

Abstract
Base excision repair, a major repair pathway in mammalian cells, is responsible for correcting DNA base damage and maintaining genomic integrity. Recent reports show that the Rad9-Rad1-Hus1 complex (9-1-1) stimulates enzymes proposed to perform a long patch-base excision repair sub-pathway (LP-BER), including DNA glycosylases, apurinic/apyrimidinic endonuclease 1 (APE1), DNA polymerase β (pol β), flap endonuclease 1 (FEN1), and DNA ligase I (LigI). However, 9-1-1 was found to produce minimal stimulation of FEN1 and LigI in the context of a complete reconstitution of LP-BER. We show here that pol β is a robust stimulator of FEN1 and a moderate stimulator of LigI. Apparently, there is a maximum possible stimulation of these two proteins such that after responding to pol β or another protein in the repair complex, only a small additional response to 9-1-1 is allowed. The 9-1-1 sliding clamp structure must serve primarily to coordinate enzyme actions rather than enhancing rate. Significantly, stimulation by the polymerase involves interaction of primer terminus-bound pol β with FEN1 and LigI. This observation provides compelling evidence that the proposed LP-BER pathway is actually employed in cells. Moreover, this pathway has been proposed to function by sequential enzyme actions in a “hit and run” mechanism. Our results imply that this mechanism is still carried out, but in the context of a multienzyme complex that remains structurally intact during the repair process.
The mammalian genome experiences constant stress from both external and internal factors that causes genomic instability. Eukaryotic cells have developed a number of DNA repair pathways that correct DNA damage before it results in permanent chromosomal alteration. Base excision repair (BER)3 is the major pathway responsible for reversing DNA damage sustained by individual nucleotide bases. Mammalian BER is initiated by DNA glycosylases, which recognize structural alteration of a nitrogenous base and excise it leaving an intact sugar-phosphate backbone with an apurinic/apyrimidinic (AP) site (1). AP sites in humans are detected by AP endonuclease 1 (APE1) that cleaves the phosphate backbone of the damaged strand, leaving a nick with a 3′-OH group and a 5′-deoxyribose phosphate (dRP) residue. The dRP-bordered nick is not a substrate for ligation. If the dRP residue is not oxidized or reduced, repair can proceed via a short patch-BER pathway, in which the dRP residue is removed by the 5′-lyase activity of DNA polymerase β (pol β), which concurrently fills in the 1-nt gap, and the resulting nick is sealed by the DNA ligase III-XRCC1 complex (2-4).
However, if the oxidative state of the dRP is altered, the lyase activity of pol β is inhibited, but the polymerase activity of pol β can still displace the oxidized or reduced dRP residue into a 2-10-nt 5′ flap intermediate, which will then be cleaved by FEN1 and subsequently joined by LigI (4-7). This process is known as long patch-base excision repair (LP-BER). Recent studies examining the relevance of the two different pathways in vitro predict a predominant role for short patch-BER in the cell as compared with LP-BER (8). Because the cell undergoes constant repair of damaged bases, it is very difficult to assess the relative use of one pathway over the other in vivo. Studies using plasmid DNA containing defined DNA damage have been used as an indirect approach to evaluate the role of the two different BER pathways in cells and the size of the DNA repair patches (9). Results from these studies have shown that repair patches of 6-12 nucleotides are generated during repair of plasmids that contain a single base lesion, at least supporting the existence of LP-BER in vivo.
LP-BER has also been proposed to proceed by either a PCNA-dependent sub-pathway involving the use of DNA pol δ/ε or a PCNA-independent sub-pathway that uses only DNA pol β. However, most LP-BER reconstitution experiments in vitro indicate that pol β works more efficiently than pol δ with the other proposed LP-BER proteins. FEN1 is known to stimulate pol β-mediated DNA synthesis on an LP-BER substrate suggesting that these two proteins interact functionally and mechanistically (10). pol β has also been shown to interact with LigI by co-immunoprecipitation experiments indicating that they might be a part of a multiprotein DNA repair complex (11).
The heterotrimeric protein complex, Rad9, Rad1, and Hus1 (the 9-1-1 complex), plays a significant role in the early recognition of DNA damage and recruiting appropriate proteins to repair sites. The 9-1-1 complex interacts with several of the proteins involved in the proposed BER pathways, including DNA glycosylases (12-14), APE1 (15), pol β (16), FEN1 (17,18), and LigI (19, 20). In a recent report (15), the 9-1-1 complex was shown to interact both physically and functionally with APE1 and pol β and to stimulate their respective activities. Stimulation of the endonuclease ensures the abasic site is recognized and cleaved off efficiently. Stimulation of nucleotide addition by pol β is expected to promote the LP-BER sub-pathway, as 9-1-1 stimulates the strand displacement activity of pol β, thereby requiring FEN1 flap cleavage before ligation to repair the site of damage. Because 9-1-1 is structurally similar to the sliding clamp PCNA, early studies were focused on determining the effects of 9-1-1 on DNA replication and repair proteins previously shown to be stimulated by PCNA. The 9-1-1 complex has been reported to stimulate both FEN1 cleavage (17, 18) and nick sealing by LigI (20) in vitro. However, the 9-1-1 clamp poorly stimulated FEN1 and LigI in the entire LP-BER-reconstituted system as compared with strong stimulation by 9-1-1 of individual cognate substrates (15). The authors (15) suggest that FEN1 and LigI evolved to respond to stimulation by PCNA and not 9-1-1 during LP-BER. The issue with this explanation is that it does not take into consideration how LP-BER would be efficiently carried out when damage-induced p21 binds and inhibits PCNA (21).
To define how 9-1-1 interacts with the components of BER, we have reconstituted the entire LP-BER pathway using purified human enzymes and substrates that simulate an abasic site created after recognition and cleavage of damaged base by a glycosylase. Similar to results of Gembka et al. (15), we observe much less stimulation of either FEN1 or LigI by 9-1-1 in the fully reconstituted system compared with 9-1-1 stimulation of FEN1 on a flap substrate or LigI on a nicked substrate alone. Our subsequent analysis of the protein-protein interactions among the various LP-BER enzymes provides insight into why the 9-1-1 clamp exhibits minimal stimulation in the reconstituted system. Moreover, our mechanistic characterization of the significant role of pol β in mediating the activities of various enzymes in the multiprotein repair complex both explains the behavior of 9-1-1 and strongly suggests the existence of the LP-BER pathway in vivo.
