

Abstract
The incorporation of cobalt into low molecular mass nitrile hydratase
(L-NHase) of Rhodococcus rhodochrous J1 has been found to depend on
the α-subunit exchange between cobalt-free L-NHase (apo-L-NHase lacking
oxidized cysteine residues) and its cobalt-containing mediator (holo-NhlAE
containing 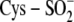 and
Cys-SO- metal ligands), this novel mode of post-translational
maturation having been named self-subunit swapping, and NhlE having been
recognized as a self-subunit swapping chaperone (Zhou, Z., Hashimoto, Y.,
Shiraki, K., and Kobayashi, M. (2008) Proc. Natl. Acad. Sci. U. S. A.
105, 14849–14854). We discovered here that cobalt was inserted into both
the cobalt-free NhlAE (apo-NhlAE) and the cobalt-free α-subunit
(apo-α-subunit) in an NhlE-dependent manner in the presence of cobalt
and dithiothreitol in vitro. Matrix-assisted laser desorption
ionization time-of-flight mass spectroscopy analysis revealed that the
non-oxidized cysteine residues in apo-NhlAE were post-translationally oxidized
after cobalt insertion. These findings suggested that NhlE has two activities,
i.e. cobalt insertion and cysteine oxidation. NhlE not only functions
as a self-subunit swapping chaperone but also a metallochaperone that includes
a redox function. Cobalt insertion and cysteine oxidation occurred under both
aerobic and anaerobic conditions when Co3+ was used as a cobalt
donor, suggesting that the oxygen atoms in the oxidized cysteines were derived
from water molecules but not from dissolved oxygen. Additionally, we isolated
apo-NhlAE after the self-subunit swapping event and found that it was recycled
for cobalt transfer into L-NHase.
and
Cys-SO- metal ligands), this novel mode of post-translational
maturation having been named self-subunit swapping, and NhlE having been
recognized as a self-subunit swapping chaperone (Zhou, Z., Hashimoto, Y.,
Shiraki, K., and Kobayashi, M. (2008) Proc. Natl. Acad. Sci. U. S. A.
105, 14849–14854). We discovered here that cobalt was inserted into both
the cobalt-free NhlAE (apo-NhlAE) and the cobalt-free α-subunit
(apo-α-subunit) in an NhlE-dependent manner in the presence of cobalt
and dithiothreitol in vitro. Matrix-assisted laser desorption
ionization time-of-flight mass spectroscopy analysis revealed that the
non-oxidized cysteine residues in apo-NhlAE were post-translationally oxidized
after cobalt insertion. These findings suggested that NhlE has two activities,
i.e. cobalt insertion and cysteine oxidation. NhlE not only functions
as a self-subunit swapping chaperone but also a metallochaperone that includes
a redox function. Cobalt insertion and cysteine oxidation occurred under both
aerobic and anaerobic conditions when Co3+ was used as a cobalt
donor, suggesting that the oxygen atoms in the oxidized cysteines were derived
from water molecules but not from dissolved oxygen. Additionally, we isolated
apo-NhlAE after the self-subunit swapping event and found that it was recycled
for cobalt transfer into L-NHase.
Nitrile hydratase (NHase3; EC 4.2.1.84) (1, 2), which is composed of α- and β-subunits, contains either a non-heme iron (3, 4) or a noncorrin cobalt ion (5–7) in a ligand environment that includes two oxidized cysteine residues (CXLC(SO2H)SC(SOH)) (8–12). The enzyme catalyzes the hydration of a nitrile to the corresponding amide followed by consecutive reactions: amide → acid → acyl-CoA by amidase (13) and acyl-CoA synthetase (14, 15), respectively. Rhodococcus rhodochrous J1 produces high and low molecular mass NHases (H-NHase and L-NHase), which exhibit different physicochemical properties and substrate specificities (1, 16). In both H- and L-NHase, cobalt acts as an active center for the production of acrylamide and nicotinamide. Acrylamide is manufactured at the industrial level not only in Japan but also in the United States and France (17, 18).
Metalloproteins have been characterized intensively for decades yet only recently have investigators focused on the mechanisms underlying biological metallocenter assembly (19). The synthesis of some metalloproteins has been found to require the participation of accessory proteins (19). An open reading frame, nhlE, which is just downstream of the structural genes of L-NHase (nhlBA), is necessary for functional L-NHase maturation (20). In contrast to the cobalt-containing cysteine-oxidized L-NHase (holo-α2β2) derived from nhlBAE, the gene product derived from nhlBA is a non-oxidized cobalt-free apo-L-NHase (20). An L-NHase maturation mediator, NhlAE (encoded by the nhlAE genes, and consisting of αe2 containing the cobalt-containing cysteine-oxidized α-subunit of L-NHase), has been discovered, and the incorporation of cobalt into L-NHase has been found to depend on the α-subunit exchange between apo-L-NHase and NhlAE. This is a novel post-translational maturation process different from general mechanisms of metallocenter biosynthesis known so far. Thus, we named it self-subunit swapping (Fig. 1(i)) (20). Compared with activators acting as metallochaperones in Fe-NHases (21) from Rhodococcus sp. N-771 (22), Pseudomonas putida 5B (23), Rhodococcus sp. N-774 (24), and so on, NhlE acts as a self-subunit swapping chaperone (Fig. 1(i)), exhibiting novel behavior for a protein in a protein complex (20).
FIGURE 1.

Incorporation of a cobalt ion into l-NHase. (i) Self-subunit swapping maturation of L-NHase. (ii) Insertion of a cobalt ion into apo-NhlAE. The cobalt ion is shown as a closed circle. The holo-α-subunit is shown in gray. Dotted lines with arrows indicate α-subunit swapping.
Metal ions in both Fe-NHase and Co-NHase are located in their α-subunits, which share a characteristic metal binding motif (CXLC(SO2H)SC(SOH)) containing two oxidized cysteine residues: cysteine-sulfinic acid (αCys-SO2H) and cysteine-sulfenic acid (αCys-SOH) (8–12). The post-translationally oxidized Cys-SO2H and Cys-SOH have deprotonated Cys-SO-2 and Cys-SO- structures, respectively (3), and the deprotonated Cys-SO-2 and Cys-SO- in the holo-α-subunit form salt bridges with two arginines of the β-subunit (which are conserved in all known Co-type and Fe-type NHases) in the holoenzyme (8–11). The apoenzyme is likely to be non-oxidized, judging from the results of previous studies on NHase (25) and the related enzyme thiocyanate hydrolase (26–28). Such cysteine oxidation has been demonstrated to occur in cobalt-containing NhlAE (holo-αe2), but not in cobalt-free NhlAE (apo-αe2), and the cysteine oxidation plays an essential role in self-subunit swapping maturation (20).
