transfer, we investigated the effects of exogenous metal ion binding on the kinetics of the proton-assisted electron transfer kAB(2) (QA−•QB−• + H+ → QA(QBH)−, where QA is the primary quinone acceptor). Zn2+ and Cd2+ bound stoichiometrically to the RC (KD ≤ 0.5 μM) and reduced the observed value of kAB(2) 10-fold and 20-fold (pH 8.0), respectively. The bound metal changed the mechanism of the kAB(2) reaction. In native RCs, kAB(2) was previously shown to be rate-limited by electron transfer based on the dependence of kAB(2) on the driving force for electron transfer. Upon addition of Zn2+ or Cd2+, kAB(2) became approximately independent of the electron driving force, implying that the rate of proton transfer was reduced (≥ 102-fold) and has become the rate-limiting step. The lack of an effect of the metal binding on the charge recombination reaction D+•QAQB−• → DQAQB suggests that the binding site is located far (>10 Å) from QB. This hypothesis is confirmed by preliminary x-ray structure analysis. The large change in the rate of proton transfer caused by the stoichiometric binding of the metal ion shows that there is one dominant site of proton entry into the RC from which proton transfer to QB−• occurs.
Keywords: bacterial photosynthesis, Rhodobacter sphaeroides, metal binding, proton-coupled electron transfer
The bacterial reaction center (RC) is a membrane-bound pigment protein complex that converts light into chemical energy through a two-electron, two-proton reduction of a bound quinone molecule QB (the secondary quinone acceptor) to a quinol molecule QBH2 (1, 2). The protons taken up to form quinol are transferred from the solvent of the cytoplasm to the bound quinone molecule. Understanding the details of proton transfer in this and other systems is important for a basic understanding of bioenergetics. This paper addresses the pathway of the proton transfer by measuring the effects of metal ion binding on proton transfer rates in the bacterial RCs from Rhodobacter sphaeroides.
The isolated RC is composed of three polypeptide subunits (L, M, and H), four bacteriochlorophylls, two bacteriopheophytins, one internally bound nonheme Fe2+, and two ubiquinone (UQ10) molecules (reviewed in refs. 1 and 2). In the RC, the light-induced electron transfer proceeds from a primary donor (a bacteriochlorophyll dimer) through a series of electron donor and acceptor molecules (a bacteriopheophytin and a primary quinone acceptor QA) to a loosely bound secondary quinone QB, which serves as a mobile electron and proton carrier (3–5), transferring electrons and protons from the RC to other components of the bioenergetic cycle.
The first electron transfer to QB (kAB(1)) does not involve direct protonation of the quinone (Eq. 1).
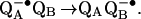 |
1 |
However, the second electron transfer (kAB(2)) is coupled to the direct protonation of the quinone (Eq. 2a). Subsequent protonation (Eq. 2b) leads to the formation of quinol.
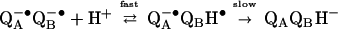 |
2a |
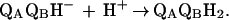 |
2b |
In native RCs from Rb. sphaeroides, the proton-coupled electron transfer reaction kAB(2) (Eq. 2a) was shown to be a two-step process in which fast protonation precedes rate-limiting electron transfer (6). The value of kAB(2) in native RCs is, therefore, not a direct measure of the rate of proton transfer. However, when the proton transfer rate is sufficiently reduced, proton transfer becomes the rate-limiting step as has been observed in the site-directed mutation of Asp-L213 to Asn (7). To determine which of the two steps in Eq. 2a is rate limiting, a driving-force assay, based on the free-energy dependence of kAB(2), has been used (6).
The pathways for proton transfer have been studied by a number of groups (8–13). Results of kAB(2) measurements on site-directed mutants had shown the importance of several amino acid residues, e.g., Glu-L212, Ser-L223, and Asp-L213, for the proton transfer reactions (Eq. 2) (reviewed in refs. 14 and 15). These residues can be connected to the external surface through a number of possible proton transfer pathways and an internal carboxylic acid cluster that have been resolved in the crystal structures of the RC (16–19). Which of these is functionally the most important pathway had not been determined.
A complementary approach to site-directed mutagenesis, to identify residues involved in proton transfer, is to assess the effect of metal binding on the kinetics of proton transfer (Eq. 2). In solution, metal ions bind to acidic and uncharged amine or imidazole groups (20). In a protein, these groups are provided by carboxylic acids and histidine residues. For example, the binding of Cu2+ has an inhibitory effect on proton transfer in carbonic anhydrase (21–23).
