

Abstract
The chemistry underlying superoxide toxicity is not fully understood. A
potential mechanism for superoxide-mediated injury involves addition to
tyrosyl radicals, to give peptide or protein hydroperoxides. The rate constant
for the reaction of tyrosyl radicals with superoxide is higher than for
dimerization, but the efficiency of superoxide addition to peptides depends on
the position of the Tyr residue. We have examined the requirements for
superoxide addition and structurally characterized the products for a range of
tyrosyl peptides exposed to a
peroxidase/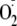 system. These included enkephalins as examples of the numerous proteins and
physiological peptides with N-terminal tyrosines. The importance of amino
groups in promoting hydroperoxide formation and effect of methionine residues
on the reaction were investigated. When tyrosine was N-terminal, the major
products were hydroperoxides that had undergone cyclization through conjugate
addition of the terminal amine. With non-N-terminal tyrosine, electron
transfer from
system. These included enkephalins as examples of the numerous proteins and
physiological peptides with N-terminal tyrosines. The importance of amino
groups in promoting hydroperoxide formation and effect of methionine residues
on the reaction were investigated. When tyrosine was N-terminal, the major
products were hydroperoxides that had undergone cyclization through conjugate
addition of the terminal amine. With non-N-terminal tyrosine, electron
transfer from
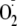 to the peptide radical prevailed. Peptides containing methionine revealed a
novel and efficient intramolecular oxygen transfer mechanism from an initial
tyrosine hydroperoxide to give a dioxygenated derivative with one oxygen on
the tyrosine and the other forming methionine sulfoxide. Exogenous amines
promoted hydroperoxide formation on tyrosyl peptides lacking a terminal amine,
without forming an adduct. These findings, plus the high hydroperoxide yields
with N-terminal tyrosine, can be explained by a mechanism in which hydrogen
bonding of
to the peptide radical prevailed. Peptides containing methionine revealed a
novel and efficient intramolecular oxygen transfer mechanism from an initial
tyrosine hydroperoxide to give a dioxygenated derivative with one oxygen on
the tyrosine and the other forming methionine sulfoxide. Exogenous amines
promoted hydroperoxide formation on tyrosyl peptides lacking a terminal amine,
without forming an adduct. These findings, plus the high hydroperoxide yields
with N-terminal tyrosine, can be explained by a mechanism in which hydrogen
bonding of
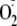 to the amine increases is oxidizing potential and alters its reactivity. If
this amine effect occurred more generally, it could increase the biological
reactivity of
to the amine increases is oxidizing potential and alters its reactivity. If
this amine effect occurred more generally, it could increase the biological
reactivity of
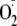 and have major implications.
and have major implications.
Free radical-mediated oxidative damage occurs in numerous diseases and is
thought to contribute to the aging process. The primary radical generated by
the reduction of oxygen is superoxide
(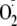 ),
a relatively benign radical that nevertheless must be removed by superoxide
dismutases (SODs)2 for
an organism to survive in an aerobic environment
(1). A number of potentially
damaging reactions of
),
a relatively benign radical that nevertheless must be removed by superoxide
dismutases (SODs)2 for
an organism to survive in an aerobic environment
(1). A number of potentially
damaging reactions of
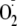 have been identified
(1–4).
One of these, which has received relatively little attention, is the addition
of
have been identified
(1–4).
One of these, which has received relatively little attention, is the addition
of
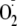 to other radicals to form hydroperoxides
(5,
6). This reaction has been
shown to occur readily with tyrosine and Tyr-containing dipeptides, resulting
in the formation of tyrosine hydroperoxides
(5–7).
Hydroperoxides are potentially damaging reactive oxygen species. Formation on
proteins can result in detrimental structural and functional changes
(8). Protein hydroperoxides are
also oxidants that can injure other biomolecules.
to other radicals to form hydroperoxides
(5,
6). This reaction has been
shown to occur readily with tyrosine and Tyr-containing dipeptides, resulting
in the formation of tyrosine hydroperoxides
(5–7).
Hydroperoxides are potentially damaging reactive oxygen species. Formation on
proteins can result in detrimental structural and functional changes
(8). Protein hydroperoxides are
also oxidants that can injure other biomolecules.
Tyrosyl radicals are generated in many physiological situations and
proteins are major targets for reactive oxidants
(9). In proteins exposed to
free radicals, regardless of the initial site of attack, the resultant radical
commonly localizes to Tyr
(10–13).
Tyrosyl radicals are also produced from tyrosyl peptides through the action of
peroxidases such as myeloperoxidase, and are generated during the catalytic
cycle of enzymes such as ribonucleotide reductase and cyclooxygenase
(14). Tyrosyl radicals undergo
a variety of subsequent reactions. They readily dimerize to form dityrosine,
which has been well documented as a product of oxidative injury
(15,
16). Another oxidative
biomarker, nitrotyrosine, is also formed via tyrosyl radicals
(4,
15,
17). However, one of their
most favored reactions is with
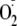 (5,
7,
18,
19). The reaction has a rate
constant several times higher than that for dimerization
(7,
20) and is favored over
dityrosine formation in situations where both tyrosyl and
(5,
7,
18,
19). The reaction has a rate
constant several times higher than that for dimerization
(7,
20) and is favored over
dityrosine formation in situations where both tyrosyl and
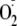 radicals are generated (7,
20).
radicals are generated (7,
20).
The reaction of
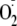 with phenoxyl radicals results in either repair of the parent phenol (reaction
2, Fig. 1b) or
addition to form a hydroperoxide (reaction 3). With tyrosine, most of the
with phenoxyl radicals results in either repair of the parent phenol (reaction
2, Fig. 1b) or
addition to form a hydroperoxide (reaction 3). With tyrosine, most of the
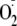 reacts by addition (7,
20). The structure of tyrosine
hydroperoxide has not been determined directly but inferred from NMR studies
of the corresponding monoxide derivative formed by slow decomposition
(7). These were shown to be
bicyclic compounds formed by conjugate addition of the amino group to the
phenol ring (HOHICA, designated I and named in full in
Fig. 1b, proposed to
arise from reactions 5 and 6).
reacts by addition (7,
20). The structure of tyrosine
hydroperoxide has not been determined directly but inferred from NMR studies
of the corresponding monoxide derivative formed by slow decomposition
(7). These were shown to be
bicyclic compounds formed by conjugate addition of the amino group to the
phenol ring (HOHICA, designated I and named in full in
Fig. 1b, proposed to
arise from reactions 5 and 6).
FIGURE 1.

a, experimental system used for the generation of superoxide and
tyrosyl (TyrO·) radicals. b, proposed mechanism for
the formation and decomposition of tyrosine hydroperoxide derivatives.
R and R′ represent OH and H, respectively, for Tyr, or
amino acid residue(s) for the peptides. Reaction 1 shows
peroxidase-mediated formation of Tyr radicals, which can either dimerize (not
shown) or react with
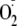 by electron transfer (reaction 2) or addition (reaction 3).
Addition results in the formation of hydroperoxides (o- and
p-isomers, only the o-isomer shown) that may exist
transiently and decompose to release 1O2 (reaction 4) or
form a stable species that can undergo conjugate addition of the terminal
amino group are shown (when R′= H, reaction 5). An equivalent
reaction is proposed for non-N-terminal Tyr (R′= amino acid residue) in
which conjugation involves the amide nitrogen. Hydrolysis of the
hydroperoxides that are modified by conjugate addition gives the corresponding
hydroxide derivatives (I,
3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid or HOHICA)
in reaction 6. c, possible alternative hydrolysis products
(mono-oxygenated derivatives). II, 3,4-dihydroxyphenylalanine
derivatives from the o-isomer; III,
4-alanyl-4-hydroxy-cyclohexadienone (HACHD) derivatives from the
p-isomer.
by electron transfer (reaction 2) or addition (reaction 3).
Addition results in the formation of hydroperoxides (o- and
p-isomers, only the o-isomer shown) that may exist
transiently and decompose to release 1O2 (reaction 4) or
form a stable species that can undergo conjugate addition of the terminal
amino group are shown (when R′= H, reaction 5). An equivalent
reaction is proposed for non-N-terminal Tyr (R′= amino acid residue) in
which conjugation involves the amide nitrogen. Hydrolysis of the
hydroperoxides that are modified by conjugate addition gives the corresponding
hydroxide derivatives (I,
3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid or HOHICA)
in reaction 6. c, possible alternative hydrolysis products
(mono-oxygenated derivatives). II, 3,4-dihydroxyphenylalanine
derivatives from the o-isomer; III,
4-alanyl-4-hydroxy-cyclohexadienone (HACHD) derivatives from the
p-isomer.