Cysteine oxidation in proteins is receiving increasing attention due to its important physiological properties. Oxidized cysteine residues Cys-SO2H or Cys-SOH are known to play roles in diverse processes, including signal transduction, oxygen metabolism and the oxidative stress response, transcription regulation, and metal coordination in various proteins such as NADH peroxidase (29, 30), peroxiredoxins (31), hydrogenase (32, 33), and so on (8–12, 26–28, 34). Among these enzymes, NHase and thiocyanate hydrolase are intriguing ones because they possess both Cys-SO2H and Cys-SOH as ligands of the metal center and neither residue plays any catalytic redox role at all (29). Although the oxidized cysteine residues in both enzymes are essential for their activities (20, 34), the mechanism(s) involved in the post-translational oxidation of both residues remains unclear.
Non-corrin cobalt enzymes like NHase are receiving increasing interest not only in bioinorganic chemistry but also in biotechnology, and their availability and remarkable chemical versatility make them invaluable catalysts in the chemical industry (6, 35–37). Although we have discovered a cobalt transporter (NhlF) that mediates cobalt uptake into the cell (38), the mechanism underlying the insertion of a cobalt into a noncorrin-cobalt-containing protein in the cell has never been clarified. In this study we discovered that cobalt was directly inserted into a non-oxidized apo-α-subunit on the addition of NhlE, which yielded a cobalt-containing cysteine-oxidized NhlAE in vitro, suggesting that NhlE acts as a metallochaperone that is crucial for both post-translational cysteine oxidation and cobalt incorporation into the α-subunit of L-NHase (Fig. 1(ii)).
EXPERIMENTAL PROCEDURES
Bacterial Strain and Plasmid Used—Rhodococcus fascians DSM43985 was used as the host for vector plasmid pREIT19, which was used for nhlBAE, nhlAE, and nhlBA-(α-A3G) expression as described previously (20).
Culture Conditions—The R. fascians DSM43985 transformants carrying pREIT-nhlAE for holo-αe2 expression were grown at 28 °C for 72 h in 2YT medium containing CoCl2·6H2O (0.1 g/liter) and kanamycin (50 μg/ml), and 0.1% (v/v) of isovaleronitrile, as an inducer, was added to the medium after incubation for 12 h. R. fascians DSM43985 transformants carrying pREIT-nhlBAE were grown under the same conditions for 96 h except that the inducer was continuously added every 24 h for a total of 4 times to increase the amount of L-NHase expressed. The R. fascians DSM43985 transformants carrying pREIT-nhlAE, pREIT-nhlBAE, and pREIT-nhlBA-(α-A3G) used for apoprotein expression were incubated under the same conditions except that CoCl2·6H2O was not added to the medium.
Protein Purification—Proteins were purified in the same manner as that described previously (20) except that 10 mm potassium phosphate buffer (KPB) (pH 7.5) containing 0.5 mm dithiothreitol (DTT) was used in all purification steps. No influence on L-NHase activity was observed under these purification conditions. The apo-L-NHases purified under these conditions were mostly of the heterotetramer form (apo-α2β2), and only little heterodimer forms (apo-αβ) were detected. Thus, the purified apo-α2β2 was used as the apo-L-NHase in this study.
In Vitro Activation of Apo-α2β2—The routine apo-α2β2 activation buffer consisted of 10 mm KPB (pH 7.5), 10 μm cobalt, and 2 mm DTT unless otherwise noted. The final concentrations of apo-α2β2 and apo-αe2 in the activation buffer were 0.1 and 0.4 mg/ml, respectively. A CoCl2 solution was used as a cobalt donor unless otherwise noted. The mixtures were incubated at 28 °C.
Purification of Resultant Proteins—Hiload 16/60 Superdex 200-pg (GE Healthcare) and Resource Q (6 ml) (GE Healthcare) columns were used to purify the resultant L-NHases and NhlAEs from mixtures in the same way as described previously (20) except that 10 mm KPB (pH 7.5) containing 0.5 mm DTT was used.
Enzyme Assay—L-NHase activity was assayed in a reaction mixture (0.5 ml) comprising 10 mm KPB (pH 7.5), 20 mm 3-cyanopyridine, and 10 μl of enzyme containing activation buffer or an appropriate amount of the enzyme. The reaction was carried out at 20 °C for 20 min and stopped by the addition of 0.5 ml of acetonitrile. The amount of nicotinamide formed in the reaction mixture was determined as described previously (20). One unit of L-NHase activity was defined as the amount of enzyme that catalyzed the release of 1 μmol of nicotinamide per min at 20 °C.
Denaturation of NhlAE and Renaturation of the α-Subunit and NhlE—The denaturation and renaturation experiments were carried out at room temperature in 20 mm Tris-HCl buffer (pH 7.5). SDS was carefully added to NhlAE, which had been mixed with 2-mercaptoethanol. The concentrations of resultant apo-αe2 (R-apo-αe2) and SDS were both 0.2 mg/ml, and that of 2-mercaptoethanol was 10% (v/v) (final, respectively). After 2 h, the α-subunit and NhlE were purified by gel filtration on a Hiload 16/60 Superdex 200-pg column equilibrated with 20 mm Tris-HCl buffer (pH 7.5) containing 1% SDS. After dialysis, the SDS remaining with the α-subunit was eliminated with a Resource Q column (6 ml) equilibrated with 20 mm Tris-HCl buffer (pH 7.5), and the protein was eluted with 0.5 liters of the buffer by increasing the concentration of KCl linearly from 0 to 0.5 m. This step was carried out twice to isolate the α-subunit. These two procedures were carried out with an AKTA purifier (GE Healthcare). Gel filtration chromatograph analysis was performed for molecular mass determination, the flow rate being 0.5 ml/min. A Superose 12 HR 10/30 column (GE Healthcare) and 0.2 m KCl-containing buffer were used for estimation of the molecular masses of the purified α-subunits and NhlE.