Utschig et al. (24) have shown that Zn2+ binds to the RC and affects the rate of transfer of the first electron kAB(1) (Eq. 1). We have confirmed their findings and have extended the work to an investigation of the effect of Zn2+ and Cd2+ binding on the proton-coupled electron transfer kAB(2) (Eq. 2a). In addition, we have investigated the driving-force dependence of kAB(2), to establish which of the two steps in Eq. 2a is rate limiting. Furthermore, we have measured the effects of the metal ions on the charge recombination rate constant kBD (D+•QAQB−• → DQAQB, where D is the primary donor), which is sensitive to the electrostatic potential near QB. By localizing the position of the bound cation, the location of the site of proton entry into the RC from which proton transfer to QB−• occurs.
METHODS
Reagents and Quinones.
The quinones Q10 (coenzyme Q10; 2,3-dimethoxy-5-methyl-6-decaisoprenyl-1,4-benzoquinone) and MQ4 (menatetrenone; 2-methyl-3-tetraisoprenyl-1,4-naphthoquinone) were obtained from Sigma. ADMNQ (2,6-dimethyl-3-undecyl-1,4,-naphthoquinone) and ATMNQ (2,6,7-trimethyl-3-undecyl-1,4,-naphthoquinone) were kindly provided by Andreas Labahn (Albert-Ludwigs Universität, Freiburg, Germany) (25). All quinones were prepared in ethanol before their use. The QB site inhibitors terbutryne and stigmatellin were obtained from Chem Service (West Chester, PA) and Fluka, respectively, and were prepared in ethanol. Cytochrome c from horse heart was obtained from Sigma and was reduced (>95%) by hydrogen gas on platinum black (Aldrich) and filtered (0.2-μM pore size acetate filter). All other reagents were of analytical grade.
Isolation and Preparation of RCs.
RCs from Rb. sphaeroides R26 were isolated in 15 mM Tris⋅HCl, pH 8, 0.025% lauryl dimethylamine-N-oxide (LDAO), 1 mM EDTA following published procedures (26) as modified by Utschig et al. (27). Both QA and QB were removed as described (28, 29) to yield RCs with ≤ 10% residual QA/RC and ≤ 0.2% residual QB/RC, as measured at 865 nm from the charge recombination rate and amplitude (28). The ratio of absorbance, A280/A800, was 1.20. Substitution of a naphthoquinone (NQ) for the native UQ at the QA site was performed as described (6, 30).
Reconstitution of the QB site was achieved by incubation of the solubilized RC solution with Q10 adsorbed to celite (diatomaceous earth, Fisher) for ≈20 min at 25°C while stirring (6) or by addition of Q10 (≈3 Q10 per RC) solubilized in 1% LDAO. Occupancy of the QB sites varied from 50% to 80% over the range of pH studied.
Transient Optical Spectroscopy.
Charge recombination rates were measured by monitoring the recovery of the donor band at 865 nm after bleaching with a single laser flash (Phase R DL2100c, 590 nm, ≈0.2 J/pulse, 0.4-μs full-width-half-max) by using a single-beam spectrophotometer (31). All measurements were performed at 21°C. To determine the D+QAQB−• → DQAQB recombination rate (kBD), the observed absorption decays were fitted to multiple exponentials by using procedures previously described (30). To measure the D+QA−• → DQA recombination rate (kAD), electron transfer from QA−• to QB was blocked by the addition of 100 μM terbutryn or 10 μM stigamatellin. For studies of the driving-force dependence of rates, NQs were substituted into the QA site. The relative occupancies of the NQ and UQ in the QA site were determined from a deconvolution of the charge recombination kinetics (6), because kAD is different for NQ and UQ (6, 30).
The rate constant (kAB(1)) for the transfer of the first electron to QB (Eq. 1) was measured by monitoring the bacteriopheophytin bandshift at 750 nm, which is sensitive to the reduction state of the quinones QA and QB (31, 32). To improve the signal-to-noise ratios, 9–36 traces were averaged.
The proton-coupled electron transfer kAB(2) (Eq. 2a) was determined by monitoring the decay of the semiquinone absorption at 450 nm after a second saturating laser flash in the presence of an external reductant (10 μM horse heart cytochrome c) (33). In RCs with a NQ added to occupy the QA site (see above), biphasic kinetics were observed with one rate corresponding to the rate observed in RCs with UQ10 occupying the QA site (kAB(2) ≈ 1,200 s−1, pH 7.5, native RCs without a heavy metal) and the other rate corresponding to RCs with a NQ occupying the QA site (kAB(2) ≈ 6,000 s−1, pH 7.5, native RCs without a heavy metal). The relative occupancies of UQ10 and NQ determined in this manner agreed with the relative occupancies determined from the deconvolution of biphasic kinetics for kAD.