EXPERIMENTAL PROCEDURES
Materials—Synthetic oligodeoxyribonucleotides, including those containing a tetrahydrofuran (THF) residue or the 5′-biotin conjugation, were prepared by Integrated DNA Technologies (Coralville, IA). Radionucleotides [γ-32P]ATP and [α-32P]dCTP were purchased from PerkinElmer Life Sciences. Substrate-labeling enzymes, the Klenow fragment of Escherichia coli DNA polymerase I (for 3′ labeling), and polynucleotide kinase (for 5′ labeling) were from Roche Applied Science. Streptavidin, ATP, and dNTPs were also purchased from Roche Applied Science. All other reagents were purchased from among the best commercially available products.
Enzymes—FEN1 (22), 9-1-1 (23), APE1 (24), pol β (25), and synthesis-defective mutant pol β (26) were prepared as described previously.
Oligodeoxyribonucleotides—Oligodeoxyribonucleotide substrates were designed to represent LP-BER intermediates. They have two adjacent primers annealed to a template. Oligodeoxyribonucleotide sequences are listed in Table 1. The primer sequences are listed 5′ to 3′, and the template sequences are listed 3′ to 5′ to facilitate visual alignment. A “Ø” in the nucleotide sequence represents a THF residue, and a “B” in the nucleotide sequence represents a biotin-conjugated strand. Downstream primers were labeled with 32P either at the 5′ or 3′ end as indicated in the figures. Labels were added at the 5′ end of strands by incubating them with T4 polynucleotide kinase and [γ-32P]ATP. Labels were added at 3′ ends of strands by annealing 20 pmol of primer to 50 pmol of template having a G overhang at the 5′ end. [α-32P]dCTP was added to the 3′ end of the primer with the Klenow fragment of E. coli DNA polymerase I. Labeled primers were purified by electrophoresis and isolated from a 15% polyacrylamide, 7 m urea denaturing gel. To anneal strands into substrates, primers were combined in annealing buffer containing 10 mm Tris-HCl (pH 8.0), 50 mm NaCl, and 1 mm DTT, heated to 95 °C for 5 min, transferred to 70 °C, and then slowly cooled to room temperature. Nicked, gapped, and 5′ flap substrates were annealed in a 1:2:4 ratio of labeled downstream primer to template to upstream primer. Substrates containing an internal THF residue were annealed in a 1:2 ratio of labeled primer to template.
TABLE 1.
Oligonucleotide sequences
Ø in the nucleotide sequence represents a THF residue, and B in the nucleotide sequence represents a biotin-conjugated strand.
| Primer | Length in nt | Sequence |
|---|---|---|
| Upstream | ||
| U1 | 20 | 5′-CGACCGTGCCAGCCTAAAAC-3′ |
| U2 | 19 | 5′-CGACCGTGCCAGCCTAAAA-3′ |
| U3 | 26 | 5′-CGCCAGGGTTTTCCCAGTCACGACCA-3′ |
| U4 | 25 | 5′-CGCCAGGGTTTTCCCAGTCACGACC-3′ |
| U5 | 24 | 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′ |
| U6 | 22 | 5′-CGCCAGGGTTTTCCCAGTCACG-3′ |
| U7 | 20 | 5′-CGCCAGGGTTTTCCCAGTCA-3′ |
| Downstream | ||
| D1 | 55 | 5′-CGACCGTGCCAGCCTAAAAØACTTGCCCGTGCCACCATCCCGACGCCACCTCCTG-3′ |
| D2 | 35 | 5′-ØCTTGCCCGTGCCACCATCCCGACGCCACCTCCTG-3′ |
| D3 | 36 | 5′-ØACTTGCCCGTGCCACCATCCCGACGCCACCTCCTG-3′ |
| D4 | 28 | 5′-GCCGTCGTTTTACAACGACGTGACTGGG-3′ |
| D5 | 35 | 5′-ACTTGCCCGTGCCACCATCCCGACGCCACCTCCTG-3′ |
| D6 | 53 | 5′-BTTCACGCCTGTTAGTTAATTCACTGGCCGTCGTTTTACAACGACGTGACTGGG-3′ |
| Template | ||
| T1 | 49 | 3′-GCGGTCCCAAAAGGGTCAGTGCTGGGCAAAATGTTGCTGCACTGACCCG-5′ |
| T2 | 56 | 3′-GCTGGCACGGTCGGATTTTGTGAACGGGCACGGTGGTAGGGCTGCGGTGGAGGACG-5′ |
| T3 | 53 | 3′-GCTGGCACGGTCGGATTTTGACGGGCACGGTGGTAGGGCTGCGGTGGAGGACG-5′ |
Enzyme Assays—For analysis of DNA pol β stimulation of FEN1 and LigI, 20 μl of reaction mixtures containing the indicated quantities of enzyme and 5 fmol of 32P-radiolabeled DNA substrate were incubated at 37 °C for 10 min. The reactions were stopped by the addition of 2× termination dye (90% formamide (v/v) with 10 mm EDTA, 0.1% bromphenol blue, and 0.1% xylene cyanole). Unless stated otherwise, the reaction buffer contained 50 mm Tris-HCl (pH 8.0), 2 mm DTT, 30 mm NaCl, 0.1 mg/ml bovine serum albumin (BSA), 8 mm MgCl2, 2 mm ATP, and 50 μm each of dATP, dCTP, dTTP, and dGTP. For the biotin-streptavidin blocking experiments, the initial reaction mixtures lacked MgCl2. The substrate was incubated with enzymes either before or after the addition of streptavidin. Streptavidin, added in a 50-fold excess over substrate, was complexed with the biotinylated substrate by placing the reaction at 37 °C for 10 min. MgCl2 was added after blocking at a concentration of 8 mm per reaction. The reaction was terminated with 2× termination dye as described above. After termination, samples were heated at 95 °C for 5 min and loaded onto a pre-heated denaturing (7 m urea) 15% polyacrylamide gel.