Matrix-assisted Laser Desorption Ionization Time-of-flight Mass Spectrometry (MALDI-TOF MS) Sequencing—MALDI-TOF MS was typically performed using a Voyager-DE STR mass spectrometer (Applied Biosystems) in the linear positive mode. The purified α-subunit derived from R-apo-αe2 was reduced with 1 mm DTT and carboxamidomethylated with 2.1 mm iodoacetamide and then diluted with 3 volumes of 50 mm Tris-HCl (pH 8.0) and treated with trypsin at 35 °C for 20 h. The details were given previously (20).
Other Analytical Methods—CD measurement, UV-Visible Measurement, and Cobalt Ion Determination Were Described Previously (20).
RESULTS
Post-translational Activation of Apo-L-NHase by Apo-αe2 in the Presence of Cobalt in Vitro—Previous work (20) has shown that apo-L-NHase is converted into holo-α2β2 after mixing with holo-αe2 through self-subunit swapping maturation. To determine whether cobalt could be directly incorporated into apo-L-NHase to yield holo-L-NHase, we mixed apo-L-NHase (apo-α2β2, see “Experimental Procedures”) with cobalt (20 μm, final) in 10 mm KPB (pH 7.5) and then measured the L-NHase activity in the mixture. As shown in Fig. 2, no significant increase in L-NHase activity was observed. However, we found that L-NHase activity increased when apo-αe2 was mixed in additionally and that the L-NHase activity significantly increased when 0.5 mm DTT was further added to this mixture (Fig. 2). These findings indicated that apo-α2β2 was post-translationally activated by apo-αe2 in the presence of cobalt and that the post-translational activation was enhanced by the addition of DTT in vitro. Subsequently, the effect of the DTT concentration on activation of apo-α2β2 by apo-αe2 in the presence of cobalt was investigated. Mixtures were incubated with various concentrations of DTT, and then the L-NHase activity in each activation mixture at 12 h was measured. The L-NHase activity in the activation mixtures became higher as the DTT concentration increased, but more than 4 mm DTT caused a decrease in activation competence (see Fig. 3A and “Discussion”). These findings suggest that a certain amount of DTT is necessary for activation of apo-α2β2 by apo-αe2 in the presence of cobalt and that the suitable concentration of DTT is 2 mm. Thereafter, the effect of the cobalt concentration on activation of apo-α2β2 was investigated with this suitable DTT concentration. The L-NHase activity in the activation mixtures reached a plateau with 10 μm cobalt added (Fig. 3B). Thus, an activation mixture containing 2 mm DTT and 10 μm cobalt provided the optimal conditions and, thus, was used in the following experiments.
FIGURE 2.

Apo-αe2-dependent activation of apo-α2β2. Apo-α2β2 was incubated in 10 mm KPB (pH 7.5) containing 20 μm CoCl2 and apo-αe2, apo-αe2 plus 0.5 mm DTT, or 0.5 mm DTT. Aliquots of the samples were removed at the indicated times and then assayed for L-NHase activity. Values represent the means ± S.D. for at least triplicate independent experiments.
FIGURE 3.

Activation of apo-α2β2 by apo-αe2 under various conditions in vitro. A, effect of the DTT concentration on apo-α2β2 activation. Apo-α2β2 was mixed with apo-αe2 in the activation buffer containing 20 μm CoCl2 and 0, 0.5, 1, 2, 4, 6, 8 or 10 mm DTT. B, effect of the cobalt concentration on apo-α2β2 activation. Apo-α2β2 was mixed with apo-αe2 in the activation buffer containing 2 mm DTT and 0, 5, 10, 20, 40, 60, or 80 μm CoCl2. Aliquots of the samples were removed at 12 h and then assayed for L-NHase activity. Values represent the means ± S.D. for at least triplicate independent experiments.
Direct Incorporation of Cobalt into Apo-αe2—Site-directed mutagenesis and N-terminal amino acid sequence analyses were performed to confirm the source of the α-subunit in the active L-NHase derived from the mixture of apo-α2β2, apo-αe2, cobalt, and DTT. As a result, the source of the α-subunit in the active L-NHase was found to be the apo-αe2 (supplemental Fig. S1 and Table S1), demonstrating that the activation of apo-α2β2 was dependent on self-subunit swapping. We have already demonstrated that self-subunit swapping occurs between apo-α2β2 and holo-αe2 but not between apo-α2β2 and apo-αe2 (20). Considering these findings, we speculated that cobalt should be first inserted into the cobalt-free α-subunit (apo-α-subunit) of apo-αe2 to yield the cobalt-containing α-subunit (holo-α-subunit), resulting in the formation of holo-αe2, and then self-subunit swapping occurs between apo-α2β2 and this holo-αe2 to yield holo-α2β2. To test this hypothesis, we mixed apo-αe2 with cobalt and DTT in the absence of apo-α2β2. The R-apo-αe2 was purified after incubation for 4 h (Fig. 4A) and then compared with apo-αe2 and holo-αe2. The results of cobalt content determination showed that R-apo-αe2 contained 0.98 ± 0.08 mol ion/mol of αe2, similar to the content of holo-αe2 (Table 1). Although R-apo-αe2, holo-αe2, and apo-αe2 showed similar CD spectra (Fig. 4B), the UV-visible spectrum of R-apo-αe2 was similar to that of holo-αe2 but not to that of apo-αe2 (Fig. 4C). It has been reported that the absorption in the 300–350-nm region for Co-NHase reflects S → Co3+ charge transfer (7, 20). As shown in Fig. 4C, an extra shoulder in the 300–350-nm region was found for R-apo-αe2 as well as holo-αe2, suggesting that the cobalt-ligand environment of R-apo-αe2 is the same as those of holo-αe2 and Co-NHases. On the other hand, we also mixed apo-α2β2 with cobalt and DTT in the absence of apo-αe2. The resultant apo-α2β2 (R-apo-α2β2) was purified after incubation for 4 h and then compared with apo-α2β2 and holo-α2β2. The UV-visible spectrum of R-apo-α2β2 was similar to that of apo-α2β2, but not to that of holo-α2β2, and no extra shoulder in the 300–350-nm region was observed (supplemental Fig. S1). In contrast to the cobalt content of holo-α2β2 (0.88 ± 0.03 mol/mol of αβ), only 0.16 ± 0.03 mol cobalt ion/mol of αβ was detected in R-apo-α2β2 (Table 1). These findings demonstrated that a cobalt ion was directly incorporated into the apo-α-subunit of apo-αe2 (Fig. 1 (ii)) but not into that of apo-α2β2 in vitro.