EPR Spectroscopy.
The light-induced EPR spectra of RC samples in the presence and in the absence of 10 mM ZnSO4 and CdSO4 were obtained on a spectrometer of local design at a microwave frequency of 9 GHz (34). The samples were concentrated to A8001cm ≈ 77, frozen in a 15×6×1-mm flat quartz cell and illuminated in the frozen state by a 500-W projector lamp (34).
RESULTS
Binding of Exogenous Metal Ions to the RC.
The measured value of kAB(2) (Eq. 2a) was used as functional assay for heavy metal binding to the RC. Most of the heavy metals compounds that were tested (MnSO4, CuSO4, ZnSO4, CdSO4, HgCl2) caused a decrease in the observed rate at 1 mM concentrations, except for FeSO4, which had no effect at this concentration. However, only the addition of CdSO4 and ZnSO4 caused a significant decrease in the observed rate at 10 μM concentrations (Fig. 1). These two metals became, therefore, the focus of our investigation. The disparate results obtained with different compounds show that the observed effect on kAB(2) is not caused by the anionic counterion (i.e., SO42−), which was the same for all of the metals tested, except HgCl2.
Figure 1.

Absorbance decay of the semiquinones at 450 nm as a function of time after the second of two laser flashes in the presence of various concentrations of ZnSO4 (a) and CdSO4 (b). From the decay, the rate constant kAB(2) was determined. Note the slowing of the kinetics with increasing cation concentrations. The pedestal at long times after the laser flash is caused by the absorbance change of the cytochrome c used to reduce the primary donor (see Materials and Methods). Conditions were: 2 μM RCs in 10 mM Tris⋅HCl (pH 7.7), 0.25% lauryl dimethylamine-N-oxide with the concentration of ZnSO4 or CdSO4 as indicated in the figure.
Measurements of Charge Recombination.
The charge recombination rates for the reactions D+QA−• → DQA (kAD) and D+QAQB−• → DQAQB (kBD) were measured at 865 nm in the presence and absence of an exogenous cation. The measured values of kAD (≈9 s−1) and kBD (≈ 0.8 s−1) were the same with or without exogenous Zn2+ or Cd2+ (Table 1). The amplitude of the kBD phase remained unchanged upon addition of Zn2+ or Cd2+. Similarly the pH profile of kBD was essentially the same with or without Zn2+ or Cd2+ (data not shown).
Table 1.
Measured rate constants for RCs in the presence and absence of metal ions (pH 8.0, T = 21°)
| RCs178 | kAD (s−1) | kBD (s−1) | kAB(1) (s−1)† | kAB(2) (s−1) |
|---|---|---|---|---|
| Native | 8.8 | 0.8 | 7,000 | 1,200 |
| Native + Zn2+ | 8.8 | 0.7 | 700 | 120 |
| Native + Cd2+ | 9.0 | 0.8 | 700 | 60 |
Errors in the rates are estimated to be ∼8% for the charge recombination rate constants kAD and kBD and ∼15% for the forward electron transfer rate constants kAB(1) and kAB(2)
Conditions for kinetic measurements as described in Materials and Methods. The samples labeled Native + Zn2+ and Native + Cd2+ were measured in the presence of 10μM ZnSO4 and CdSO4, respectively.
This is the observed rate constant using a single exponential fit to the data. This reaction can be better fitted with a sum of two exponentials (24), but the effect of the metal binding is clearly shown by using this simple analysis.
Measurements of the First Electron Transfer Rate kAB(1).
The measured rates of transfer for the first electron to QB (kAB(1), Eq. 1), measured at 750 nm, were reduced ≈10-fold upon addition of 10 μM Zn2+ or Cd2+ (Table 1). The slower observed rate constant was independent of the metal concentration above 10 μM. At cation concentrations below 10 μM, we could deconvolute the observed kinetics into two phases, one phase at 7,000 s−1 (the rate observed without exogenous cations) and one phase at 700 s−1 (the rate observed at ≥10 μM concentration). From the dependence of the amplitude of the slow phase with cation concentration, we estimated a dissociation constant (KD) of ≤0.5 μM for Zn2+ and Cd2+ (data not shown).
Measurements of the Proton-Coupled Electron Transfer Rate kAB(2).