Binding Assay—Purified FEN1 (1 or 2 ng) and pol β (1 or 2 ng) were allowed to bind together in a coupling buffer consisting of 25 mm HEPES (pH 7.5), 100 mm NaCl, 1 mm EDTA, 1 mm DTT, and 10% glycerol for 2 h at 4 ° C (in a 1:1 or 2:1 ratio, as stated in Fig. 4). In some cases a 100-fold excess of BSA was added to the reaction to rule out nonspecific binding of FEN1 and DNA pol β to each other. The bound protein complexes were then added to a spin column containing the coupling gel to which the specific antibody (100 μg of either anti-FEN1 polyclonal antibody (Abcam, ab17993), anti-pol β monoclonal antibody (Abcam, ab3181), or glyceraldehyde-3-phosphate dehydrogenase (Santa Cruz Biotechnology, sc-25778)) was immobilized and allowed to bind overnight at 4 °C with gentle end-over mixing. Following binding to the antibody column, the proteins were released using elution buffer (ProFound co-immunoprecipitation kit, Pierce). The immunoprecipitates were separated on precast SDS-polyacrylamide gels (8-16%; Bio-Rad). Western blot analysis was performed with anti-FEN1 polyclonal antibody or anti-pol β monoclonal antibody.
FIGURE 4.

pol β structurally interacts with FEN1. Binding and co-immunoprecipitation experiments were performed as described under “Experimental Procedures.” A and C contain immunoprecipitates probed on a Western blot with antibody against pol β. B and D contain immunoprecipitates probed on a Western blot with antibody against FEN1. Lane 1 in all four panels contains IgG alone; lane 2 contains an IP with antibody against FEN1 (A), IP with antibody against pol β (B), and 25 μg of HeLa extract (C and D); lane 3 contains an IP with antibody against polβ (A and D) and IP with antibody against FEN1 (B and C); lane 4 contains an IP with antibody against glyceraldehyde-3-phosphate dehydrogenase (nonspecific IgG) (A and B); lanes 5-7 contain IP with antibody against FEN1 (A) and pol β (B). The lanes also specify the proteins and the ratio at which they were bound together in the binding buffer (for A and B). IB, immunoblot; ML, molecular ladder.
Co-immunoprecipitation—Immunoprecipitation was performed using the protocol described in the ProFound co-immunoprecipitation kit (Pierce). Briefly 200 μg of specific antibody was immobilized on the antibody coupling gel overnight at 4 °C with gentle end-over mixing. Four mg of HeLa cell lysate was added to the spin column containing the immobilized antibody and allowed to bind for 4-5 h at 4 °C, after which the co-immunoprecipitation complex was released by elution buffer. The immunoprecipitates were separated on precast SDS-polyacrylamide gels (8-16%; Bio-Rad). Western blot analysis was performed with anti-FEN1 polyclonal antibody or anti-pol β monoclonal antibody.
Electrophoretic Mobility Shift Assay (EMSA)—Binding efficiency of pol β to different substrates was measured using EMSA. Increasing concentrations of pol β (0.01-100 fmol per reaction) were incubated with 1 fmol of various 5′-labeled substrates for 10 min on ice in a reaction buffer consisting of 50 mm Tris-HCl (pH 8.0), 2 mm DTT, 30 mm NaCl, 0.1 mg/ml BSA, and 5% glycerol. The reactions were loaded on pre-run 5% polyacrylamide gels in 0.5× TBE (Invitrogen). Gels were subjected to electrophoresis for 30-45 min at 150 V.
Gel Analysis—Each experiment was performed at least in triplicate, and the presented data are the average of the percentage of cleavage/ligation observed. After gel electrophoresis, the denaturing gels were transferred to Whatman filter paper, wrapped in Saran plastic, and dried on a vacuum gel drier (Savant, Bio-Rad). Dried gels were then exposed to a phosphor screen and analyzed by PhosphorImaging (GE Healthcare). Scanned images were analyzed by using ImageQuant version 5.0 software.
Dissociation Constant (Kd) Measurements—After gel shift analysis, curves were fit using nonlinear least squares regression of the hyperbolic Equation 1,
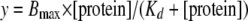 |
(Eq.1) |
where y is the percent of oligonucleotide bound; [protein] is the concentration of protein in nanomolar; Bmax is the maximum binding, and Kd is the equilibrium constant.
RESULTS
Reconstitution of LP-BER—To examine the interactions of the 9-1-1 DNA clamp with other proteins involved in LP-BER, we used a 56-nt duplex DNA substrate containing an internal THF residue to reconstitute LP-BER in vitro. Fig. 1A shows the activity of APE1, pol β, FEN1, and LigI on the substrate. Fig. 1A, lane 1, derives from a solution containing 5 fmol of the THF substrate alone. In Fig. 1A, the designations SM and RP signify that the starting material (SM) substrate and the repaired product (RP) migrate to the same position. Fig. 1A, lane 2, derives from a reaction in which the THF substrate was incubated with 3 fmol of APE1, resulting in virtually 100% cleavage. The incised product is denoted by IP in Fig. 1. Fig. 1A, lane 3, represents a reaction with 20 fmol of pol β in addition to APE1. The downstream primer may have been displaced, but because it was not cleaved, it migrated to the same position as in Fig. 1A, lane 2. For Fig. 1A, lane 4, the reaction contained 1 fmol of FEN1 in addition to pol β and APE1. Strand displacement synthesis of the cleaved primer by pol β formed flap substrates that were cleaved by FEN1 and appear as a ladder of smaller products below the incision product of APE1 cleavage. Fig. 1A, lanes 5-7, show regeneration of the full-length repaired product upon addition of increasing amounts LigI. LigI has very low activity on a 5′-THF nick substrate (data not shown and Ref. 27), so the full-length product should not have resulted from ligation of the APE1 cleavage product. To prove that the full-length product generated in Fig. 1A, lanes 5-7, was fully repaired, we incubated the LP-BER enzymes with a THF-substrate containing a PstI restriction endonuclease site centered on the THF lesion. When the THF site is present, PstI cannot cleave. When the THF has been replaced with the correct nucleotide, the substrate is cleaved, giving a product distinct from the APE1 cleavage product. When this substrate was incubated with APE1, pol β, FEN1, and LigI and then exposed to PstI, all of the product was cleaved, indicating that the product was fully repaired duplex DNA (data not shown).