FIGURE 4.

Characterization of purified holo-αe2, apo-αe2, R-apo-αe2, and R-(α+e2). SDS/PAGE (A), far-UV CD (B), and UV-visible (C) spectra of the purified holo-αe2, apo-αe2, R-apo-αe2 and R-(α+e2).
TABLE 1.
Characterization of the purified NhlAEs, L-NHases, α-subunits, R-NhlAEs, R-L-NHases, and R-α-subunit Values represent the means ± S.D. for at least triplicate independent experiments.
| Protein | Cobalt content | Specific activity |
|---|---|---|
| mol of ions/mol of protein | units/mg | |
| Apo-αe2 | 0.02 ± 0.01/αe2 | 0 |
| R-apo-αe2 | 0.98 ± 0.08/αe2 | 0 |
| Holo-αe2 | 0.85 ± 0.03/αe2 | 0 |
| Apo-α2β2 | 0.02 ± 0.01/αβ | 4.16 ± 0.42 |
| R-apo-α2β2 | 0.16 ± 0.03/αβ | 20.6 ± 3.4 |
| Holo-α2β2 | 0.88 ± 0.03/αβ | 345 ± 12 |
| Holo-α | 0.84 ± 0.03/α | 0 |
| Apo-α | 0.02 ± 0.01/α | 0 |
| R-apo-α | 0.14 ± 0.03/α | 0 |
| R-(α+e2) | 0.90 ± 0.05/αe2 | 0 |
| R-holo-αe2 | 0.05 ± 0.02/αe2 | 0 |
| R-apo-α2β2a | 0.95 ± 0.07/αβ | 270 ± 10 |
The resultant L-NHase purified from the mixture of R-holo-αe2, apo-α2β2, cobalt, and DTT
Cysteine Oxidation in R-apo-αe2—We discovered here that cobalt was directly inserted into apo-αe2 (Fig. 4 and Table 1). This finding permitted us to speculate that the cysteine residues could be oxidized in R-apo-αe2. We analyzed R-apo-αe2 by MALDI-TOF MS. During such analysis cysteine residues and Cys-114-SO- (if present) would be reduced and carboxamidomethylated (CAM) as well as disulfide-bonded cysteine residues. The α-subunit (R-apo-α-subunit) isolated from R-apo-αe2 was treated with trypsin after reduction and carboxamidomethylation. The molecular mass of the tryptic peptide containing all metal ligand residues, 83EMGVGGMQGEEMVVLENTGTVHNMVVCTLCSCYPWPVLGLPPNWYK128 (EK46), was measured. The MALDI-TOF MS spectrum of EK46 (Fig. 5) of the R-apo-α-subunit was compared with those of the apo-α-subunit isolated from apo-αe2 and the holo-α-subunit isolated from holo-αe2 (20). Considering the mass peak with an m/z of 5242.4 (Fig. 5, inset) corresponding to the calculated m/z value of the [M+H]+ ion of EK46 with three CAM-cysteines and the mass peak with an m/z of 5217.9 (Fig. 5, inset) corresponding to the calculated m/z value of the [M+H]+ ion of EK46 with two CAM-cysteines and one Cys-SO2H, Cys-112-SO-2 was suggested to exist in holo-αe2 but not in apo-αe2 (20). In the mass spectrum of EK46 of the R-apo-α-subunit, the magnitude of the m/z 5242 peak corresponding to EK46 with three CAM-cysteines showed a dramatic decrease, and an intense peak at m/z 5218 corresponding to EK46 with two CAM-cysteines and one Cys-SO2H was observed (Fig. 5), suggesting that Cys-112 in apo-αe2 was oxidized to Cys-112-SO-2 in R-apo-αe2. Although the occurrence of Cys-114-SOH oxidation has not been confirmed because of the chemical instability (34), this finding strongly suggests that oxidized cysteine residues (αCys-112-SO-2 and αCys-114-SO-) exist in R-apo-αe2.
FIGURE 5.

MALDI-TOF MS spectra of the metal-binding peptide, EK46, of R-apo-αe2. The mass peaks with an m/z value of 5242 correspond to the [M+H]+ ion of EK46 with three CAM-cysteines (calculated m/z value = 5243.1); the mass peaks with m/z values of 5218 correspond to the [M+H]+ ion of EK46 with one Cys-SO2H and two CAM-cysteines (calculated m/z value = 5218.1). The inset shows the MALDI-TOF MS spectra of the metal-binding peptide, EK46, of apo-αe2 (top) and holo-αe2 (bottom) (20).
Necessity of NhlE for Cobalt Incorporation into the α-Subunit—To determine whether cobalt could be directly inserted into the apo-α-subunit in the absence of NhlE or not, we separated the α-subunit and NhlE through denaturation and renaturation (20). The resultant α-subunits (holo-α and apo-α, derived from holo-αe2 and apo-αe2, respectively) were found to each be a monomer, respectively, whereas the resultant NhlE was found to be a dimer (e2) (Fig. 6A). We mixed the apo-α-subunit with cobalt and DTT as described above and then purified the R-apo-α after incubation for 4 h. As a result, the cobalt content (0.14 ± 0.03/α) and the UV-visible spectrum of R-apo-α were found to be similar to those of apo-α but not to those of holo-α (Table 1 and Fig. 6B). On the other hand, from the mixture containing apo-α and e2 (molar ratio of apo-α to e2, 1:1) in the presence of cobalt and DTT after incubation for 4 h, cobalt-containing NhlAE (R-(α+e2)) was successfully isolated. The cobalt content (0.90 ± 0.05/αe2) and CD and UV-visible spectra of R-(α+e2) are similar to those of holo-αe2 (Table 1 and Fig. 4, B and C), and R-(α+e2) was also able to convert apo-L-NHase to holo-L-NHase (data not shown). These findings demonstrated that NlhE is necessary for the conversion of the apo-α-subunit into the holo-α-subunit.
FIGURE 6.