The rate of transfer for the second electron to QB (kAB(2), Eq. 2a), after the second saturating laser flash at 450 nm, was measured in native RCs to be 1,200 s−1 at pH 8 (Table 1). Upon addition of 10 μM Zn2+ or Cd2+, kAB(2) decreased to a limiting value of 120 s−1 and 60 s−1, respectively (Table 1). The effect of the metal on the rate was observed immediately after addition without an incubation period. The fraction of the sample exhibiting the slower rate depended on the concentration of the cations (Fig. 2) and allowed us to estimate a dissociation constant (KD) of ≤0.5 μM for Zn2+ and Cd2+, which is, within experimental error, the same as that determined from the kAB(1) measurements.
Figure 2.

Fraction of slow phase (Aslow)of kAB(2) as a function of the concentration of added ZnSO4 (●) or CdSO4 (■). The kinetics of Fig. 2 were decomposed into two components: ΔA450nm(t) = Afast exp(−kfastt) + Aslow exp(−kslowt), where kfast = 1,200 s−1 (the observed rate without the addition of the metal) and kslow = 120 s−1 or 60 s−1 (the observed rate upon addition of high concentrations of ZnSO4 or CdSO4, respectively). The solid curve is a fit to a standard binding equation with KD = 0.3 μM. Conditions were the same as in Fig. 1.
The decrease in the value of kAB(2) caused by the addition of exogenous Zn2+ or Cd2+ was eliminated upon addition of EDTA (a strong chelater of cationic metals) to a concentration of twice the exogenous metal concentration. Further addition of Zn2+ or Cd2+ to twice that of the EDTA concentration led again to a reduced value of kAB(2) to 120 s−1 or 60 s−1, respectively.
The reduced value of kAB(2) to 60 s−1 upon addition of 10 μM Cd2+ could be increased to 120 s−1 upon addition of 50 μM Zn2+, showing that Zn2+ had replaced Cd2+.
The pH profile of kAB(2) was measured in a mixture of 2 mM Hepes, 2 mM Ches, 2 mM Mes in 0.025% lauryl dimethylamine-N-oxide buffer from pH 7 to 9.5 and in 0.04% maltoside from pH 5 to 9. The same values of kAB(2) were observed in either detergent at the same pH. In the presence of Zn2+ or Cd2+, the value of kAB(2) decreased with increasing pH with a slope proportional to [H+]0.5 over the measured pH ranges (data not shown). This pH dependence differs from the native behavior where kAB(2) decreases with pH proportional to [H+]0.3 below pH 5 and [H+]0.9 above pH 8.5.
Dependence of the Proton-Coupled Electron Transfer Rates on the Driving Force for Electron Transfer.
The driving force for electron transfer was changed by replacing the native Q10 in the QA site with a series of NQs that have different redox protentials while retaining the native Q10 in the QB site. The experimental results on native RCs without a bound Zn2+ or Cd2+ showed an increase in the observed rate upon increasing the driving force for electron transfer. In the presence of Zn2+ and Cd2+, kAB(2) became approximately independent of the driving force for electron transfer (Fig. 3). The dependence of kAB(2) on the driving force was restored to either sample by addition of EDTA to twice the concentration of that of the exogenous metal. Upon further addition of Zn2+ and Cd2+ to twice the EDTA concentration, kAB(2) became again approximately independent of the driving force.
Figure 3.

The rate constant of proton-coupled second electron transfer, kAB(2), in RCs as a function of the change in redox free energy (driving force) for electron transfer in the absence and in the presence of 10 μM Zn2+ and Cd2+. Note that kAB(2) in the absence of added metals show a “Marcus”-like dependence on the electron driving force characteristic of a rate-limited electron transfer reaction as has been reported (6), whereas in the presence of Zn2+ or Cd2+, kAB(2) is approximately independent of the electron driving force, showing that proton transfer (Eq. 3) has become rate limiting. Quinones substituted into the QA site were from left to right: MQ0, menadione (2-methyl-1,4-naphthoquinone); Q10, coenzyme Q10; MQ4, menatetrenone (2-methyl-3-tetraisoprenyl-1,4-naphthoquinone); ADMNQ, 2,6-dimethyl-3-undecyl-1,4,-naphthoquinone; and ATMNQ, 2,6,7-trimethyl-3-undecyl-1,4,-naphthoquinone. Conditions were the same as in Fig. 1.
EPR Spectroscopy.
The light-induced EPR specta of RCs frozen in the dark were measured in the presence and absence of Zn2+ and Cd2+. All spectra were characteristic of the broad (≈300 G) Fe2+-Q−• complex at g = 1.8 with a ratio of Fe2+-Q−•/D+• of 1.0 ± 0.1 (34) (data not shown).