Hydroperoxide formation has been observed with peptides but only when tyrosine is N-terminal or the reaction is promoted by amino compounds (5). The amine effect has implications for hydroperoxide formation on proteins, but the mechanism is not understood. It has also been postulated that the repair mechanism involves singlet oxygen release from an intermediate (reaction 4) rather than electron transfer (reaction 2) (18), but this has not been studied experimentally.
The objectives of this investigation were to determine the structures of
the hydroperoxide and any other superoxide addition products, and to
understand the mechanism of formation, using a range of synthetic and
physiological tyrosyl peptides. These include the opioids Leu- and
Met-Enkephalin (Leu-Enk, YGGFL; and Met-Enk, YGGFM, respectively) and
Endomorphin 2 (Endo2, YPFF). The opioids have a free N-terminal Tyr that is
essential for activity and are potential physiological targets for
inactivation by
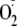 addition. We also investigated whether the presence of a Met residue (as in
Met-Enk) influences Tyr-hydroperoxide formation on the peptide and whether
addition. We also investigated whether the presence of a Met residue (as in
Met-Enk) influences Tyr-hydroperoxide formation on the peptide and whether
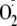 addition results in the formation of methionine sulfoxide. If so, this could
be a physiological mechanism for production of methionine sulfoxide, which is
one of the most prevalent products of oxidative stress
(21,
22).
addition results in the formation of methionine sulfoxide. If so, this could
be a physiological mechanism for production of methionine sulfoxide, which is
one of the most prevalent products of oxidative stress
(21,
22).
Peptides were exposed to a xanthine oxidase (XO) system to generate
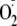 and hydrogen peroxide (H2O2) plus horseradish peroxidase
(HRP) to catalyze the reaction of H2O2 with the peptide
to give the tyrosyl radical (Fig.
1a). Products were analyzed using a general hydroperoxide
assay (Fe2+/xylenol orange or FOX assay) and by liquid
chromatography/electrospray mass spectrometry (LC/MS). We have obtained
structural information on the hydroperoxides, identified a mechanism of rapid
intramolecular oxidation of Met residues via a hydroperoxide intermediate, and
provide an explanation for why amino groups facilitate the addition of
and hydrogen peroxide (H2O2) plus horseradish peroxidase
(HRP) to catalyze the reaction of H2O2 with the peptide
to give the tyrosyl radical (Fig.
1a). Products were analyzed using a general hydroperoxide
assay (Fe2+/xylenol orange or FOX assay) and by liquid
chromatography/electrospray mass spectrometry (LC/MS). We have obtained
structural information on the hydroperoxides, identified a mechanism of rapid
intramolecular oxidation of Met residues via a hydroperoxide intermediate, and
provide an explanation for why amino groups facilitate the addition of
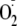 to the tyrosyl radical.
to the tyrosyl radical.
EXPERIMENTAL PROCEDURES
Reagents—Water was purified by running through a Milli-Q system (Millipore) so that its resistivity was greater than 18 mΩ-cm. All reagents and enzymes were purchased from Sigma, unless otherwise indicated. HOCl solutions were prepared from commercial bleach (Janola) and were standardized spectrophotometrically (using ε292 nm = 350 m–1 cm–1). The peptides: YGGFM, Boc-YGGFM, GYGGFM, MEVDPIGHLY, RFYVVM, and YSFKDMGLGR were purchased from Bachem (Bubendorf) and Tyr-Met (YM) and Met-Tyr (MY) were custom synthesized by Genscript (NJ). All peptides were >98% pure. Deuterium oxide (99.9%) was obtained from Cambridge Isotope Laboratories. Anthracene-9,10-diyldiethyl sulfate (EAS) was a generous gift of Prof. Paolo Di Mascio.
The Tyr-para-hydroperoxide derivative of Gly-Tyr-Gly (Gly-Tyr-Gly-OOH) was a generous gift of Prof. Michael Davies. It was generated by the reaction of Gly-Tyr-Gly with singlet oxygen, where singlet oxygen was generated in situ by irradiation in the presence of Rose-Bengal (23). The concentration of Gly-Tyr-Gly-OOH was 500 μm (as measured by the FOX assay) in the presence of 2.5 mm Gly-Tyr-Gly.
YM-sulfoxide (YM-S=O) was prepared by oxidation of YM by 1 m equivalent HOCl. No other components were detected by LC/MS indicating ∼100% conversion.
The concentrations of stock solutions of H2O2 were determined iodometrically. Concentrations in solutions prepared from the stock were confirmed spectrophotometrically (ε(H2O2)240 nm = 43.6 m–1 cm–1). Stock solutions of XO were prepared by dilution of an ammonium sulfate suspension with 50 mm phosphate buffer, pH 7.4, and spinning through a G-25 Sephadex column to remove the ammonium sulfate. The activity of XO was measured by the cytochrome c assay and by quantifying H2O2 formation using the FOX assay. Enzyme and acetaldehyde stock solutions were prepared fresh daily and stored on ice.
Peroxidase-mediated Oxidation of Tyrosine-containing Peptides in the
Presence of Superoxide—Reaction mixtures consisted of acetaldehyde
(1 mm unless stated otherwise), XO (typically 0.001 unit/ml), HRP
(typically 140 nm), and 0.2 mm peptide in 50
mm phosphate buffer plus 50 μm
diethylenetriaminepentaacetic acid (except for the FOX assay experiments).
This amount of XO with 1 mm acetaldehyde corresponds to an initial
rate of 2.8 μm/min
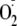 and total production of 36 μm H2O2 over a
30-min reaction period. Reactions were started by addition of XO. They were
carried out at 20–25 °C, typically for 30 min for LC/MS, 10 min for
FOX and dimer analyses, and stopped by adding 20 μg/ml catalase to remove
residual H2O2. When necessary the enzymes were removed
from the reaction mixtures by ultracentrifugation using 10–30-kDa cutoff
microconcentrators (Amicon Microcon). Samples were protected from light to
avoid photochemical reactions.
and total production of 36 μm H2O2 over a
30-min reaction period. Reactions were started by addition of XO. They were
carried out at 20–25 °C, typically for 30 min for LC/MS, 10 min for
FOX and dimer analyses, and stopped by adding 20 μg/ml catalase to remove
residual H2O2. When necessary the enzymes were removed
from the reaction mixtures by ultracentrifugation using 10–30-kDa cutoff
microconcentrators (Amicon Microcon). Samples were protected from light to
avoid photochemical reactions.
Dimer Quantification—Dimers were measured fluorimetrically (excitation 325 nm, emission 400 nm) with a Hitachi F-4500 fluorescence spectrofluorimeter as described in Ref. 5. Results are expressed as relative fluorescence and are related to concentrations using calibration curves obtained by generating the dimer from the relevant peptide in at least 20-fold excess and known concentrations of H2O2 in the presence of HRP. Dimer concentrations were corroborated by measuring A315 at pH 8.0 using ε = 5,080 m–1 cm–1.
Hydroperoxide Quantification—Hydroperoxides were analyzed using a modified FOX method (5, 24) standardized against H2O2, under similar conditions as for dimer quantification.
Liquid Chromatography (LC)-Electrospray Ionization (ESI)-Mass Spectrometry (MS)—LC-ESI-MS and LC-ESI-MS/MS analyses were performed with a Thermo Finnigan LCQ Deca XP Plus ion trap mass spectrometer (San Jose, CA) coupled to a Surveyor HPLC system and PDA detector. Positive ion mode was used for all peptides and negative ion mode for p-hydroxyphenylacetic acid (HPA) and EAS derivatives. Data were analyzed using Finnigan Xcalibur, Thermo Finnigan Qual Browser 1.3, and High Chem Mass Frontier 3.0 programs. Fragmentation patterns were analyzed using Bioworks Browser 3.1 and peptide fragment ions were assigned and discussed based on the Roepstorff-Fohlman nomenclature. Further details on chromatography conditions and detection are given under supplementary Methods.