Characterization of purified holo-α, apo-α, NhlE, and R-apo-α. A, molecular mass and structure determination of the α-subunits and NhlE derived from apo-αe2 and holo-αe2 by gel filtration on a Superose 12 HR 10/30 column. Each of nhlA and nhlE (DDBJ accession numbers X64360 (nhlA) and D83695 (nhlE)) would encode proteins of 207 amino acids (22.8 kDa) and 148 amino acids (16.9 kDa), respectively. Marker proteins: i, glutamate dehydrogenase (yeast) (290 kDa); ii, lactate dehydrogenase (pig heart) (142 kDa); iii, enolase (yeast) (67 kDa); iv, myokinase (yeast) (32 kDa); v, cytochrome c (horse heart) (12.4 kDa). Values represent the means ± S.D. for at least triplicate independent experiments. B, UV-visible spectra of the purified holo-α, apo-α, and R-apo-α.
Effects of Aerobic and Anaerobic Conditions on Activation of Apo-α2β2 by Apo-αe2 in the Presence of Co2+ and Co3+—To confirm the effect of dissolved oxygen on the post-translational activation of apo-α2β2 by apo-αe2 in the presence of cobalt and DTT, the same experiment was carried out under anaerobic conditions, for which most of the dissolved oxygen was removed with argon. Although apo-α2β2 was activated by apo-αe2 in the presence of cobalt and DTT under aerobic conditions (245 ± 6 units/mg), it was hardly activated under anaerobic conditions (48.3 ± 4.5 units/mg) (Table 2). On the other hand, apo-α2β2 activation by holo-αe2 through self-subunit swapping under anaerobic conditions was similar to that under aerobic conditions, suggesting that self-subunit swapping between apo-α2β2 and holo-αe2 occurred irrespective of the presence or not of dissolved oxygen. Subsequently, [Co(NH3)6]Cl3(Co3+ donor) was used instead of CoCl2 (Co2+ donor) to investigate the effect of the cobalt redox state on activation of apo-α2β2 by apo-αe2 in the presence of DTT under aerobic and anaerobic conditions. As shown in Table 2, activation of apo-α2β2 by apo-αe2 with Co3+ was observed under both aerobic and anaerobic conditions and was similar to that with Co2+ under aerobic conditions.
TABLE 2.
Effects of aerobic and anaerobic conditions on activation of apo-α2β2 by apo-αe2 and holo-αe2 The mixtures were incubated in KPB (pH 7.5) at 28°C under aerobic or anaerobic conditions. [Co(NH3)6]Cl3 and CoCl2 solutions were used as Co3+ and Co2+ donors, respectively. The concentrations of apo-α2β2 and holo-αe2 in the mixture were 0.1 and 0.4 mg/ml, respectively. Aliquots of the samples were removed at 12 h and then assayed for L-NHase activity. Values represent the means ± S.D. for at least triplicate independent experiments.
| Mixture | Conditions | L-NHase activity |
|---|---|---|
| units/mg | ||
| Apo-α2β2 + apo-αe2 + Co2+ + DTT | Aerobic | 245 ± 6 |
| Apo-α2β2 + apo-αe2 + Co2+ + DTT | Anaerobic | 48.3 ± 4.5 |
| Apo-α2β2 + holo-αe2 | Aerobic | 335 ± 12 |
| Apo-α2β2 + holo-αe2 | Anaerobic | 330 ± 8 |
| Apo-α2β2 + apo-αe2 + Co3+ + DTT | Aerobic | 246 ± 10 |
| Apo-α2β2 + apo-αe2 + Co3+ + DTT | Anaerobic | 243 ± 8 |
Recycling of Used NhlAE—After holo-αe2 post-translationally activates apo-α2β2 through self-subunit swapping, during which the holo-α-subunit of holo-αe2 and the apo-α-subunit of apo-α2β2 are exchanged, cobalt-containing and cysteine-oxidized holo-α2β2 and non-cysteine-oxidized cobalt-free αe2 (R-holo-αe2) are formed (20). Here, we have the following question: Can R-holo-αe2 be recycled for cobalt insertion into L-NHase? When apo-α2β2 has been changed into holo-α2β2 completely after incubation with more than 2-fold of holo-αe2, the αe2 purified from the mixture after self-subunit swapping under these conditions comprises a mixture of apo-αe2 (used holo-αe2: R-holo-αe2) and unused holo-αe2 (20). To isolate only R-holo-αe2, we mixed holo-αe2 with apo-α2β2 in the ratio of 1:4 (0.1 and 0.4 mg/ml, respectively) in 10 mm KPB (pH 7.5) and then purified the resulting αe2 in the mixture after 12 h of incubation at 28 °C (Fig. 7A). The results of cobalt content determination (0.05 ± 0.02 mol/mol αe2) (Table 1) and UV-visible spectrum analysis (supplemental Fig. S2) indicated that almost all of the purified αe2 consisted of R-holo-αe2. Subsequently, post-translational activation of apo-α2β2 by R-holo-αe2 was investigated. As shown in Fig. 7B, activation of apo-α2β2 was not observed in a mixture of apo-α2β2 and R-holo-αe2, this being consistent with that self-subunit swapping does not occur between apo-α2β2 and apo-αe2 (20). In the presence of cobalt and DTT, however, R-holo-αe2 increased the L-NHase activity of apo-α2β2 to a level similar to that in the case of apo-αe2 (Fig. 7B). The enzyme activity and cobalt content (270 ± 10 units/mg and 0.95 ± 0.07/αβ) of the resultant L-NHase purified from the mixture of R-holo-αe2, apo-α2β2, cobalt, and DTT after 12 h were almost identical to those (275 ± 8 units/mg and 0.96 ± 0.07/αβ) with apo-αe2 instead of R-holo-αe2 (Table 1), demonstrating that almost all of the apo-α2β2 was converted to holo-α2β2. These findings suggested that NhlAE once used would be recycled for post-translational maturation of L-NHase.
FIGURE 7.

Recycling of NhlAE once used for cobalt incorporation into l-NHase. A, SDS/PAGE of the purified R-holo-αe2 (lane 1) and a mixture of apo-α2β2 with holo-αe2 in KPB (pH 7.5) at 12 h (lane 2) and 0 h (lane 3). B, activation of apo-α2β2 by R-holo-αe2 and apo-αe2. Aliquots of the samples were removed at the indicated times and then assayed for L-NHase activity. Values represent the means ± S.D. for at least triplicate independent experiments.