DISCUSSION
We investigated the effects of Zn2+ and Cd2+ binding to RCs on the transfer rate of the first electron, kAB(1) (Eq. 1), and on the rate of the proton-assisted second electron transfer, kAB(2) (Eq. 2a). By localizing the binding site of Zn2+ and Cd2+ we identified the point of entry of the protons and the start of the proton transfer pathway(s) to QB−•.
The Effect of Zn2+ and Cd2+ Binding on kAB(1).
The rate of the first electron reduction kAB(1) (Eq. 1) was reduced ≈10-fold upon the binding of Zn2+ or Cd2+. These findings confirm the results of Utschig et al. (24) on the effect of Zn2+ binding and show that a similar effect is found upon binding of Cd2+.
The reaction mechanism of kAB(1) in isolated RCs involves a slow rate-limiting gating step that involves the movement of QB (18) before electron transfer (35). Thus, the decreased rate upon binding Zn2+ or Cd2+ implies a slowing down of the conformational gating step. The movement of QB into the active position requires a rotation of the quinone head group and a displacement of several water molecules (17–19). The possibility that bound Zn2+ or Cd2+ directly hindered the quinone rotation and movement is excluded because a decrease in kBD would be expected as discussed in a later section. A possible explanation advanced by Utschig et al. (24) is that cation binding alters protein conformation, thereby affecting protein dynamics that are necessary for QB−• formation. One possibility is that bound Zn2+ or Cd2+ hinders the movement of water out of the RC through a contiguous water chain that was observed in the crystal structures of Rb. sphaeroides (16, 18, 19), thereby indirectly hindering the movement of QB into its active position. Another possible explanation is that QB reduction is slowed as a consequence of a slowing of the rate of proton uptake and/or redistribution of the protons that stabilize the semiquinone (36–40).
The Effect of Zn2+ and Cd2+ Binding on kAB(2).
The rate of the proton-coupled electron transfer kAB(2) (Eq. 2a) was reduced ≈10-fold and ≈20-fold upon addition of Zn2+ or Cd2+, respectively. The mechanism of the kAB(2) reaction was deduced from the dependence of the observed rate on the driving force for electron transfer (6). For RCs in the absence of cations, kAB(2) depends on the electron transfer driving force (Fig. 3) consistent with a rate-limiting electron transfer after a fast proton transfer (kH+ ≫ kAB(2), i.e., ≥ 104 s−1) as was previously reported (6). In the presence of Zn2+ or Cd2+, kAB(2) is approximately independent of the driving force (Fig. 3), implying a change in the mechanism of the proton-coupled electron transfer. This conclusion is further supported by the change in the pH dependence of kAB(2). The rate of proton transfer (first step of Eq. 2a) now has become the rate-limiting step for the reaction (i.e., kAB(2) = kH+ ⩬ 102 s−1), i.e.,
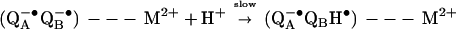 |
3 |
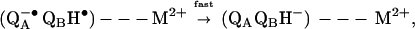 |
where M2+ is either Zn2+ or Cd2+. Thus, the rate of proton transfer to QB−• is reduced from kH+ ≥ 104 s−1 without a bound metal ion to kH+ ⩬ 102 s−1 with a bound metal ion (i.e., a ≥102-fold reduction). The reduced rate of proton transfer upon stoichiometric binding of Zn2+ or Cd2+ implies that there is one dominant site of proton entry into the RC that is blocked by the bound metal ion.
In RCs with a bound Zn2+ or Cd2+, kAB(2) is a measure of the rate of proton transfer, which enables one to trace the proton transfer pathway from QB−• to the surface of the RC by measuring kAB(2) in a series of site-directed mutant RCs. These studies should provide insight into the rates of proton conduction in the RC.
Characterization and Localization of the Binding Site of Zn2+ and Cd2+: Identification of the Dominant Proton Transfer Pathway.
The competitive replacement of Cd2+ by Zn2+ shows that both metal ions bind to the same or to nearby positions on the RC. The stoichiometry of the kinetic effects was found to be ≈1 metal cation per RC. The dissociation constants for Zn2+ and Cd2+ were determined to be KD ≤ 0.5 μM.
We now turn to a discussion of the location of the Zn2+ and Cd2+ binding site(s). The light-induced, low-temperature EPR spectrum, which is characteristic of the Fe2+-Q−• complex, excludes the possibility that either Zn2+ or Cd2+ displaces the Fe2+ in the interior of the RC. The measured recombination kinetics, kBD, in the presence of Zn2+ and Cd2+ show that there are no electrostatic or structural changes near QB. Thus, a direct interaction between QB−• and the bound Zn2+ or Cd2+ is excluded.