Acid Hydrolysis—Peptides were lyophilized and vapor phase hydrolyzed with 6 m HCl containing 1% (w/v) phenol, plus 50 μl of mercaptoacetic acid when products were analyzed for the recovery of 3,4-dihydroxyphenylalanine (25). After hydrolysis, the residual HCl was evaporated and the samples were redis-solved in water and analyzed by LC-MS.
Quantification of Met-Enk and Leu-Enk Products—Yields of Leu-Enk hydroperoxide and Met-Enk and Gly-Met-Enk dioxides were quantified by calibrating the LC/MS peak integral for each species. Each compound was generated using the XO/HRP system and purified by LC with the same setup as for MS analysis. For Met-Enk and Gly-Met-Enk, the product peak was collected, concentrated, and a sample reinjected to check for purity. When necessary the purification step was repeated until no contaminants were detectable. The concentrations of the enkephalin dioxides in the purified solutions were established on the basis of Phe content. An aliquot of each solution was hydrolyzed and the amount of Phe determined by LC/MS using selective ion monitoring. A standard curve was created with authentic Phe, which was identified from its retention time and fragmentation pattern. Controls using internal standards of authentic Phe showed nearly 100% recovery under the applied conditions. Acid hydrolysis was >90% efficient based on Phe recovery from the parent enkephalin hydrolyzed under the same conditions. This gave the Phe content and hence the dioxide concentration in the pure sample. A known amount of pure dioxide was injected into the LC/MS to calibrate its peak integral and this calibration was used to quantify dioxide formation under experimental conditions. The instrument response was corrected by use of check standard of Leu-Enk before each set of runs.
For Leu-Enk, the hydroperoxide was not stable enough to use the same procedure. Instead, the entire Leu-Enk-OOH peak from each of two chromatographic runs was collected and the combined sample was hydrolyzed and analyzed for Phe content as above. The Phe content in half the hydrolysate was related to the mean peak integral for the original samples, using the same time interval for collection and integration. This calibration was then used to quantify the hydroperoxide peak from experimental samples that were separated at the same time as the calibration sample.
RESULTS
Reactions of Superoxide with Peptide Radicals
It has been observed (5,
26,
27) that radicals generated on
Tyr and tyrosyl dipeptides react with
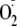 to form hydroperoxides (5,
26,
27). This occurs in
competition with dimerization of the radicals, and with Tyr and the XO/HRP
system as in Fig. 1a,
more hydroperoxide than dityrosine was formed
(5). We extended these
observations to the enkephalins and related peptides, and found that they were
all oxidized by HRP and H2O2. Yields of dimer formation
in the XO/HRP/acetaldehyde systems were 3–4-fold higher when SOD was
present, indicating that in the absence of SOD, the peptide radicals reacted
with
to form hydroperoxides (5,
26,
27). This occurs in
competition with dimerization of the radicals, and with Tyr and the XO/HRP
system as in Fig. 1a,
more hydroperoxide than dityrosine was formed
(5). We extended these
observations to the enkephalins and related peptides, and found that they were
all oxidized by HRP and H2O2. Yields of dimer formation
in the XO/HRP/acetaldehyde systems were 3–4-fold higher when SOD was
present, indicating that in the absence of SOD, the peptide radicals reacted
with
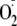 in preference to dimerization (Fig.
2a). Using the FOX assay
(24) to detect hydroperoxides,
a positive response was obtained with Tyr-Gly (YG) but not Gly-Tyr (GY)
(Fig. 2b), in
agreement with previous observations
(5) that hydroperoxide
formation requires an N-terminal Tyr. Substantial
in preference to dimerization (Fig.
2a). Using the FOX assay
(24) to detect hydroperoxides,
a positive response was obtained with Tyr-Gly (YG) but not Gly-Tyr (GY)
(Fig. 2b), in
agreement with previous observations
(5) that hydroperoxide
formation requires an N-terminal Tyr. Substantial
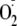 -dependent
hydroperoxide formation was observed with Leu-Enk and Endo2, but despite
having an N-terminal Tyr, Met-Enk gave no detectable hydroperoxide by FOX
analysis (Fig.
2b).
-dependent
hydroperoxide formation was observed with Leu-Enk and Endo2, but despite
having an N-terminal Tyr, Met-Enk gave no detectable hydroperoxide by FOX
analysis (Fig.
2b).
FIGURE 2.

a, dimer; and b, peroxide formation from Tyr-containing peptides. Samples were incubated with HRP, acetaldehyde, and XO and the respective peptide as described under “Experimental Procedures.” These peptides were shown to be good substrates for HRP, with initial rates of dimer formation measured under similar conditions of 2–16 times that of Tyr. Dimers were detected by fluorescence and hydroperoxides by the FOX assay. Zero time blanks were subtracted and error bars represent the range of means from two experiments. Peroxide yields are expressed as H2O2 equivalents using a calibration curve that was developed with known amounts of H2O2. However, it should be noted that isotope tracer studies have demonstrated that tyrosine hydroperoxide concentrations are underestimated by the FOX assay and are approximately 6 times higher than when expressed as H2O2 equivalents (5).
It should be noted that the FOX assay was calibrated with H2O2 as no hydroperoxide standards are available, and this results in underestimation of hydroperoxide yields. The version of the assay used here gives a 6-fold lower response to tyrosine hydroperoxide than H2O2 (5). Assuming that tyrosyl peptides behave similarly, the data in Fig. 2 indicate that the enkephalins gave more hydroperoxide than dimer. For Leu-Enk, this was confirmed by direct analysis (see below).
Product Analysis and Structural Characterization of Hydroperoxides
Tyr-Gly and Peptides with N-terminal Tyr Residues—To establish the structures of the peptide hydroperoxides directly, the products of the reactions of Leu-Enk, YG, and Endo2 were analyzed by LC/MS. Each gave, in addition to the dimer, a major product with molecular mass corresponding to the native peptide + 32 (potentially the hydroperoxide) and a product at a mass of the native peptide + 16 (monoxide) (Fig. 3). In some cases two peaks of equivalent mass and fragmentation pattern were evident, presumably representing o- and p-isomers. Formation of both species was strongly inhibited by SOD. Based on peak integrals and assuming the two species had similar MS characteristics, more dioxides than monoxides were present. The monoxides are assumed to have arisen from hydrolysis of the hydroperoxides during the reaction and sample processing (as in Ref. 7 and shown below in Fig. 6).
FIGURE 3.

LC/MS detection of the oxygenated products from (a) Leu-Enk (YGGFL), (b) YG, and (c) GY. Samples were incubated with HRP, acetaldehyde, and XO as described under “Experimental Procedures”; chromatography conditions are described under supplementary Methods. Bottom chromatograms represent the native peptides, top chromatograms the monoxide and dioxide derivatives, all obtained by extraction from the total ion chromatograms. The peaks with dashed lines correspond to reactions carried out in the presence of SOD. SOD also inhibited dioxide and monoxide formation with the other peptides. Dioxide and monoxide species were also detected for Endo2 (YPFF) under similar conditions using selective ion monitoring.
FIGURE 6.

Intermolecular reaction of Leu-Enk hydroperoxide (Leu-Enk-OOH) with (a) Met and (b) Met-Enk. Decay of Leu-Enk-OOH (10 μm in 50 mm phosphate buffer) was measured in the presence and absence of 200 μm methionine by LC/MS, or 200 μm Met-Enk by the FOX assay. Diethylenetriaminepentaacetic acid (50 μm) was present for a but not b. Leu-Enk-OOH was generated using the conditions described under “Experimental Procedures.” After 30 min incubation, 20 μg/ml catalase was added and the enzymes were removed by centrifugation using an Amicon 10-kDa cutoff filter. The reaction of Leu-Enk-OOH with Met-Enk was also monitored by LC/MS, where a similar second-order rate constant was obtained (k = 0.18 m–1 s–1). Loss of the hydroperoxide was accompanied by increases in the Leu-Enk monoxide and Met-Enk sulfoxide peaks at similar rates. The rate of the reaction was also investigated at lower Met-Enk concentrations. The obtained pseudo-first ordered rate constants together with the fits of the pseudo-first order kinetic traces to a single exponential curve indicate that the overall reaction is indeed second-order (first-order for both [Leu-Enk-OOH] and [Met-Enk]).