DISCUSSION
Reactive cysteine residues in proteins are well known to be critical components in redox signaling (29). Cys-SOH oxidation has been found in various redox sensor and regulating proteins (29–31). Depending on the environment, Cys-SOH in proteins sometimes provides a metastable oxidized form and at other times it is a fleeting intermediate giving rise to more stable disulfide, Cys-SO2H, and sulfenyl-amide forms (29). Cys-SOH and Cys-SO2H serve as catalytically essential redox centers, transient intermediates during peroxide reduction, sensors for the intracellular redox status, catalytic active sites, etc. (29, 30, 32, 33). Compared with various proteins containing reactive cysteine residues, NHase and thiocyanate hydrolase have both post-translational oxidized Cys-SOH and Cys-SO2H in their active sites (8–12, 26–28); both cysteines play an important role in the catalytic reaction but do not play any catalytic redox role (29). Several research groups have presented chemical and crystallographic evidence that Cys-SOH and Cys-SO2H are essential for Fe- and Co-NHases and thiocyanate hydrolase activity (8–12, 25, 26, 35). However, an enzyme catalyzing the cysteine oxidation reaction has never been reported. We discovered here that the non-oxidized cysteine residues in the apo-α-subunit of apo-αe2 are oxidized during cobalt insertion (Fig. 5). Once the cobalt is incorporated into NhlAE, it can never be removed through dialysis, even during NhlAE denaturation to isolate the holo-α-subunit (20). These findings suggest that the insertion of cobalt into the apo-α-subunit of αe2 is associated with cysteine oxidation; cobalt coordination is essential for the cysteine oxidation, and cysteine oxidation would increase the metal binding affinity. Taking the above together with the findings that the α-subunit in apo-L-NHase and the α-subunit itself were recalcitrant as to direct binding of cobalt (Table 1), we demonstrated here that αe2 is a vital complex for cobalt incorporation into the α-subunit of L-NHase and that NhlE is involved in the oxidation of cysteine ligands in the α-subunit during αe2 coordination with cobalt. To the best of our knowledge there have been no reports on protein-dependent post-translational cysteine oxidation in vitro. Although the Cys-SO2H in peroxiredoxine has been reported to be a reversible intermediate that is reduced to Cys-SOH through sulfiredoxin (39), this redox reaction occurs through a protein repair system rather than a protein-dependent post-translational maturation system. We discovered that NhlE facilitates Cys-SOH and Cys-SO2H oxidation post-translationally and for the first time succeeded in making cysteine residues in a protein undergo oxidation to stable Cys-SOH and Cys-SO2H in a protein-dependent manner in vitro, whereas enhancement of the spontaneous oxidation of cysteine residues in the α-subunit of Fe-NHase in Rhodococcus sp. N-771 in the presence of ferric citrate in vitro has been observed (34).
Cobalt exists as a non-corrin low-spin Co3+ ion in H-NHase in R. rhodochrous J1 (40) and NHase in P. putida NRRL-18668 (7), suggesting that the holo-α-subunit of L-NHase (holo-α2β2) and the holo-α-subunit of holo-αe2 (which is the source of that of holo-α2β2) should also have a non-corrin low-spin Co3+ ion. Therefore, the post-translational oxidation of the cysteine residues was associated with the oxidation of Co2+ to Co3+, because Co2+ was used as the cobalt donor in the in vitro experiments. Two possible sources of the oxygen atoms in the oxidized cysteine residues are considered: dissolved oxygen and a water molecule. Apo-α2β2 was activated by apo-αe2 in the presence of Co3+ and DTT under both aerobic and anaerobic conditions (Table 2), suggesting that the cysteines were oxidized irrespective of the presence of dissolved oxygen. On the other hand, apo-α2β2 was activated by apo-αe2 in the presence or not of Co2+ and DTT under aerobic conditions but not under anaerobic conditions (Table 2), demonstrating that dissolved oxygen was necessary for the oxidation of Co2+ to Co3+. Considering these findings, we would like to propose that the oxygen atoms in the oxidized cysteine residues are derived from water molecules, although the oxygen atoms in the cysteine residues modified through oxidation using an inhibitor (2-cyano-2-propyl hydroperoxide) for Fe-NHase and spontaneous oxidation are proposed to come from water molecules (41) and dissolved oxygen (34), respectively.
The crystal structure of the apo-NHase in Pseudonocardia thermophila JCM3059 has shown that the oxidation of cysteine residues in the active center does not occur, but electron density connecting Cys-108 and Cys-113 of the α-subunit was observed, indicating the formation of a disulfide bond between the two residues (25). Considering this finding, we speculate that a disulfide bond should also exist between the corresponding Cys-109 and Cys-114 in the α-subunit of apo-αe2 and that the cobalt incorporation would be blocked by the disulfide bond. An unusually strong reducing agent, DTT, because of its high conformational propensity to form a six-member ring with an internal disulfide bond, would act as an antioxidant for the reduction of the disulfide bond between Cys-109 and Cys-114, and the reduced forms of the resultant cysteine residues might be a necessary state at the beginning of cobalt insertion into apo-αe2. This hypothesis is supported by the finding that L-NHase activity was significantly increased in the presence of two other reducing agents, mercaptoethanol and glutathione, instead of DTT (data not shown). Because we were not able to quantify the holo-αe2-derived from apo-αe2 in the presence of cobalt and DTT, L-NHase activity was measured in place of the holo-αe2 formation after the conversion (Fig. 1(ii)) coupled with self-subunit swapping (Fig. 1(i)). A low concentration (<2 mm) of DTT would not be able to reduce all of the disulfide bonds completely. On the other hand, a high concentration (more than 4 mm) of DTT would inhibit L-NHase activity during the L-NHase assay. In the cases of 4, 6, and 8 mm DTT used for activation of apo-α2β2, the final DTT concentrations in the L-NHase assay buffer were 0.08, 0.12, and 0.16 mm, respectively. In fact, the L-NHase activity was assayed using holo-α2β2 in the presence of DTT, the following levels being exhibited: 0.05 mm, 92%; 0.1 mm, 85%. These results strongly support inhibition of the L-NHase activity by a high concentration of DTT. In addition, a high concentration of DTT might also inhibit the cysteine residue oxidation.