FIGURE 1.

Reconstitution of LP-BER. A, intact, THF-containing substrate is repaired efficiently by LP-BER in vitro. Reactions were performed as described under “Experimental Procedures.” All lanes contain 5 fmol of substrate, which were generated by annealing primers D1 and T2 in a 1:2 ratio (depicted in the figure). The asterisk denotes the position of the radiolabel. The Ø represents an abasic site. Lane 1 contains only substrate, and the lone band marks the migration of both the starting material (SM) and the repaired product (RP). APE1, when present, was added at 3 fmol per reaction. The incision product (IP) of APE1 is denoted in the figure. polβ, when present, was added at 20 fmol per reaction. FEN1, when present, was added at 1 fmol per reaction. The three quantities of LigI added to lanes 5-7 were 1, 5, and 25 fmol, respectively. B, 9-1-1 displays low level stimulation of FEN1 and ligase in a reconstituted LP-BER system. Reactions were run as described under “Experimental Procedures.” APE1, when present, was added at 3 fmol per reaction. pol β, when present, was added at 20 fmol per reaction. 9-1-1 was added at either 250 or 500 fmol as denoted by the triangles. The amount of FEN1 and LigI (in fmol) used in each reaction is noted above each lane.
After determining the optimum concentrations of enzymes required to efficiently repair the pre-made LP-BER substrate, we limited the FEN1 or LigI concentration in a fully reconstituted system, which diminished repair efficiency. Purified 9-1-1 complex was then added to determine whether it could stimulate FEN1 or LigI to restore efficiency (Fig. 1B). Gembka et al. (15) had previously reported that the 9-1-1 complex did not significantly stimulate FEN1 and LigI activities when present as a part of the fully reconstituted system. This contrasted with high level stimulation observed when the enzymes were tested in isolation on cognate substrates. Similar to observations by Gembka et al. (15), on using limiting concentrations of either FEN1 or LigI, we saw only a 1.5-2-fold stimulation of full-length product formation (Fig. 1B, lanes 4, 5, 7, and 8). Altering buffer conditions, reaction times, or enzyme concentrations did not increase the ability of 9-1-1 to stimulate the latter steps of LP-BER. A reaction mixture containing optimum amounts of FEN1 and LigI (Fig. 1B, lane 9) showed robust repair, proving that stimulation of these two enzyme components by 9-1-1 would have greatly improved overall repair efficiency.
pol β Stimulates FEN1 Cleavage on a Variety of Substrates—There could be two possible scenarios whereby 9-1-1 is unable to significantly stimulate the activities of FEN1 and LigI in a fully reconstituted system. First, there are other proteins in the multienzyme repair complex that strongly stimulate FEN1 and LigI, such that further stimulation by 9-1-1 is not possible. In such a situation, the activities of FEN1 and LigI have achieved a maximum, such that presence of any additional stimulatory factor can have no effect. Second, in the fully reconstituted system, another repair protein disrupts the interaction between 9-1-1 and FEN1 or LigI. This might occur because of steric interference between proteins attempting to bind at the upstream-downstream primer junction or because of competition between 9-1-1 and a second enzyme for binding to FEN1 or LigI.
Exploring these possibilities, we discovered that pol β stimulates flap cleavage by FEN1. The substrate employed was a 5-nt nick-flap with a completely annealed upstream primer that reaches to the base of the flap (Fig. 2A). The 9-1-1 clamp stimulated FEN1 on this nick-flap substrate about 9-fold (Fig. 2A, lanes 4 and 5). In the absence of 9-1-1, pol β produced a 12-fold stimulation of flap cleavage (Fig. 2A, lanes 6 and 7).
FIGURE 2.


pol β stimulates FEN1 cleavage on a variety of substrates. Reactions were performed as described under “Experimental Procedures” either in the presence (A) or absence (B) of dNTPs. All lanes represent reactions containing 5 fmol of 5-nt nick-flap substrate, which was generated by annealing primers D4, T1, and U4 in a 1:2:4 ratio (depicted in the figure). There are no missing nucleotides between the 3′ end of the upstream primer and base of the flap. The asterisk indicates the location of the radiolabel. Lane 1 in each panel (A and B) contains only substrate, and the lone band marks its migration. Lane 2 derives from a reaction containing substrate plus 20 fmol of pol β. In lanes 3-9 the reaction also contained 1 fmol of FEN1. The two quantities of 9-1-1 used were 250 and 500 fmol. + in the 9-1-1 row signifies the presence of 500 fmol of 9-1-1. The two quantities of pol β used were 10 and 20 fmol. C, cleavage by FEN1 (1 fmol) was assayed in the presence of increasing amounts of wild-type pol β (2.5, 5, 10, and 20 fmol) in the presence of dNTPs. The 5-nt nick-flap (D4:T1:U4) (filled squares), 5-nt double flap (D4:T1:U3) (filled triangles), and the 3-nt nick THF flap (D2:T3:U1) (filled circles) are depicted in the graph. Addition of dCTP to the reaction buffer containing the 5-nt nick-flap is graphically represented by the open squares, and addition of dTTP to the reaction buffer containing the 3-nt nick THF-flap is represented by the open circles. The inset in the graph represents reactions containing 5 fmol of the double flap substrate, in the presence of 50 amol per reaction of FEN1, and increasing concentrations of pol β (2.5, 5, 10, and 20 fmol), open triangles. D, cleavage by FEN1 (1 fmol) was assayed in the presence of increasing amounts of polymerase-defective/5′-dRP lyase-active D256A pol β mutant (2.5, 5, 10, and 20 fmol) in the presence of dNTPs. Graphical representations are identical to those described in C. E, FEN1 cleavage (75 amol) was assayed in the presence of either 20 fmol of pol β or 500 fmol of 9-1-1 using the 5-nt nick-flap, 5-nt double flap, or the 3-nt THF-flap substrate. In experiments graphically represented by the black bar, 75 amol of FEN1 and 20 fmol of pol β were preincubated at 37 °C with the specific substrates for 2 min, followed by addition of 500 fmol of 9-1-1, which was further incubated for 10 min at 37 °C. Error bars indicate the standard deviation from a minimum of three independent experiments.