The yield of Leu-Enk hydroperoxide was measured under the conditions of Fig. 3a with the peak integral calibrated on the basis of the Phe content of the purified product (see “Experimental Procedures”). For three independent experiments, 10.7 μm (S.D. 0.5) hydroperoxide and 4.5 μm dimer (S.D. 0.4) were formed. These accounted for the majority of the total Leu-Enk loss. This result shows that the hydroperoxide was the major product in our systems.
MS/MS fragment ions were assigned for the dioxide and monoxide products. As shown for YG, the parent peptide gave the expected a1 fragment for Tyr (Fig. 4a). The monoxide (Fig. 4b) gave a1 and b1 fragment ions with the extra oxygen attached to the Tyr residue, and the dioxide (Fig. 4c) gave an a1 fragment containing both oxygens. Where there was loss of H2O from the monoxide, H2O2 was lost from the dioxide, implying that the two differ by an -OH or -OOH at the same site. Similar features were evident in the fragmentation patterns of the dioxide and monoxide products for Leu-Enk and Endo2 (not shown) and fits obtained using the Bioworks Browser (as for Met-Enk in Fig. S5) were consistent with the extra oxygens in these peptides being attached to the Tyr.
FIGURE 4.

Fragmentation patterns of (a) YG, (b) YG monoxide, and (c) YG dioxide. YG was treated and analyzed as described in the legend to Fig. 3. Proposed structures and fragment assignments are discussed in the text.
YG monoxide had a very similar fragmentation pattern to the monoxide formed by exposing free Tyr to the XO/HRP system (HOHICA in Fig. S1a). Both showed the same a1 fragment with an extra oxygen and the m/z 134 peak (representing loss of water from the a1 fragment and thus a lack of aromaticity, see later). The m/z 134 peak, plus the absence of the peak representing ammonia loss (presumably reflecting lack of a free amino group), are key features that discriminate these structures from other theoretical alternatives with the same mass (II and III in Fig. 1c: data in Fig. S1, b and c). As HOHICA (I in Fig. 1b) has been characterized as a bicyclic compound (7), we conclude that YG dioxide has a hydroperoxide on the Tyr ring that has undergone the same conjugate addition (Fig. 1b, reaction 5).
Gly-Tyr—Although GY gave no detectable hydroperoxide in Fig. 2b, low yields of dioxide and monoxide derivatives were detected by LC/MS (Fig. 3c). The fragmentation patterns (Fig. S2) show a hydroperoxide group, or corresponding -OH, located on the Tyr, and also suggest (as discussed below for peptides containing Met) that the Tyr ring is modified by conjugate addition of the amide nitrogen.
Intramolecular Oxygen Transfer in Peptides Containing Tyr and Met
The situation was different for peptides containing Met. We investigated
dipeptides MY, YM, as well as Met-Enk and Gly-Met-Enk. These peptides produced
dimers when exposed to the XO/HRP system, and dimerization was enhanced when
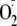 was removed with SOD (Table 1
and Fig. 2a). LC/MS
analysis (Fig. 5,
a–d, and
Table 1) showed that a
dioxygenated M + 32 + H+ species was produced from all the
peptides. Formation of the dioxygenated species required
was removed with SOD (Table 1
and Fig. 2a). LC/MS
analysis (Fig. 5,
a–d, and
Table 1) showed that a
dioxygenated M + 32 + H+ species was produced from all the
peptides. Formation of the dioxygenated species required
 as it was suppressed by SOD. However, none of the peptides gave a positive
response in the FOX assay (Table
1 and Fig.
2b), implying that the two-oxygen addition products were
not hydroperoxides.
as it was suppressed by SOD. However, none of the peptides gave a positive
response in the FOX assay (Table
1 and Fig.
2b), implying that the two-oxygen addition products were
not hydroperoxides.
TABLE 1.
Methionyl peptides examined for dioxide formation
Dimer concentrations were measured by spectrofluorimetry and hydroperoxide concentrations by the FOX assay. Peptides were separated by LC/MS using chromatographic conditions given under supplementary Methods and the [M + 32 + H]+ peaks were detected as shown in Fig. 5 for YM, MY, Met-Enk, Gly-Met-Enk, and YM-S = O.
|
Amino acid sequence
|
Molecular mass
|
RTaof [M + H]+
|
RT of dioxide
|
Dimer
|
Hydroperoxide (H2O2 eq. μm)
|
|||
|---|---|---|---|---|---|---|---|---|
| No SOD | SOD | |||||||
| g/mol | min | μm | ||||||
| YM | 312.4 | 17.9 | 12.7, 14.0 | 3.8 ± 0.1 | 7.0 ± 0.1 | 0.23 ± 0.006 | ||
| MY | 312.4 | 17.4 | 12.6, 14.7 | 5.41 ± 0.1 | 6.9 ± 0.2 | <0.2 | ||
| YGGFM | 573.7 | 16.1 | 11.3, 12.5 | 3.3 ± 0.1 | 8.6 ± 0.1 | <0.2 | ||
| GYGGFM | 630.7 | 13.3 | 10.2, 11.0 | 8.0 ± 0.4 | 10.5 ± 0.4 | <0.2 | ||
| Boc-YGGFM | 673.8 | 14.1 | 9.6 | 5.8 ± 0.2 | 11.4 ± 0.5 | <0.2 | ||
| RFYVVM | 814.0 | 8.4, 10.6 | 7.5, 9.6 | 10.7 ± 0.8 | 17.6 ± 0.4 | <0.2 | ||
| YSFKDMGLGR | 1244.4 | 10.7 | 9.4 | 1.0 ± 0.2 | 2.7 ± 0.2 | <0.2 | ||
| MEVDPIGHLY | 1173.4 | 18.6 | 12.3 | 3.3 ± 0.3 | 7.8 ± 0.3 | <0.2 | ||
| YM-S = O | 361.1 | 15.93 | 13.53, 14.07 | 2.55 ± 0.09 | 4.15 ± 0.05 | 0.32 ± 0.002 | ||
RT retention time. Conditions as described under “Experimental Procedures.”
FIGURE 5.

LC/MS detection of the dioxide products from (a) Met-Enk, (b) Gly-Met-Enk, (c) YM, (d) MY, and (g) YM-S=O and fragmentation patterns of (e) YM, (f) YM dioxide, and (h) YM-S=O dioxide. Chromatograms represent the native peptides (dashed lines) and dioxides (solid lines) obtained by selective ion monitoring (a and c) or by extraction from the total ion chromatograms (b, d, and g). Amounts of products (left ordinate axis) are shown as relative abundance compared with the parent peptide set as 100%. Reaction conditions are described under “Experimental Procedures” and chromatographic conditions under supplementary Methods. Dioxide yields were inhibited >90% by 10μg/ml SOD. Some peptide-monoxide derivatives were also detected, but except for YM where a small amount of Tyr-OH was detected, it was due to sulfoxide impurity in the authentic sample. A small amount of YM-S=O monoxide (i.e. YM dioxide) was also detected in the YM-S=O system that is presumably due to the hydrolysis of the YM-S=O hydroperoxide (in analogy with the YG or Leu-Enk hydroperoxides, see Fig. 3).
Yields of dioxygenated products were quantified for Met-Enk and Gly-Met-Enk by calibrating peak integrals for each dioxide based on Phe content (see “Experimental Procedures”). Using this calibration, under the conditions of Fig. 5, 7 ± 2 μm Met-Enk-dioxide and 1.8 ± 0.4 μm Gly-Met-Enk-dioxide were formed (n = 4). Dimer concentrations measured in the same solutions were 3.3 (S.D. 0.2) and 10.4 (S.D. 0.4) μm, respectively. The sum of the dioxide and dimers closely accounted for the loss of the corresponding native peptide. This establishes that the dioxide is the major product with Met-Enk. It also confirms that the position of the Tyr dictates the efficiency of dioxide formation as it does for the hydroperoxides and is consistent with the dioxides arising from a hydroperoxide intermediate.
Structural information was obtained by comparing the dioxide fragmentation patterns to those of the parent peptides. YM gave the expected a1 and y1 fragments and an M – 46 + H+ peak characteristic of methionyl peptides (Fig. 5e). For YM dioxide, the y1 fragment (m/z 166) shows the presence of an extra oxygen, and there is a peak at m/z = 281 representing loss of 64 mass units (–HS(=O)-CH3, a characteristic loss for sulfoxides (28). This is clear evidence that one of the oxygens is present as Met sulfoxide (Fig. 5f). There is a peak at m/z 134, as seen with YG monoxide and HOHICA, and no peak corresponding to ammonia loss. From this fragmentation pattern, we conclude that the other oxygen is located on the Tyr, which has undergone conjugate addition.