The αe2 complex once used was demonstrated to be recycled in vitro during post-translational maturation of L-NHase (Fig. 7 and Table 1). If the αe2 complex is recycled in vivo, the recycling system involved would allow sufficient maturation of the L-NHase even in the case of a small amount of αe2. In fact, the SDS/PAGE patterns of cell-free extracts in the previous study (20) showed that the amount of NhlAE is lower than that of L-NHase and that the molar ratio of the purified holo-αe2 to holo-α2β2 is about 2:3 (data not shown) when nhlBAE (containing both the L-NHase and NhlAE genes) is used for L-NHase and NhlAE expression. In addition, the self-subunit swapping kinetics appear not to be fast; however, the rate of activation of apo-α2β2 by holo-αe2 becomes faster as the holo-αe2 concentration increases (20). The recycling of the used NhlAE for cobalt transfer would provide a sufficient quantity of holo-αe2 for activation of apo-α2β2 in vivo, resulting in efficient self-subunit swapping maturation.
Because cobalt coordination and oxidation of cysteine residues, both of which are essential for L-NHase activation (1, 20), occur in NlhAE but not in apo-L-NHase, NhlAE is a vital complex in the process of functional L-NHase biogenesis. The structural genes for the α- and β-subunits of L-NHase are on the order of β and α; the NhlE gene (nhlE) is located just downstream that of the α-subunit gene (nhlA) of L-NHase, and NhlE lacks any known metal binding motifs but exhibits significant sequence similarity to the β-subunit of L-NHase (27% identity). This gene organization of L-NHase and NhlE allows NhlE to easily form a complex with the apo-α-subunit after translation.
In R. rhodochrous J1, a cobalt transporter (NhlF) (38) and Mg2+ uptake systems would be involved in the uptake of cobalt into cells (1). Taking the above together with the results described in this study, we propose a model for the overall process of cobalt incorporation into this noncorrin-cobalt-containing enzyme, L-NHase. As shown in Fig. 8, cobalt is transported into R. rhodochrous J1 cells by NhlF and Mg2+ uptake systems and then inserted into apo-αe2, resulting in cobalt- and oxidized cysteine-containing holo-αe2 (Fig. 1(ii)) followed by the formation of holo-α2β2 through replacement of the cobalt-free and non-oxidized α-subunit in apo-α2β2 with the cobalt-containing and oxidized cysteine-containing α-subunit in holo-αe2 (Fig. 1(i)). During the biogenesis of L-NHase, αe2 could be recycled for further biogenesis of L-NHase. In the present study, for the first time we demonstrated that NhlE acts not only as a chaperone for self-subunit swapping (Fig. 1(i)) but also as a metallochaperone that is crucial for both cobalt insertion and post-translational cysteine oxidation (Fig. 1(ii)). The discovery of this novel function of NhlE will help us to clarify the mechanism underlying this post-translational cysteine oxidation.
FIGURE 8.

A proposed model for the process of cobalt incorporation into l-NHase. Mg2+ uptake systems are designated as Mg2+ uptake systems (which transport cations such as Co2+, Zn2+, and Mg2+ into the cell). The cobalt ion is shown as a closed circle. MUS, Mg2+ uptake system.
Supplementary Material
Acknowledgments
We thank Dr. T. Sakashita and T. Cui for the amino acid sequence analysis and protein purification, respectively. We also thank Dr. K. Shiraki for the CD spectra analysis and T. Kanetou, I. Sagawa, K. Yamamoto, and Y. Fukuta for the MALDI-TOF MS analysis.
This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2 and Table S1.
Footnotes
The abbreviations used are: NHase, nitrile hydratase; DTT, dithiothreitol; MALDI-TOF MS, matrix-assisted laser desorption ionization time-of-flight mass spectrometry; R-apo-αe2, resultant apo-αe2; KPB, potassium phosphate buffer; CAM, carboxamidomethylated.
References
- 1.Kobayashi, M., and Shimizu, S. (1998) Nat. Biotechnol. 16 733-736 [DOI] [PubMed] [Google Scholar]
- 2.Endo, I., Odaka, M., and Yohda, M. (1999) Trends Biotechnol. 17 244-248 [DOI] [PubMed] [Google Scholar]
- 3.Noguchi, T., Nojiri, M., Takei, K., Odaka, M., and Kamiya, N. (2003) Biochemistry 42 11642-11650 [DOI] [PubMed] [Google Scholar]
- 4.Greene, S. N., and Richards, N. G. (2006) Inorg. Chem. 45 17-36 [DOI] [PubMed] [Google Scholar]
- 5.Komeda, H., Kobayashi, M., and Shimizu, S. (1996) J. Biol. Chem. 271 15796-15802 [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi, M., and Shimizu, S. (1999) Eur. J. Biochem. 261 1-9 [DOI] [PubMed] [Google Scholar]
- 7.Payne, M. S., Wu, S., Fallon, R. D., Tudor, G., Stieglitz, B., Turner, I. M., Jr., and Nelson, M. J. (1997) Biochemistry 36 5447-5454 [DOI] [PubMed] [Google Scholar]
- 8.Hourai, S., Miki, M., Takashima, Y., Mitsuda, S., and Yanagi, K. (2003) Biochem. Biophys. Res. Commun. 312 340-345 [DOI] [PubMed] [Google Scholar]
- 9.Miyanaga, A., Fushinobu, S., Ito, K., and Wakagi, T. (2001) Biochem. Biophys. Res. Commun. 288 1169-1174 [DOI] [PubMed] [Google Scholar]
- 10.Nagashima, S., Nakasako, M., Dohmae, N., Tsujimura, M., Takio, K., Odaka, M., Yohda, M., Kamiya, N., and Endo, I. (1998) Nat. Struct. Biol. 5 347-351 [DOI] [PubMed] [Google Scholar]