The most likely location(s) of the metal ions would be a surface accessible region that is rich in His, Glu, and/or Asp residues. There are three surface accessible His residues (H68, H126, H128) forming a cluster located ≈20 Å from the QB binding site at the surface of the H subunit (Fig. 4); this cluster previously was proposed as a possible Zn2+ binding location by Utschig et al. (24). His-H68 is located near one of the termini of the possible proton transfer pathway P1 (Fig. 4). His-H126 and His-H128 are located closer to P3 (Fig. 4).
Figure 4.

Part of the RC structure near the secondary quinone, QB binding site, as determined for the QB− state by Stowell et al. (18). Possible proton transfer pathways (P1–P3) proposed by Abresch et al. (19) are shown by dashed lines. One carbonyl oxygen of QB is located near Ser-L223 and the backbone NH of Ile-L224 (not shown); the other carbonyl oxygen of QB is located near His-L190. Nearby are two carboxylic acid groups Asp-L213 and Glu-L212 that have been implicated in proton transfer to reduced QB (reactions 2a and 2b, respectively) (8–13) and to which the proton transfer pathways lead. Also shown are a His cluster (consisting of H68, H126, and H128) and a carboxylic acid cluster (consisting of Asp-L213, Asp-L210, Asp-M17, Glu-H173, Asp-H170, and Asp-M124).
There are also several surface accessible carboxylic acid residues (Asp-L210, Asp-M17, Asp-H124, Glu-H224, Asp- M240). Three of these carboxylic acid groups are components of a larger cluster of carboxylic acid residues located ≈10 Å from QB and near P3 (Fig. 4), which was proposed to act as a local proton reservoir (13).
The most direct way of determining the binding location of Zn2+ and Cd2+ is by x-ray diffraction. Preliminary x-ray diffraction results have been obtained by H. L. Axelrod, E. C. Abresch, M.L.P., M.Y.O., and G.F. (unpublished work), which show that Cd2+ and Zn2+ are ligated to His-H126, His-H128, and Asp-H124 (see Fig. 4). Thus, this region of the RC surface has been identified as being crucial for fast physiological proton transfer from solution to the bound QB−•.
The Mechanism of Proton Conduction in the Bacterial RC.
The binding of Cd2+ or Zn2+ to the surface accessible region on the H-subunit (His-H126, His-H128, and Asp-H124) results in a drastic reduction (≥ 102-fold) in the rate of proton transfer. The simplest explanation for the inhibition of proton transfer is that one or more of these three residues function as proton donors. The binding of Cd2+ in the vicinity of P3 reduces their effectiveness as a source of protons.
An alternate explanation is that the binding of the metal ion creates a barrier for proton entry into one of the proton transfer pathways that have been proposed (8–15, 19, 41–43). In this view, the bound cation may electrostatically hinder proton uptake. The pathway most likely to be involved is P3 (19) (Fig. 4). Yet another explanation is that the metal ion binding affects the protein dynamics as has been postulated to be the cause for the changed kinetic for kAB(1) (24).
SUMMARY
The results of the binding of Zn2+ and Cd2+ to RCs from Rb. sphaeroides can be summarized as follows:
(i) Zn2+ and Cd2+ bind nearly stoichiometrically at or near the same position on the RC with a dissociation constant KD ≤ 0.5 μM.
(ii) The first electron transfer rate, kAB(1) (Eq. 1), is reduced ≈10-fold, implying a slowing down of the conformational gating step.
(iii) The proton transfer rate to QB−• is reduced ≫ 102-fold, making it the rate-limiting step in the kAB(2) reaction (Eq. 2a).
(iv) The large reduction (≫ 102-fold) in the rate of proton transfer upon stoichiometric binding implies that there is one dominant proton entry point into the RC.
(v) Preliminary x-ray studies localized the Cd2+ and Zn2+ binding site near His-H126, His-H128, and Asp-H124.
(vi) The simplest explanation of the inhibitory effects of Zn2+ and Cd2+ on the proton transfer rate is that their binding to the histidine and aspartic acid residues reduces their effectiveness as proton donors.
Acknowledgments
We thank Herb Axelrod and Ed Abresch for permission to quote their unpublished results, Roger Isaacson and Ed Abresch for technical assistance, and Andreas Labahn for the ADMNQ (2,6-dimethyl-3-undecyl-1,4,-naphthoquinone) and ATMNQ (2,6,7-trimethyl-3-undecyl-1,4,-naphthoquinone). This work was supported by the National Science Foundation (NSF MCB94–16652) and National Institutes of Health (GM 41637 and GM 13191).