Fragmentation of MY-dioxide also gave a major peak at M – 64 + H+ (Fig. S3b), indicating that it is also the sulfoxide derivative. The pattern is more complex than for YM-dioxide, with no clear y1 peak. However, successive loss of two waters from both the parent ion and M – 64 + H+ peak implies a labile -OH group in addition to the carboxyl group on the Tyr. As discussed under Fig. S3, possible Tyr-OH structures with the requisite mass include a 3,4-dihydroxyphenylalanine derivative or its p-equivalent (II and III in Fig. 1c) and a conjugate addition product (I in Fig. 1b). Only I and III would have a labile -OH. Acid hydrolysis of the dioxides purified from MY and Gly-Met-Enk gave no detectable 3,4-dihydroxyphenylalanine or III under conditions where they should have been detected (for details, see Fig. S4). On this basis we propose that the Tyr is most likely modified by conjugate addition, possibly with the amide nitrogen.
When YM was first converted to its sulfoxide derivative then exposed to the
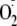 /H2O2/HRP
system, the major product (in addition to the dimer) had a molecular mass
corresponding to [YM + 48 + H+]. This represents the addition of
two extra oxygens to YM-S=O (Fig.
5g). The formation of YM-trioxide was greatly inhibited
by SOD.
/H2O2/HRP
system, the major product (in addition to the dimer) had a molecular mass
corresponding to [YM + 48 + H+]. This represents the addition of
two extra oxygens to YM-S=O (Fig.
5g). The formation of YM-trioxide was greatly inhibited
by SOD.
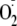 -dependent
formation of a hydroperoxide was detected by the FOX assay
(Table 1). This, together with
the obtained fragmentation pattern (Fig.
5h) indicates that YM-trioxide is the Tyr hydroperoxide
derivative of YM-S=O.
-dependent
formation of a hydroperoxide was detected by the FOX assay
(Table 1). This, together with
the obtained fragmentation pattern (Fig.
5h) indicates that YM-trioxide is the Tyr hydroperoxide
derivative of YM-S=O.
We propose that Met oxidation occurs via intramolecular transfer of one oxygen from an initial tyrosine hydroperoxide derivative. In support of this, there was little or no formation of products in which only the Tyr or Met residue of the peptide was oxidized. As only a small fraction of the parent peptide was modified, this would be expected if the reaction was intermolecular. Further evidence against intermolecular oxygen transfer is that the reaction of preformed Leu-Enk-OOH with Met or Met-Enk occurred over hours (Fig. 6).
The effect of the relative positions of the Tyr and Met residues on the oxygen transfer reaction was investigated using the peptides shown in Table 1. All produced dimers when exposed to the XO/HRP system (Table 1). This reaction was enhanced in the presence of SOD, but no detectable hydroperoxides were formed. In each case, a dioxygenated product was detected by LC/MS (Table 1) with a fragmentation pattern showing loss of 64 mass units characteristic of methionine sulfoxide and other features consistent with localization of the other oxygen on the Tyr residue. The closeness of fit of the observed fragments with those simulated by the Bioworks Browser for the structure representing the best fit is illustrated for Met-Enk in Fig. S5. We propose, therefore, that in peptides containing Tyr and Met, formation of a hydroperoxide on the Tyr is rapidly followed by intramolecular oxygen transfer to the Met (shown for YM in Fig. 7) and this can operate when these residues are adjacent or up to at least 9 residues apart.
FIGURE 7.

Proposed mechanism for the formation of Tyr-Met-dioxide. The Tyr residue is oxidized in a peroxidase-mediated one-electron oxidation reaction by H2O2 to give the phenoxyl radical, which adds to superoxide to form the hydroperoxide intermediate. The hydroperoxide undergoes conjugate addition of the amine nitrogen to the Tyr ring and the Met residue is oxidized to its sulfoxide via intramolecular oxygen transfer from the Tyr hydroperoxide. The reaction is shown for the ortho hydroxyl isomer; an equivalent reaction can be written for the para form.
No Evidence for Singlet Oxygen Release
The low yields of hydroperoxides or other major
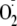 -dependent
products with peptides in which the Tyr was not N-terminal, combined with
previous observations that less of these peptides were consumed in the
reaction (5), are consistent
with
-dependent
products with peptides in which the Tyr was not N-terminal, combined with
previous observations that less of these peptides were consumed in the
reaction (5), are consistent
with
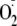 reacting with the peptide radicals to repair the Tyr. Two mechanisms have been
proposed: repair via electron transfer
(19) or release of
1O2 from an unstable hydroperoxide intermediate
(7,
18) (reactions 2 or
4 in Fig. 1b,
respectively). Production of 1O2 was investigated using
anthracene-9,10-diyldiethyl sulfate as a trap
(29) and LC/MS to quantify the
endoperoxide product. The detection limit, established with
1O2 generated from hypochlorous acid and
H2O2, was 0.5 μm (Fig. S6). We maximized
product yields from the XO/HRP system for GY and YG and reasoned that if the
YG radical reacted with
reacting with the peptide radicals to repair the Tyr. Two mechanisms have been
proposed: repair via electron transfer
(19) or release of
1O2 from an unstable hydroperoxide intermediate
(7,
18) (reactions 2 or
4 in Fig. 1b,
respectively). Production of 1O2 was investigated using
anthracene-9,10-diyldiethyl sulfate as a trap
(29) and LC/MS to quantify the
endoperoxide product. The detection limit, established with
1O2 generated from hypochlorous acid and
H2O2, was 0.5 μm (Fig. S6). We maximized
product yields from the XO/HRP system for GY and YG and reasoned that if the
YG radical reacted with
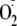 mainly by addition and GY released 1O2, the difference
in hydroperoxide yield between the two should theoretically be detected with
GY as 1O2. (This is argued in more detail in Fig. S6.)
As shown in Fig. S6c, 1O2 production was below
the detection limit under conditions where the difference in hydroperoxide
production was at least 15μm (measured in
H2O2 equivalents, and for reasons given above most
likely in excess of 50 μm). At most this would represent only a
few percent of the theoretical yield. Therefore, we conclude that the release
of 1O2 plays little if any role in the repair pathway
for non-N-terminal Tyr.
mainly by addition and GY released 1O2, the difference
in hydroperoxide yield between the two should theoretically be detected with
GY as 1O2. (This is argued in more detail in Fig. S6.)
As shown in Fig. S6c, 1O2 production was below
the detection limit under conditions where the difference in hydroperoxide
production was at least 15μm (measured in
H2O2 equivalents, and for reasons given above most
likely in excess of 50 μm). At most this would represent only a
few percent of the theoretical yield. Therefore, we conclude that the release
of 1O2 plays little if any role in the repair pathway
for non-N-terminal Tyr.
Enhanced Superoxide Addition to Tyrosyl Radicals by Exogenous Amines
One mechanism proposed to explain the higher yields of hydroperoxide on N-terminal Tyr is that formation is facilitated by conjugate addition of the free amine (18). This was tested by adding exogenous amines to tyrosyl peptides and identifying the products using LC/MS. Lys and ethanolamine have been shown to enhance hydroperoxide yields on GY, measured by the FOX assay (5). If this were due to conjugate addition of the amine, a higher molecular weight product would be formed. With GY, both Lys and ethanolamine gave a concentration-dependent increase in hydroperoxide yield (Fig. 8). However, the hydroperoxide had the same chromatographic mobility, mass, and fragmentation pattern as the GY-OOH formed in the absence of amine (Fig. 8a, inset). There was no evidence of a product arising from intermolecular conjugate addition. Furthermore, Lys enhanced hydroperoxide formation on HPA without forming an addition product (Fig. 8b), excluding any requirement for a peptide nitrogen. The hydroperoxide yield with YG, which was already much higher in the absence of amines, was not affected by adding Lys.
FIGURE 8.