- 11.Huang, W., Jia, J., Cummings, J., Nelson, M., Schneider, G., and Lindqvist, Y. (1997) Structure 5 691-699 [DOI] [PubMed] [Google Scholar]
- 12.Stevens, J. M., Belghazi, M., Jaouen, M., Bonnet, D., Schmitter, J. M., Mansuy, D., Sari, M. A., and Artaud, I. (2003) J. Mass Spectrom. 38 955-961 [DOI] [PubMed] [Google Scholar]
- 13.Kobayashi, M., Fujiwara, Y., Goda, M., Komeda, H., and Shimizu, S. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 11986-11991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hashimoto, Y., Hosaka, H., Oinuma, K., Goda, M., Higashibata, H., and Kobayashi, M. (2005) J. Biol. Chem. 208 8660-8667 [DOI] [PubMed] [Google Scholar]
- 15.Abe, T., Hashimoto, Y., Hosaka, H., Tomita-Yokotani, K., and Kobayashi, M. (2008) J. Biol. Chem. 283 11312-11321 [DOI] [PubMed] [Google Scholar]
- 16.Komeda, H., Kobayashi, M., and Shimizu, S. (1996) Proc. Natl. Acad. Sci. U. S. A. 93 4267-4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi, M., Nagasawa, T., and Yamada, H. (1992) Trends Biotechnol. 10 402-408 [DOI] [PubMed] [Google Scholar]
- 18.Zhou, Z., Hashimoto, Y., and Kobayashi, M. (2005) Actinomycetologica 19 18-26 [Google Scholar]
- 19.Kuchar, J., and Hausinger, R. P. (2004) Chem. Rev. 104 509-525 [DOI] [PubMed] [Google Scholar]
- 20.Zhou, Z., Hashimoto, Y., Shiraki, K., and Kobayashi, M. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 14849-14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu, J., Zheng, Y., Yamagishi, H., Odaka, M., Tsyjimura, M., Maeda, M., and Indo, I. (2003) FEBS Lett. 553 391-396 [DOI] [PubMed] [Google Scholar]
- 22.Nojiri, M., Yohda, M., Odaka, M., Matsushita, Y., Tsujimura, M., Yoshida, T., Dohmae, N., Takio, K., and Endo, I. (1999) J. Biochem. 125 696-704 [DOI] [PubMed] [Google Scholar]
- 23.Wu, S., Fallon, R., and Payne, M. (1997) Appl. Microbiol. Biotechnol. 48 704-708 [DOI] [PubMed] [Google Scholar]
- 24.Hashimoto, Y., Nishiyama, M., Horinouchi, S., and Beppu, T. (1994) Biosci. Biotechnol. Biochem. 58 1859-1865 [DOI] [PubMed] [Google Scholar]
- 25.Miyanaga, A., Fushinobu, S., Ito, K., Shoun, H., and Wakagi, T. (2004) Eur. J. Biochem. 271 429-438 [DOI] [PubMed] [Google Scholar]
- 26.Kataoka, S., Arakawa, T., Hori, S., Katayama, Y., Hara, Y., Matsushita, Y., Nakayama, H., Yohda, M., Nyunoya, H., Dohmae, N., Maeda, M., and Odaka, M. (2006) FEBS Lett. 580 4667-4672 [DOI] [PubMed] [Google Scholar]
- 27.Katayama, Y., Hashimoto, K., Nakayama, H., Mino, H., Nojiri, M., Ono, T. A., Nyunoya, H., Yohda, M., Takio, K., and Odaka, M. (2006) J. Am. Chem. Soc. 128 728-729 [DOI] [PubMed] [Google Scholar]
- 28.Arakawa, T., Kawano, Y., Kataoka, S., Katayama, Y., Kamiya, N., Yohda, M., and Odaka, M. (2007) J. Mol. Biol. 366 1497-1509 [DOI] [PubMed] [Google Scholar]
- 29.Claiborne, A., Yeh, J. I., Mallett, T. C., Luba, J., Crane, E. J., Charrier, V., and Parsonage, D. (1999) Biochemistry 38 15407-15416 [DOI] [PubMed] [Google Scholar]
- 30.Poole, L. B., Karplus, P. A., and Claiborne, A. (2004) Annu. Rev. Pharmacol. Toxicol. 44 325-347 [DOI] [PubMed] [Google Scholar]
- 31.Nakamura, T., Yamamoto, T., Abe, M., Matsumura, H., Hagihara, Y., Goto, T., Yamaguchi, T., and Inoue, T. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 6238-6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volbeda, A., Martin, L., Cavazza, C., Matho, M., Faber, B. W., Roseboom, W., Albracht, S. P., Garcin, E., Rousset, M., and Fontecilla-Camps, J. C. (2005) J. Biol. Inorg. Chem. 10 239-249 [DOI] [PubMed] [Google Scholar]
- 33.Ogata, H., Hirota, S., Nakahara, A., Komori, H., Shibata, N., Kato, T., Kano, K., and Higuchi, Y. (2005) Structure 13 1635-1642 [DOI] [PubMed] [Google Scholar]
- 34.Murakami, T., Nojiri, M., Nakayama, H., Odaka, M., Yohda, M., Dohmae, N., Takio, K., Nagamune, T., and Endo, I. (2000) Protein Sci. 9 1024-1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai, J., Han, C., Hu, T., Zhang, J., Wu, D., Wang, F., Liu, Y., Ding, J., Chen, K., Yue, J., Shen, X., and Jiang, H. (2006) Protein Science 15 2071-2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chae, P. S., Kim, M. S., Jeung, C. S., Lee, S. D., Park, H., Lee, S., and Suh, J. (2005) J. Am. Chem. Soc. 127 2396-2397 [DOI] [PubMed] [Google Scholar]
- 37.Deng, H., Callender, R., Zhu, J., Nguyen, K. T., and Pei, D. (2002) Biochemistry 41 10563-10569 [DOI] [PubMed] [Google Scholar]
- 38.Komeda, H., Kobayashi, M., and Shimizu, S. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 36-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jönsson, T. J., Tsang, A. W., Lowther, W. T., and Furdui, C. M. (2008) J. Biol. Chem. 283 22890-22894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brennan, B. A., Alms, G., Nelson, M. J., Durney, L. T., and Scarrow, R. C. (1996) J. Am. Chem. Soc. 118 9194-9195 [Google Scholar]
- 41.Tsujimura, M., Odaka, M., Nakayama, H., Dohmae, N., Koshino, H., Asami, T., Hoshino, M., Takio, K., Yoshida, S., Maeda, M., and Endo, I. (2003) J. Am. Chem. Soc. 125 11532-11538 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.