ABBREVIATIONS
- D
primary donor
- QA
primary quinone acceptor
- QB
secondary quinone acceptor
- Q
quinone molecule
- Q10
coenzyme Q10 (2,3-dimethoxy-5-methyl-6-decaisoprenyl-1,4-benzoquinone)
- RC
reaction center
- NQ
naphthoquinone
- UQ
ubiquinone
Note Added in Proof
The proposal that the slow rate of proton transfer (Eq. 3) is caused by disruption of the proton donor(s) or blockage of the proton transfer pathway is supported by recent results from the effect of mutations on the observed rate. Replacement of either Asp-M17 or Asp-L210 with Asn resulted in an additional ∼10-fold reduction in the observed rate (at 1 mM Cd2+) from that observed in native RCs. Because the mutation sites are located close to P3 (Fig. 4), these results show that proton transfer in the presence of Cd2+ proceeds through P3 or a pathway near P3. In addition to Zn2+ and Cd2+, we found that Co2+ and Ni2+ reduced kAB(2) by ∼40-fold and ∼100-fold, respectively.
References
- 1.Breton J, Vermeglio A, editors. The Photosynthetic Bacterial Reaction Center, Structure, and Dynamics. New York: Plenum; 1988. [Google Scholar]
- 2.Feher G, Allen J P, Okamura M Y, Rees D C. Nature (London) 1989;339:111–116. [Google Scholar]
- 3.Crofts A R, Wraight C A. Biochim Biophys Acta. 1983;726:149–185. [Google Scholar]
- 4.Maroti P, Wraight C A. In: Current Research in Photosynthesis. Baltscheffsky M, editor. Vol. 1. Boston: Kluwer; 1990. pp. 1.165–1.168. [Google Scholar]
- 5.Gunner M. Curr Topics Bioenerg. 1991;16:319–367. [Google Scholar]
- 6.Graige M S, Paddock M L, Bruce J M, Feher G, Okamura M Y. J Am Chem Soc. 1996a;118:9005–9016. [Google Scholar]
- 7.Paddock M L, Senft M E, Graige M S, Ronsey S H, Toranchik T, Feher G, Okamura M Y. Photosynth Res. 1998;55:281–291. [Google Scholar]
- 8.Paddock M L, Rongey S H, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1989;86:6602–6606. doi: 10.1073/pnas.86.17.6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takahashi E, Wraight C A. Biochim Biophys Acta. 1990;1020:107–111. [Google Scholar]
- 10.Takahashi E, Wraight C A. Biochemistry. 1992;31:855–866. doi: 10.1021/bi00118a031. [DOI] [PubMed] [Google Scholar]
- 11.Paddock M L, Rongey S H, McPherson P H, Juth A, Feher G, Okamura M Y. Biochemistry. 1994;33:734–745. doi: 10.1021/bi00169a015. [DOI] [PubMed] [Google Scholar]
- 12.Baciou L, Michel H. Biochemistry. 1995;34:7967–7972. doi: 10.1021/bi00025a001. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi E, Wraight C A. Proc Natl Acad Sci USA. 1996;93:2640–2645. doi: 10.1073/pnas.93.7.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takahashi E, Wraight C A. In: Advances in Molecular and Cell Biology. Barbar J, editor. Greenwich, CT: JAI Press; 1994. pp. 197–251. [Google Scholar]
- 15.Okamura M Y, Feher G. In: Anoxygenic Photosynthetic Bacteria. Blankenship R E, Madigan M T, Bauer C E, editors. Dordrecht, The Netherlands: Kluwer; 1995. pp. 577–594. [Google Scholar]
- 16.Ermler U, Fritzsch G, Buchanan S K, Michel H. Structure (London) 1994;2:925–936. doi: 10.1016/s0969-2126(94)00094-8. [DOI] [PubMed] [Google Scholar]
- 17.Lancaster C R D, Michel H, Honig B, Gunner M R. Biophys J. 1996;70:2469–2492. doi: 10.1016/S0006-3495(96)79820-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stowell M H B, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 19.Abresch E C, Paddock M L, Stowell M H B, McPhillips T M, Axelrod H L, Soltis S M, Rees D C, Okamura M Y, Feher G. Photosynth Res. 1998;55:119–125. [Google Scholar]
- 20.Lewis J, Wilkins L, editors. Modern Coordination Chemistry. New York: Interscience; 1960. [Google Scholar]
- 21.Eriksson A E, Jones T A, Liljas A. In: Zinc Enzymes. Bertini I, Luchinat C, Maret W, Zeppezauer M, editors. Boston: Birkhauser; 1986. pp. 317–328. [Google Scholar]
- 22.Tu C, Wynns G C, Silverman D N. J Biol Chem. 1981;256:9466–9470. [PubMed] [Google Scholar]
- 23.Hurt J D, Tu C, Laipis P J, Silverman D N. J Biol Chem. 1997;272:13512–13518. doi: 10.1074/jbc.272.21.13512. [DOI] [PubMed] [Google Scholar]
- 24.Utschig L M, Ohigashi Y, Thurnauer M C, Tiede D M. Biochemistry. 1998;37:8278–8281. doi: 10.1021/bi980395n. [DOI] [PubMed] [Google Scholar]
- 25.Schmid, R., Goebel, F., Warnecke, A. & Labahn, A. (1999) J. Chem. Soc., Perkin Trans., in press.