To address whether pol β stimulation of FEN1 is synthesis-dependent, we performed a FEN1 cleavage reaction on the same 5-nt nick-flap substrate with pol β but in the absence of dNTPs (Fig. 2B). A control experiment containing a synthesis-competent substrate and high levels of pol β in the absence of dNTPs showed no synthesis (data not shown). In the absence of synthesis and strand displacement activity, pol β stimulated FEN1 cleavage nearly 8-fold (Fig. 2B, lanes 6 and 7). In the presence of 9-1-1 and pol β, FEN1 cleavage activity was stimulated to about 11-fold (Fig. 2B, lanes 8 and 9). Evidently, synthesis adds an additional increment to a substantial basal level of stimulation. The link with synthesis was unexpected because the initial 5-nt flap configuration was already a substrate for FEN1. A possible explanation for the improved cleavage is that strand displacement by pol β should produce a 1-nt 3′ tail in addition to the 5′ flap. This double flap configuration is a better substrate for FEN1 cleavage than the original nick-flap (28). When 9-1-1 and pol β were both added to the FEN1 cleavage reaction (Fig. 2A, lanes 8 and 9), there was only a slight additional stimulation producing an overall increase of 15-fold. The effect of 9-1-1 may have been directly on FEN1, or it may have altered pol β to make the polymerase more effective in stimulating FEN1.
Because pol β differentially stimulated FEN1 cleavage in the presence and absence of dNTPs, we used different substrate structures to examine the relationship between synthesis and stimulation. The experiments employed either the wild-type pol β or the polymerase-defective/5′-dRP lyase-active D256A mutant pol β. The Asp-256 is critical for the nucleotidyl transfer mechanism; its mutation to alanine abolishes the polymerase activity of pol β. Fig. 2C shows a graphical representation of conversion of substrates into products by FEN1 in the presence of different concentrations of wild-type pol β. FEN1 was stimulated about 12-fold on the 5-nt nick-flap substrate as in Fig. 2A (straight line, filled squares). On addition of dCTP, pol β added one nucleotide to the upstream primer complementary to the G nucleotide on the template, forming a double flap substrate. pol β stimulated FEN1 cleavage to a higher extent; 17-fold on the newly created double flap substrate (Fig. 2C, dotted line, open squares).
Surprisingly, with a pre-made double flap substrate, we observed only a 1.5-fold stimulation by pol β (Fig. 2C, straight line, filled triangle). We realized that the high FEN1 activity in this reaction consumed so much substrate that additional stimulation could not be properly detected. To address this issue we lowered the FEN1 concentration to 50 amol/reaction, so that only 2-3% of the substrate was consumed in the absence of pol β. Nevertheless, pol β was unable to stimulate FEN1 cleavage beyond 2-fold (Fig. 2C, inset, dotted line, open triangles). One possible explanation is that the 3′ flap hindered the ability of pol β to bind to the substrate and stimulate FEN1. Another is that the double flap is such an excellent substrate for FEN1 that only a moderate additional stimulation is possible.
We also tested pol β stimulation on a 3-nt flap substrate containing a THF residue on the 5′ end of the flap. This substrate simulates an intermediate of LP-BER. pol β stimulated FEN1 product formation to approximately the same extent (11-fold) as on the nick-flap substrate (Fig. 2C, straight line, filled circle). On addition of only dTTP to the reaction, the template sequence allowed pol β to add a single T to create a double flap, which stimulated FEN1 cleavage activity by ∼15-fold (Fig. 2C, dotted line, open circle). These data suggest that pol β can synthesize on a nick-flap to create the FEN1 preferred double flap substrate, enhancing stimulation of cleavage.
Similar experiments as described in Fig. 2C were performed using the polymerase-defective/5′dRP lyase-active D256A pol β mutant protein (Fig. 2D). The mutant protein stimulated FEN1 cleavage both on the 5-nt nick-flap (Fig. 2D, straight line, filled squares) and the 3-nt THF nick-flap substrates (straight line, filled circles) to approximately the same extent as in Fig. 2B (7- and 6-fold, respectively). As previously shown in Fig. 2C, the pre-made double flap experienced only modest stimulation in FEN1 cleavage by pol β (straight line, filled triangles). Moreover, lowering the amount of FEN1 in the reaction did not substantially improve the stimulation (Fig. 2C, inset, dotted line, open triangles). On addition of dCTP or dTTP to the reactions containing the 5-nt nick-flap or 3-nt THF nick-flap substrate, respectively, the polymerization-defective pol β, unable to add nucleotides, could not create a double flap substrate. Without creating the FEN1 preferred substrate, we observed the same level of FEN1 stimulation as on the nick-flap substrate (Fig. 2D, 5-nt nick-flap + dCTP, dotted line, open squares; 3-nt nick THF flap + dTTP, dotted line, open circles). The same set of reactions as described in Fig. 2D were performed in the absence of dNTPs and yielded the same results (data not shown).
In experiments described in Fig. 2E, three different substrates were incubated with FEN1 alone, FEN1 and 9-1-1, FEN1 and pol β, or FEN1, 9-1-1, and pol β for a period of 10 min at 37 °C before terminating the reaction. Product formation on the 5-nt nick-flap, 5-nt double flap, and 3-nt nick THF flap showed only a slight increase above individual stimulations when both 9-1-1 and pol β were incubated along with FEN1 in the reaction (Fig. 2E, FEN1 + pol β + 9-1-1). Because FEN1 and pol β are likely to be interacting in a stimulatory configuration in the minimal reconstitution system, the additional effects of 9-1-1 may be minimal. To test this hypothesis, we measured the stimulation of FEN1 by 9-1-1 after pol β had already interacted with FEN1. When FEN1 and pol β were preincubated with the substrates for 2 min at 37 °C, before adding 9-1-1 and allowing the reaction to proceed for another 10 min before termination, we observed a similar level of stimulation in the FEN1 cleavage activity as when all the three proteins were present in the reaction (Fig. 2E, FEN1 + pol β, 9-1-1). Overall, our results indicate that once pol β stimulates FEN1 to a certain threshold, it cannot be further stimulated by 9-1-1.