Increases in superoxide-dependent hydroperoxide formation in the
presence of amines as detected by LC/MS. a, formation of GY-OOH
in the presence of Lys in normal (○) or deuterated (□) water. Data
points represent the peak areas of the GY-OOH relative to control with no
amine (mean ± S.D. of triplicates). All experiments were performed at
least twice. Inset, chromatograms represent the relative abundance of
GY-OOH at 0 mm (solid line) and 10 mm
(dashed line) Lys and were recorded by selective ion monitoring at
m/z = 271. b, relative peak areas of the dioxide
derivatives of HPA (○) and YG (□) at different Lys concentrations and
GY (⋄) at different ethanolamine concentrations. Reaction conditions were
as described under “Experimental Procedures” except:
[acetaldehyde] = 2.5 mm, [peptide] = 1 mm, and [Lys] or
[ethanolamine] = 0–10 mm. Before quantification the enzymes
were removed using 10-kDa microconcentrators. As controls, Lys had no effect
on the rates of production of
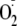 or H2O2 by XO as measured with the cytochrome c
and FOX assays, respectively. Lys and ethanolamine also did not affect the
rate of the HRP-catalyzed oxidation of GY by H2O2 or the
yields of dimer in the actual samples. In deuterated solvents pD was measured
using a glass electrode by adding 0.4 pH values to the pH meter reading. The
activity of XO was measured in D2O when the reactions were carried
out in deuterated media.
or H2O2 by XO as measured with the cytochrome c
and FOX assays, respectively. Lys and ethanolamine also did not affect the
rate of the HRP-catalyzed oxidation of GY by H2O2 or the
yields of dimer in the actual samples. In deuterated solvents pD was measured
using a glass electrode by adding 0.4 pH values to the pH meter reading. The
activity of XO was measured in D2O when the reactions were carried
out in deuterated media.
These observations suggest an alternative mechanism by which the proximity
of amino groups promotes
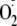 addition to the tyrosyl radical. It is unlikely to be a salt effect or general
acid catalysis as enhancement was observed in 50 mm phosphate
buffer and was not seen when Lys was added to YG. A possible explanation is
that protonated amine groups increase the electrophilicity of
addition to the tyrosyl radical. It is unlikely to be a salt effect or general
acid catalysis as enhancement was observed in 50 mm phosphate
buffer and was not seen when Lys was added to YG. A possible explanation is
that protonated amine groups increase the electrophilicity of
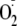 via H-bonding, and this favors the addition reaction. To test this mechanism,
the reaction was carried out in D2O, in which proton exchange
results in deuteration of the Lys amino group. Deuterated Lys, which forms
much weaker hydrogen bonds, gave correspondingly less enhancement of
hydroperoxide formation (Fig.
8a).
via H-bonding, and this favors the addition reaction. To test this mechanism,
the reaction was carried out in D2O, in which proton exchange
results in deuteration of the Lys amino group. Deuterated Lys, which forms
much weaker hydrogen bonds, gave correspondingly less enhancement of
hydroperoxide formation (Fig.
8a).
DISCUSSION
This study has focused on
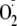 addition to tyrosyl radicals and the relevance of this mechanism to oxidative
modification of peptides and proteins. We have characterized the products with
physiological peptides and present evidence that the preferential formation of
hydroperoxides on the N-terminal Tyr residues is due to adjacent amino groups
interacting with
addition to tyrosyl radicals and the relevance of this mechanism to oxidative
modification of peptides and proteins. We have characterized the products with
physiological peptides and present evidence that the preferential formation of
hydroperoxides on the N-terminal Tyr residues is due to adjacent amino groups
interacting with
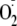 to promote the addition reaction. We have also identified a novel mechanism of
rapid intramolecular transfer of an oxygen from an initial tyrosine
hydroperoxide to a Met residue to form the sulfoxide. The biological relevance
of
to promote the addition reaction. We have also identified a novel mechanism of
rapid intramolecular transfer of an oxygen from an initial tyrosine
hydroperoxide to a Met residue to form the sulfoxide. The biological relevance
of
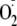 addition products was demonstrated by showing that Leu-Enk and Endomorphin 2
were converted to hydroperoxides and Met-Enk to a dioxide when exposed to a
peroxidase/
addition products was demonstrated by showing that Leu-Enk and Endomorphin 2
were converted to hydroperoxides and Met-Enk to a dioxide when exposed to a
peroxidase/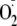 system. With the HRP/XO system we used, more hydroperoxide or dioxide was
formed than dimer. In theory this system generates a maximum of ∼0.5
system. With the HRP/XO system we used, more hydroperoxide or dioxide was
formed than dimer. In theory this system generates a maximum of ∼0.5
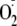 radicals per Tyr
radical,3 so in
situations where
radicals per Tyr
radical,3 so in
situations where
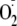 production is high, such as the surroundings of neutrophils, the hydroperoxide
would be even more favored. Therefore, superoxide addition to tyrosyl radicals
should occur physiologically and could result in inactivation of enkephalins
at sites of inflammation. Indeed, stimulated neutrophils use myeloperoxidase
and
production is high, such as the surroundings of neutrophils, the hydroperoxide
would be even more favored. Therefore, superoxide addition to tyrosyl radicals
should occur physiologically and could result in inactivation of enkephalins
at sites of inflammation. Indeed, stimulated neutrophils use myeloperoxidase
and
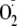 to oxidize enkephalins by this
mechanism.4
to oxidize enkephalins by this
mechanism.4
Structural Characterization of Hydroperoxides as Conjugate Addition Products—The structures of hydroperoxides on N-terminal Tyr residues and the equivalent monoxide on Met-containing peptides were confirmed as bicyclic conjugate addition products. Those detected in much lower yields when Tyr was not N-terminal also appeared to be conjugates, possibly with the amide nitrogen although definite characterization is still required. This is different from the unconjugated hydroperoxides generated by 1O2 on peptides such as GYG (23), but equivalent conjugate addition of an amide nitrogen has been observed for photooxygenation of N-acetyltyramine (30).
Mechanism of Superoxide Addition Versus Radical Repair—Our
data, along with earlier findings
(5), indicate that peptides
with non-N-terminal Tyr undergo more efficient radical repair. Others have
observed for a range of phenoxyl radicals that the contribution of repair
relative to
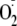 addition decreases progressively with increasing reduction potential of the
phenol (19). This was
explained by the repair reaction occurring by electron transfer (which, in
accordance with the Marcus theory, is favored when the difference in reduction
potential between the two couples is high). On this basis, Tyr and
Tyr-peptides, which all have a reduction potential
(PhO·/PhO–) of ∼0.64 V, should react
predominantly by electron transfer. The small amount of
addition decreases progressively with increasing reduction potential of the
phenol (19). This was
explained by the repair reaction occurring by electron transfer (which, in
accordance with the Marcus theory, is favored when the difference in reduction
potential between the two couples is high). On this basis, Tyr and
Tyr-peptides, which all have a reduction potential
(PhO·/PhO–) of ∼0.64 V, should react
predominantly by electron transfer. The small amount of
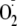 addition to non-N-terminal Tyr peptides fits well with this relationship, but
the efficient addition to N-terminal Tyr is anomalous. To explain this, von
Sonntag and co-workers (18)
proposed that rather than competition between addition and electron transfer,
the phenoxyl and
addition to non-N-terminal Tyr peptides fits well with this relationship, but
the efficient addition to N-terminal Tyr is anomalous. To explain this, von
Sonntag and co-workers (18)
proposed that rather than competition between addition and electron transfer,
the phenoxyl and
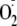 radicals combine to form a transient intermediate that either breaks down to
release oxygen or converts to a stable hydroperoxide
(Fig. 1b, reactions
4 and 5, respectively). The driving force for stabilization of
the hydroperoxide was proposed to be conjugate addition of the terminal amine.
Our data do not support this mechanism on two counts. We saw no formation of
1O2, which should have been released in reaction
4 to obey the spin conservation rule. Also, if conjugate addition were
the stabilizing factor, enhancement of hydroperoxide formation by exogenous
amines should be associated with conjugation of the amine to the tyrosyl ring.
This was not seen with GY or HPA even though the amines increased
hydroperoxide yields.
radicals combine to form a transient intermediate that either breaks down to
release oxygen or converts to a stable hydroperoxide
(Fig. 1b, reactions
4 and 5, respectively). The driving force for stabilization of
the hydroperoxide was proposed to be conjugate addition of the terminal amine.