- 26.Isaacson R A, Lendzian F, Abresch E C, Lubitz W, Feher G. Biophys J. 1995;69:311–322. doi: 10.1016/S0006-3495(95)79936-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Utschig L M, Greenfield S R, Tang J, Laible P D, Thurnauer M C. Biochemistry. 1997;36:8548–8558. doi: 10.1021/bi9630319. [DOI] [PubMed] [Google Scholar]
- 28.Okamura M Y, Isaacson R A, Feher G. Proc Natl Acad Sci USA. 1975;72:3491–3495. doi: 10.1073/pnas.72.9.3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodbury N W, Parson W W, Gunner M R, Prince R C, Dutton P L. Biochim Biophys Acta. 1986;851:6–22. doi: 10.1016/0005-2728(86)90243-4. [DOI] [PubMed] [Google Scholar]
- 30.Labahn A, Bruce J M, Okamura M Y, Feher G. Chem Phys. 1995;197:355–366. [Google Scholar]
- 31.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1984;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 32.Vermeglio A, Clayton R K. Biochim Biophys Acta. 1977;461:159–165. doi: 10.1016/0005-2728(77)90078-0. [DOI] [PubMed] [Google Scholar]
- 33.Kleinfeld D, Okamura M Y, Feher G. Biochim Biophys Acta. 1985;809:291–310. doi: 10.1016/0005-2728(85)90179-3. [DOI] [PubMed] [Google Scholar]
- 34.Butler W F, Calvo R, Fredkin D R, Isaacson R A, Okamura M Y, Feher G. Biophys J. 1984;45:947–973. doi: 10.1016/S0006-3495(84)84241-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graige M S, Feher G, Okamura M Y. Proc Natl Acad Sci USA. 1998;95:11679–11684. doi: 10.1073/pnas.95.20.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miksovska J, Kálmán L, Schiffer M, Maróti P, Sebban P, Hanson D K. Biochemistry. 1997;36:12216–12226. doi: 10.1021/bi970442w. [DOI] [PubMed] [Google Scholar]
- 37.McPherson P H, Okamura M Y, Feher G. Biochim Biophys Acta. 1988;934:348–368. doi: 10.1016/0005-2728(93)90116-w. [DOI] [PubMed] [Google Scholar]
- 38.Maroti P, Wraight C A. Biochim Biophys Acta. 1988;934:314–328. [Google Scholar]
- 39.Nabedryk E, Breton J, Hienerwadel R, Fogel C, Mäntele W, Paddock M L, Okamura M Y. Biochemistry. 1995;34:14722–14732. doi: 10.1021/bi00045a013. [DOI] [PubMed] [Google Scholar]
- 40.Maroti P, Wraight C A. Biophys J. 1997;73:367–381. doi: 10.1016/S0006-3495(97)78077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebban P, Maróti P, Hanson D K. Biochimie. 1995;77:677–694. doi: 10.1016/0300-9084(96)88183-1. [DOI] [PubMed] [Google Scholar]
- 42.Beroza P, Fredkin D R, Okamura M Y, Feher G. In: The Photosynthetic Bacterial Reaction Center II. Breton J, Vermeglio A, editors. New York: Plenum; 1992. pp. 363–374. [Google Scholar]
- 43.Allen J P, Feher G, Yeates T O, Komiya H, Rees D C. Proc Natl Acad Sci USA. 1987;84:6162–6166. doi: 10.1073/pnas.84.17.6162. [DOI] [PMC free article] [PubMed] [Google Scholar]