pol β Binds the 3′ End of the Upstream Primer and Interacts with the Flap-bound FEN1, Stimulating FEN1 Activity—Our results (Fig. 2, A-D) show that pol β stimulated FEN1 activity on a nick-flap substrate both in the presence and absence of synthesis. To verify whether interaction of pol β and FEN1 on the substrate was important for the increased rate of cleavage, we designed flap substrates with varying gap sizes. Assuming pol β binds the primer terminus and FEN1 binds the flap, a progressively larger gap would be anticipated to strain or break interactions producing the stimulation. A related report on FEN1 interaction with pol β suggested that FEN1 cleavage during LP-BER creates a 1-nt gap that augments the strand displacement activity of pol β (10). In other words, when pol β is positioned just upstream of the base of the flap it may promote FEN1 cleavage 1 nt into the annealed downstream primer leaving a 1-nt gap that is filled by pol β. When our reactions were carried out in the presence of dNTPs (Fig. 3A), stimulation of FEN1 cleavage by pol β was significant on all of the substrates. On increasing the amount of pol β the formation of FEN1 cleavage products increased steadily to a range of 8-12-fold stimulation. Presumably, pol β loaded on the 3′ end of the upstream primer and extended it. This would move pol β closer to the flap base so that it could interact with the flap-bound FEN1 for stimulation. These results provided an important control showing that when synthesis converted all of the substrates to the same configuration there was a similar degree of stimulation. Significantly, in the absence of dNTPs and synthesis, the range of stimulation by pol β varied from 2-fold on a 5-nt gap flap substrate to 9-fold on the 5-nt nick-flap substrate. Moreover, the difference in fold stimulation with and without dNTPs was greatest when using a substrate with a 5-nt gap flap. These results are consistent with the interpretation that the increasing gap size progressively weakened interactions between the polymerase and FEN1 that are important for stimulation. We conclude that the stimulation mechanism involves direct interaction of a pol β bound at the primer terminus and a FEN1 bound at the flap.
FIGURE 3.

pol β binds 3′ end of the upstream primer and interacts with the flap-bound FEN1, stimulating FEN1 activity. A, cleavage by FEN1 (2 fmol) on various substrates was assayed in the presence of increasing amounts of pol β (2.5, 5, 10, and 20 fmol) either in the presence or absence of dNTPs as described under “Experimental Procedures.” The 5-nt gap-flap (D4:T1:U7), 3-nt gap-flap (D4:T1:U6), 1-nt gap-flap (D4:T1:U5), and the nick-flap (D4:T1:U4) are depicted above the figure. The asterisk indicates location of the radiolabel. The fold stimulation of FEN1 cleavage by pol β is indicated by the bar graphs on different substrates in the presence and absence of dNTPs. B, reactions containing either 0.2 fmol of FEN1 or 25 fmol of FEN1 were assayed in the presence of increasing concentrations of pol β (2.5, 5, 10, and 20 fmol) using either a 5-nt nick-flap (D4:T1:U4) or a flap substrate lacking the upstream primer (D4:T1). Asterisk indicates the position of the radiolabel. Error bars indicate the standard deviation from a minimum of three independent experiments.
Removal of the upstream primer forms a pseudo-Y structure, a poor but usable substrate for FEN1-directed cleavage (29-31). Because pol β bound and synthesized on the upstream primer, we needed to know whether the lack of an upstream primer would eliminate the observed pol β stimulation of FEN1. At a low level of FEN1, we observed a significant stimulation of cleavage as the concentration of pol β was increased with a substrate having an upstream primer (Fig. 3B, lanes 3-6). However, in the substrate lacking the upstream primer, we did not observe any FEN1 cleavage (Fig. 3B, lanes 15-18). It has been previously reported that in substrates lacking an upstream primer, an increased concentration of FEN1 is required for observable cleavage (32). Adding a 50-fold excess of FEN1 cleaved the 5-nt nick-flap to almost 95% completion, and because the initial cleavage was so high we did not observe any additional stimulation by pol β (Fig. 3B, lanes 9-12). Increasing the FEN1 concentration resulted in cleavage of a small fraction of the pseudo-Y substrate, and adding increasing amounts of pol β to the reaction did not stimulate FEN1 beyond the basal cleavage observed (Fig. 3B, lanes 21-24). This showed that pol β cannot bind to FEN1 directly from solution and stimulate its activity and that the mere presence of pol β cannot substitute for an upstream primer. We conclude that stimulation only appears possible when pol β is bound to the 3′ end of the upstream primer and interacts structurally with FEN1.
Because we observed different levels of stimulation of FEN1 cleavage by pol β on the substrates examined above, we considered that pol β stimulation may have been less effective on certain substrates because it bound poorly to those substrates. The binding constants of pol β to the various substrates used in our studies were determined by EMSA gels. From the results summarized in Table 2, interaction of pol β with the various substrates displayed similar nanomolar binding constants with differences less than 2-fold. Among the observed binding affinities, pol β showed the highest value on a 5-nt gap-flap substrate and the least on the double flap substrate. These values imply that pol β was able to effectively bind to all the different substrates, but depending on the size of the gap, the polymerase was unable to interact properly with the flap-bound FEN1. This difference in pol β-FEN1 interaction was much greater in the absence of dNTPs because pol β was unable to alter the substrate structure to favor productive polymerase-nuclease interaction.
TABLE 2.
Relative binding constants
| Substrate | Kda |
|---|---|
| nM | |
| 5-nt gap-flap | 0.36 ± 0.04 |
| 3-nt gap-flap | 0.41 ± 0.19 |
| 1-nt gap-flap | 0.55 ± 0.9 |
| Nick flap | 0.60 ± 0.12 |
| Double flap | 0.70 ± 0.21 |
Apparent dissociation constants measured by electrophoretic mobility gel shift assay. Results are the average of two independent experiments.