Our data do not support this mechanism on two counts. We saw no formation of
1O2, which should have been released in reaction
4 to obey the spin conservation rule. Also, if conjugate addition were
the stabilizing factor, enhancement of hydroperoxide formation by exogenous
amines should be associated with conjugation of the amine to the tyrosyl ring.
This was not seen with GY or HPA even though the amines increased
hydroperoxide yields.
We propose an alternative mechanism that accounts for the Tyr anomaly. In
this mechanism the two radicals react either by electron transfer or radical
addition as proposed (19), but
hydrogen bonding to an amine group alters the reactivity of
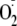 so as to favor the addition pathway. This mechanism is consistent with our
findings that Lys promoted less hydroperoxide formation when it was
deuterated. It is also supported by experimental evidence of hydrogen bonding
of
so as to favor the addition pathway. This mechanism is consistent with our
findings that Lys promoted less hydroperoxide formation when it was
deuterated. It is also supported by experimental evidence of hydrogen bonding
of
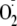 to protonated amines (31,
32), and calculations showing
that hydrogen bonding to N-H groups alters its spin and charge densities
(31,
33). The effect of hydrogen
bonding is to increase the electrophilicity and reduction potential of the
O2/
to protonated amines (31,
32), and calculations showing
that hydrogen bonding to N-H groups alters its spin and charge densities
(31,
33). The effect of hydrogen
bonding is to increase the electrophilicity and reduction potential of the
O2/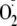 couple. The impact on the
couple. The impact on the
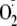 /phenoxyl
radical reaction would be to decrease the potential difference between the two
redox couples, and as reasoned above
(19), favor the radical
addition pathway. Thus, promoting
/phenoxyl
radical reaction would be to decrease the potential difference between the two
redox couples, and as reasoned above
(19), favor the radical
addition pathway. Thus, promoting
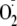 addition through hydrogen bonding to the terminal amino group would explain
the Tyr anomaly. The greater hydroperoxide formation with N-terminal Tyr than
with exogenous amines implies that hydrogen bonding provides more effective
promotion of the addition pathway when the reaction is intramolecular. This
could be explained by a lowering of transitional entropy
(34).
addition through hydrogen bonding to the terminal amino group would explain
the Tyr anomaly. The greater hydroperoxide formation with N-terminal Tyr than
with exogenous amines implies that hydrogen bonding provides more effective
promotion of the addition pathway when the reaction is intramolecular. This
could be explained by a lowering of transitional entropy
(34).
The amine effect could represent rate enhancement of the radical-radical
reaction or a change in preference for addition over electron transfer. As the
rates are almost diffusion controlled and rate constants show only minor
differences between phenols that add or transfer electrons
(19), the latter explanation
is more likely. It is further supported by our pulse radiolysis measurements
of similar rate constants for the reaction of
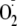 with free Tyr and Tyr peptides (1.5 × 109
m–1 s–1) regardless of whether or
not they form
hydroperoxides.5
Although our results fit with this mechanism, we cannot exclude the
possibility that there is initial formation of an associative intermediate
between superoxide and tyrosyl radicals and then interaction of this
intermediate with an amine to favor its breakdown by addition rather than
electron transfer.
with free Tyr and Tyr peptides (1.5 × 109
m–1 s–1) regardless of whether or
not they form
hydroperoxides.5
Although our results fit with this mechanism, we cannot exclude the
possibility that there is initial formation of an associative intermediate
between superoxide and tyrosyl radicals and then interaction of this
intermediate with an amine to favor its breakdown by addition rather than
electron transfer.
Reaction with
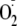 should be a favored reaction for tyrosyl radicals generated on proteins and a
likely route for formation of protein hydroperoxides. Interaction with amino
groups should have a major influence on where addition as against repair of
the Tyr residue occurs, with peroxides formed not only when Tyr is N-terminal
but also when it is favorably aligned to amino groups that could promote the
reaction.
should be a favored reaction for tyrosyl radicals generated on proteins and a
likely route for formation of protein hydroperoxides. Interaction with amino
groups should have a major influence on where addition as against repair of
the Tyr residue occurs, with peroxides formed not only when Tyr is N-terminal
but also when it is favorably aligned to amino groups that could promote the
reaction.
Intramolecular Oxygen Transfer to Methionine—The finding that peptides containing Met did not form stable hydroperoxides was unexpected, but led us to identify an intramolecular oxygen transfer mechanism for methionine sulfoxide formation. We propose that the reaction proceeds through an initial hydroperoxide. The formation of a stable hydroperoxide on preformed YM-S=O, when the oxygen transfer route is blocked, plus the 5-fold higher yield of intramolecular oxygen transfer product with Met-Enk than Gly-Met-Enk provide support for this mechanism. Also, oxidation of free methionine by Leu-Enk hydroperoxide was much too slow for intermolecular transfer to account for the reaction. Electron transfer between Met and Tyr residues has been observed in situations where Met residues are oxidized by a 1-electron mechanism (10, 12, 13). The electron transfer is in the opposite direction to oxygen transfer, regenerating Met from its radical and forming a Tyr radical. Our findings are not explained by this process and, as far as we are aware, represent a previously unrecognized mechanism of oxygen transfer. It has some similarity to the oxidation of Met residues in apoA by lipid hydroperoxides, which is facilitated by the presence of both reactants in high density lipoprotein (35, 36). We observed oxygen transfer in peptides with little discrimination for the relative positions of the Tyr and Met. The chances of it occurring in proteins must therefore be high. Proximity to Tyr in the tertiary structure might give preference to modification of particular Met residues.
There has been one other study of the reaction of
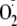 with the tyrosyl radical of Met-Enk
(20). Using radiolytic
methods, these authors measured a reaction rate with
with the tyrosyl radical of Met-Enk
(20). Using radiolytic
methods, these authors measured a reaction rate with
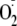 similar to that reported for other phenoxyl radicals. They detected a
dioxygenated product but did not show MS analysis and assumed it to be a
hydroperoxide. They concluded that no oxygenated products were formed from
N-terminal blocked Boc-Met-Enk, although the main peak in the mass spectrum of
their reaction mixture (m/z 706.2, which they did not
comment on) has the mass of a dioxygenated species.
similar to that reported for other phenoxyl radicals. They detected a
dioxygenated product but did not show MS analysis and assumed it to be a
hydroperoxide. They concluded that no oxygenated products were formed from
N-terminal blocked Boc-Met-Enk, although the main peak in the mass spectrum of
their reaction mixture (m/z 706.2, which they did not
comment on) has the mass of a dioxygenated species.
Physiological Significance—Superoxide and tyrosyl radicals
are among the most prevalent radicals generated biologically during oxidative
stress. Reaction between the two is highly favored. We have shown with
tyrosine and the enkephalins that this results in the formation of additional
products in greater yields than dityrosine derivatives, the tyrosine oxidation
products that generally receive most attention in relation to oxidative
injury. We have characterized the tyrosine hydroperoxides formed and proposed
a mechanism involving hydrogen bonding to amino groups that explains why
formation is favored on N-terminal Tyr. The repair reaction that predominates
when these conditions are not met does not release 1O2.
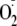 addition to tyrosyl peptides that also contain Met and subsequent
intramolecular oxygen transfer may represent an unrecognized mechanism of
methionine sulfoxide formation in proteins under conditions of oxidative
stress. Interaction of
addition to tyrosyl peptides that also contain Met and subsequent
intramolecular oxygen transfer may represent an unrecognized mechanism of
methionine sulfoxide formation in proteins under conditions of oxidative
stress. Interaction of
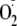 with neighboring amines could be important for determining structural
selectivity of
with neighboring amines could be important for determining structural
selectivity of
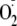 addition to Tyr residues on proteins. Moreover, the proposed mechanism,
whereby the electrophilicity of
addition to Tyr residues on proteins. Moreover, the proposed mechanism,
whereby the electrophilicity of
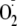 is increased through hydrogen bonding, has wide biological implications. This
would bring the reactivity of
is increased through hydrogen bonding, has wide biological implications. This
would bring the reactivity of
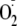 closer to that of the more oxidizing hydroperoxyl radical and should increase
its ability to react with more biological substrates. The relevance of this
mechanism to
closer to that of the more oxidizing hydroperoxyl radical and should increase
its ability to react with more biological substrates. The relevance of this
mechanism to
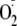 biology warrants further investigation.
biology warrants further investigation.