pol β Physically Interacts with FEN1—Because pol β stimulated FEN1 cleavage, we were interested in knowing whether pol β can bind directly to FEN1 in the absence of polymer substrate both in vitro and in situ. Human purified pol β and FEN1 were incubated in a coupling buffer, followed by immunoprecipitation of one protein using antibody bound to a spin column. If the two proteins physically interact, the interacting protein will be eluted with the immunoprecipitated protein. Binding can be verified by analyzing the immunoprecipitates on a Western blot using an antibody against the interacting protein. After immunoprecipitation of purified FEN1 and probing the immunoprecipitate on a Western blot with antibody against pol β, we detected an interaction (Fig. 4A). Incubating a higher ratio of FEN1:pol β did not increase the amount of pol β that was immunoprecipitated along with FEN1 (Fig. 4A, compare lanes 6 and 7). This suggests that there is a strong interaction between FEN1 and pol β such that pol β interacted maximally with FEN1 in a 1:1 ratio. If the two proteins interacted nonspecifically, the presence of a 100-fold excess of BSA should have prevented co-precipitation. However, even with a large excess of BSA, the two proteins co-precipitated (Fig. 4A, lane 5). There was no pol β band in the lane from an immunoprecipitation of FEN1 with nonspecific antibody. Immunoprecipitation of FEN1 alone also did not yield a pol β band, indicating that the purified FEN1 protein was not contaminated with pol β. The same set of experiments as described in Fig. 4A was carried out, except that the precipitation was performed with antibody against pol β. The Western blot was probed with antibody against FEN1, with results also demonstrating co-precipitation (Fig. 4B). Because the binding assay in vitro would also detect interactions that might not actually occur in the cell, we performed the same immunoprecipitations with HeLa cell extracts. Results with the extracts showed that the two proteins appear to interact in situ (Fig. 4, C and D). Pretreatment of the cellular extracts with DNase I or ethidium bromide did not change the interaction patterns of FEN1 and pol β (data not shown). Of course, results obtained in situ do not rule out the possibility that the proteins are tethered by other proteins in a larger complex.
pol β Appears to Stabilize FEN1 Binding to Its Cleavage Site—FEN1 is a tracking enzyme that enters at the 5′ end of a flap and slides to the flap base for cleavage (29, 33, 34). Blocking the flap 5′ end using biotin-conjugated streptavidin inhibits FEN1 cleavage (33). Binding of pol β to the 3′-OH end of an upstream primer was important for its interaction with FEN1 and subsequent stimulation of cleavage activity. Because the mode of binding of pol β was significant, we considered whether pol β can circumvent the tracking requirement of FEN1 by binding it directly from solution to the base of a flap in a way that it is productive for cleavage. To address this question, we used a 30-nt flap substrate with a 5′-biotin. In the absence of streptavidin conjugation, we observed an approximate 11-fold stimulation of FEN1 cleavage by pol β (Fig. 5, Control). Conjugation of streptavidin to the 5′ end of the flap prior to the addition of FEN1, pol β, and MgCl2 greatly inhibited the cleavage activity of FEN1 consistent with effective blocking of tracking (Fig. 5, Block, FEN1 + pol β). The addition of pol β did not relieve the inhibitory effect of streptavidin, proving that pol β cannot productively bind FEN1 by overcoming the tracking mechanism.
FIGURE 5.

pol β stabilizes FEN1 binding to its cleavage site. Reactions were performed with 5 fmol of 30-nt flap substrate (D6:T1:U3) containing a biotin label at the 5′ end of the downstream primer. Cleavage by FEN1 (2 fmol) was measured in the presence of varying concentrations of pol β (5, 10, and 20 fmol). Streptavidin was conjugated to the biotinylated 5′ end according to “Experimental Procedures.” Reactions were incubated at 37 °C for 15 min. The conversion of substrate to product (percent) was determined by quantitating the substrate and the product on a denaturing polyacrylamide gel by PhosphorImager analysis. Error bars indicate the standard deviation from a minimum of three independent experiments.
This result allowed us to assess whether pol β tethers the FEN1 to the flap base in a manner that promotes cleavage. Exposing FEN1 to the flap substrate in the absence of MgCl2 allowed FEN1 to track down the flap and bind but not cleave at the flap base. Conjugation of streptavidin following binding of FEN1 ensured that additional FEN1 molecules could not track down the flap and stimulate cleavage. Addition of pol β and MgCl2 significantly increased cleavage product formation (7-fold), compared with addition of only MgCl2 (Fig. 5, FEN1, Block, pol β, compare cleavage at zero versus a high level of pol β). This result suggests that pol β effectively stimulated cleavage of the substrates that contained FEN1 molecules that were trapped onto the flap prior to 5′ end blocking. The level of FEN1 cleavage products was slightly diminished as compared with the control reaction because FEN1 could no longer recycle and load onto other flap substrate molecules that were already blocked.
When pol β was preincubated with the flap substrate prior to blockage and subsequent addition of FEN1 and MgCl2 (Fig. 5, pol β, Block, FEN1) we saw an inhibition of FEN1 cleavage similar to the condition with streptavidin blockage prior to the addition of either enzyme (Fig. 5, Block, FEN1, pol β). Alternatively, when FEN1 and pol β were prebound to the flap before conjugating streptavidin to the 5′ end of the flap, we observed a 10-fold increase in MgCl2-activated conversion of substrate to product (Fig. 5, FEN1 + pol β, Block), compared with the experiment performed in the absence of pol β.
Based on these results, pol β can stimulate cleavage activity of FEN1 molecules by two mechanisms. In the first mechanism, polymerase added after the block can increase cleavage by FEN1 trapped at the base of a flap. Because the pol β cannot increase the number of FEN1 molecules at the flap base in this situation, it must activate a higher percentage of the trapped nuclease. In the second mechanism, the additional component of stimulation measured when pol β was added before the block is consistent with an increase in the number of FEN1 molecules at the flap base before the block traps them there. A reasonable explanation is that FEN1 interaction with pol β bound at the primer terminus tethers more FEN1 at the flap base. In this latter case, the pol β would then increase both the number and active percentage of trapped FEN1 molecules, leading to the maximum amount of cleavage possible with trapped FEN1.