Supplementary Material
Acknowledgments
We are grateful to Henk Wang and Tim Harwood who performed some of the initial enkephalin analyses and Prof. Paolo Di Mascio for his generous gift of EAS.
This work was supported by the Marsden Fund and the Health Research Council of New Zealand and used equipment provided by the National Research Centre for Growth and Development.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S6 and additional data.
Footnotes
The abbreviations used are: SOD, superoxide dismutase; YM, tyrosinemethionine; YM-S=O, tyrosine-methionine sulfoxide; MY, methionine-tyrosine; YG, tyrosine-glycine; GY, glycine-tyrosine; YGGFM, Met-Enk, methionine-enkephalin; YGGFL, Leu-Enk, leucine-enkephalin; YPFF, Endo2, endomorphin 2; GYGGFM, Gly-Met-Enk, glycine-methionine-enkephalin; Boc-YGGFM, tert-butoxycarbonyl-methionine-enkephalin; RFYVVM, thrombospondin-1 (1016–1021); YSFKDMGLGR, human C5a anaphylatoxin (Tyr65, Phe67)-C5a (65–74); MEVDPIGHLY, MAGE-3 antigen (167–176); HPA, p-hydroxyphenylacetic acid; HOHICA, 3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid; HACHD, 4-alanyl-4-hydroxy-cyclohexadienone; XO, xanthine oxidase; HRP, horseradish peroxidase; LC/MS, liquid chromatography/electrospray mass spectrometry; ESI, electrospray ionization.
The reduction of oxygen by xanthine oxidase is ∼30% by a 1-electron
route to
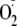 ,
with the remainder reduced directly to H2O2. If all the
H2O2 reacted with Tyr to generate Tyr radicals and the
,
with the remainder reduced directly to H2O2. If all the
H2O2 reacted with Tyr to generate Tyr radicals and the
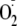 did not dismutate to give more H2O2, then there would be
approximately 70 Tyr radicals generated per 30
did not dismutate to give more H2O2, then there would be
approximately 70 Tyr radicals generated per 30
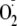 .
.
P. Nagy, A. J. Kettle, and C. C. Winterbourn, unpublished results.
P. Nagy and T. Nauser, unpublished data.
References
- 1.Imlay, J. A. (2008) Annu. Rev. Biochem. 77 755–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winterbourn, C. C. (2008) Nat. Chem. Biol. 4 278–286 [DOI] [PubMed] [Google Scholar]
- 3.Liochev, S. I., and Fridovich, I. (1999) IUBMB Life 48 157–161 [DOI] [PubMed] [Google Scholar]
- 4.Beckman, J. S., and Koppenol, W. H. (1996) Am. J. Physiol. 271 C1424–C1437 [DOI] [PubMed] [Google Scholar]
- 5.Winterbourn, C. C., Parsons-Mair, H. N., Gebicki, S., Gebicki, J. M., and Davies, M. J. (2004) Biochem. J. 381 241–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winterbourn, C. C., and Kettle, A. J. (2003) Biochem. Biophys. Res. Commun. 305 729–736 [DOI] [PubMed] [Google Scholar]
- 7.Jin, F., Leitich, J., and von Sonntag, C. (1993) J. Chem. Soc. Perkin Trans. II, 1583–1588
- 8.Davies, M. J. (2005) Biochim. Biophys. Acta 1703 93–109 [DOI] [PubMed] [Google Scholar]
- 9.Dean, R. T., Fu, S. L., Stocker, R., and Davies, M. J. (1997) Biochem. J. 324 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bobrowski, K., Wierzchowski, K. L., Holcman, J., and Ciurak, M. (1990) Int. J. Radiat. Biol. 57 919–932 [DOI] [PubMed] [Google Scholar]
- 11.Garrison, W. M. (1987) Chem. Rev. 87 381–398 [Google Scholar]
- 12.Prutz, W. A., Siebert, F., Butler, J., Land, E. J., Menez, A., and Montenaygarestier, T. (1982) Biochim. Biophys. Acta 705 139–149 [Google Scholar]
- 13.Zhang, H., Zielonka, J., Sikora, A., Joseph, J., Xu, Y., and Kalyanaraman, B. (2008) Arch. Biochem. Biophys. 10.1016/j.abb.2008.11.018 [DOI] [PMC free article] [PubMed]
- 14.Stubbe, J. A., and van der Donk, W. A. (1998) Chem. Rev. 98 705–762 [DOI] [PubMed] [Google Scholar]
- 15.Beal, M. F. (2002) Free Radic. Biol. Med. 32 797–803 [DOI] [PubMed] [Google Scholar]
- 16.Heinecke, J. W., Li, W., Francis, G. A., and Goldstein, J. A. (1993) J. Clin. Investig. 91 2866–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Souza, J. M., Peluffo, G., and Radi, R. (2008) Free Radic. Biol. Med. 45 357–366 [DOI] [PubMed] [Google Scholar]
- 18.d'Allesandro, N., Bianchi, G., Fang, X., Jin, F., Schuchmann, H.-P., and von Sonntag, C. (2000) J. Chem. Soc. Perkin Trans. II, 1862–1867
- 19.Jonsson, M., Lind, T., Reitberger, T. E., Eriksen, T. E., and Merenyi, G. (1993) J. Phys. Chem. 97 8229–8233 [Google Scholar]
- 20.Mozziconacci, O., Mirkowski, J., Rusconi, F., Pernot, P., Bobrowski, K., and Houee-Levin, C. (2007) Free Radic. Biol. Med. 43 229–240 [DOI] [PubMed] [Google Scholar]
- 21.Stadtman, E. R., and Levine, R. L. (2003) Amino Acids 25 207–218 [DOI] [PubMed] [Google Scholar]
- 22.Moskovitz, J., Flescher, E., Berlett, B. S., Azare, J., Poston, J. M., and Stadtman, E. R. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 14071–14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wright, A., Bubb, W. A., Hawkins, C. L., and Davies, M. J. (2002) Photochem. Photobiol. 76 35–46 [DOI] [PubMed] [Google Scholar]
- 24.Wolff, S. P. (1994) Methods Enzymol. 233 182–189 [DOI] [PubMed] [Google Scholar]
- 25.Gieseg, S. P., Simpson, J. A., Charlton, T. S., Duncan, M. W., and Dean, R. T. (1993) Biochemistry 32 4780–4786 [DOI] [PubMed] [Google Scholar]
- 26.Winterbourn, C. C., Pichorner, H., and Kettle, A. J. (1997) Arch. Biochem. Biophys. 338 15–21 [DOI] [PubMed] [Google Scholar]
- 27.Pichorner, H., Metodiewa, D., and Winterbourn, C. C. (1995) Arch. Biochem. Biophys. 323 429–437 [DOI] [PubMed] [Google Scholar]
- 28.Jiang, X. Y., Smith, J. B., and Abraham, E. C. (1996) J. Mass Spectrom. 31 1309–1310 [Google Scholar]
- 29.Miyamoto, S., Martinez, G. R., Rettori, D., Augusto, O., Medeiros, M. H. G., and Di Mascio, P. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 293–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito, I., Chujo, Y., Shimazu, H., Yamane, M., Matsuura, T., and Cahnmann, H. J. (1975) J. Am. Chem. Soc. 97 5272–5277 [DOI] [PubMed] [Google Scholar]
- 31.Villamena, F. A., Xia, S., Merle, J. K., Lauricella, R., Tuccio, B., Hadad, C. M., and Zweier, J. L. (2007) J. Am. Chem. Soc. 129 8177–8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ohkubo, K., Kitaguchi, H., and Fukuzumi, S. (2006) J. Phys. Chem. A 110 11613–11616 [DOI] [PubMed] [Google Scholar]
- 33.Field, S. M., and Villamena, F. A. (2008) Chem. Res. Toxicol. 21 1923–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bruice, T. C. (1976) Annu. Rev. Biochem. 45 331–373 [DOI] [PubMed] [Google Scholar]
- 35.Garner, B., Witting, P. K., Waldeck, A. R., Christison, J. K., Raftery, M., and Stocker, P. (1998) J. Biol. Chem. 273 6080–6087 [DOI] [PubMed] [Google Scholar]
- 36.Garner, B., Waldeck, A. R., Witting, P. K., Rye, K. A., and Stocker, P. (1998) J. Biol. Chem. 273 6088–6095 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.