Nick Ligation by DNA LigI Is Stimulated by Various LP-BER Proteins—As with FEN1, the 9-1-1 complex also failed to significantly stimulate LigI in a fully reconstituted system compared with the robust stimulation on cognate substrates in isolation (15). Accordingly, we questioned whether other proteins in the LP-BER multienzyme complex stimulated LigI. Here we used a nicked substrate, similar in sequence to the substrate used to reconstitute LP-BER, but without a THF at the 5′ side of the nick. We tested three LP-BER components for their ability to stimulate LigI. LigI alone at a concentration of 2 fmol/reaction joined a very small amount of the downstream primer (20-mer) to the upstream primer (35-mer) to generate the 55-nt ligation product (Fig. 6, lane 2). Titrating in 10 or 20 fmol of APE1 (Fig. 6, lanes 3 and 4, respectively) stimulated product formation to a maximum of 10-fold. On adding 250 or 500 fmol of 9-1-1 (Fig. 6, lanes 5 and 6, respectively), LigI approximately experienced 9-fold stimulation of activity. Stimulation of LigI activity by APE1 and 9-1-1 has been previously reported, and we found similar levels of increased activity (15, 27). Surprisingly, on adding 10 or 20 fmol of pol β, we also observed a modest stimulation of 2.5-fold (Fig. 6, lanes 7 and 8, respectively). Adding the highest amount of APE1 and pol β to the reaction further stimulated the product formation to a total of 16-fold (Fig. 6, lane 9). The combination of 9-1-1 and pol β did not show a very significant cumulative effect (Fig. 6, lane 10). APE1 and 9-1-1 together maximally increased ligation by 16-fold (Fig. 6, lane 11). In the presence of all three stimulatory proteins (APE1, 9-1-1, and pol β), with LigI it is clear that the maximum possible enhancement is 16-fold, approximately the same as with APE1 and pol β alone (Fig. 6, lane 12). Our results suggest that APE1 and pol β together are capable of stimulating LigI activity to the maximal extent and any further addition of 9-1-1 cannot enhance the product formation.
FIGURE 6.

Nick ligation by DNA LigI is stimulated by various LP-BER proteins. The nicked substrate (D5:T2:U1) was used to show stimulation of LigI by various LP-BER components. When present, LigI was added at a concentration of 2 fmol per reaction. APE1 was added at 5 and 10 fmol. 9-1-1 was added at a concentration of 250 or 500 fmol, and pol β was added at a concentration of 10 and 20 fmol. + in the reaction lane signifies the addition of 10 fmol of APE1, 500 fmol of 9-1-1, and 20 fmol of pol β.
9-1-1 Complex Stimulates LigI in the Absence of APE1—To perform LP-BER without APE1, we used a nicked substrate with a THF residue on the 5′ end of the downstream primer. This substrate resembled the LP-BER intermediate formed after APE1 recognized and cleaved the substrate containing the internal abasic site. Because the substrate is already in a nicked form, we could eliminate APE1 from the reconstitution system. Using similar reaction conditions to that described in Fig. 1B, we limited the FEN1 and LigI concentration and added purified 9-1-1 complex to observe stimulation of either function. Lane 1 represents a reaction containing 5 fmol of the starting substrate (Fig. 7). Five fmol of APE1 was added to the reaction to show that it had no effect on the substrate and hence can be eliminated from the subsequent LP-BER reconstitution experiments using the nicked THF substrate (Fig. 7, lane 2). Interestingly, when we eliminated APE1 from the reactions and used limiting concentrations of either FEN1 or LigI, we observed nearly 4.5-6-fold stimulation in LigI activity upon the addition of 9-1-1 (Fig. 7, lanes 4, 5, 7, and 8). Although this increase in ligase activity is slightly lower than the observed stimulation by 9-1-1 on LigI when used in isolation, it was significantly higher than that observed in the presence of the entire reconstituted system. This supports the conclusion that LigI was being maximally stimulated primarily by APE1. Moreover, the LigI was stimulated nearly to an allowable maximum so that 9-1-1 could not stimulate it further.
FIGURE 7.

9-1-1 complex stimulates LigI in the absence of APE1. Reactions were performed as described under “Experimental Procedures.” All lanes contain 5 fmol of substrate, which was generated by annealing primers D2, T2, and U2 in a 1:2:4 ratio (depicted in the figure). APE1, when present, was added at 3 fmol per reaction. pol β, when present, was added at 20 fmol per reaction. 9-1-1 was added at either 250 or 500 fmol as denoted by the triangles. The amount of FEN1 and LigI (in fmol) used in each reaction is noted above each lane.
DISCUSSION
Although there is evidence that cells employ short and long patch pathways for repair of base damage, the actual relative utilization of one pathway versus the other is not clear. Currently, there are two hypotheses addressing the pathway decision. First, when the abasic site is either oxidized or reduced, pol β cannot eliminate the modified sugar by manifesting 5′-dRP lyase activity. pol β would then perform strand displacement synthesis to create a flap cleaved by FEN1 to make a nick sealed by LigI (35). The ability of pol β to remove the dRP then determines the pathway. Second, the ATP concentration near the abasic site also appears to direct the pathway choice. The XRCC1 (x-ray cross-complementing protein 1) promotes pol β strand displacement synthesis during energy depletion, thereby choosing to process the repair via the long patch pathway (36). However, during energy abundance short patch BER is the preferred mechanism as ligase III promotes ligation to a fully repaired product by preventing strand displacement synthesis (36).
The present results clarify the hierarchy of interaction and stimulation of the various proteins involved in LP-BER. Our studies complement the important observation made by Gemka et al. (15) that the extensive stimulation of FEN1 and LigI by 9-1-1 observed with the individual proteins was greatly attenuated in a fully reconstituted LP-BER system. Even in the presence of limiting concentration of either of the proteins, inclusion of 9-1-1 resulted in only a slight increase in formation of the fully repaired product (Fig. 1B). To understand the reason, we examined the influence of other proteins in the multienzyme complex on these two proteins.
The 9-1-1 clamp forms a complex with pol β and stimulates the rate of nucleotide addition. This stimulation is specific only to pol β and not the replication polymerases such as pol δ and pol α (16). Synthesis by pol β to add a nucleotide in the space occupied by a THF creates an intermediate that resists further synthesis (10, 37). FEN1 cleavage on these substrates removes the THF barrier, creating a 1-nt gap for pol β to bind and perform strand displacement synthesis. Thus, by cleavage, FEN1 can stimulate pol β synthesis on unfavorable substrates (10).
Our current results show that on both the THF-nick and THF-flap intermediates, FEN1 cleavage is promoted by the presence of pol β (Fig. 2). High level stimulations of 11- and 12-fold were observed with the THF-nick and THF-flap intermediates, respectively. pol β increased FEN1 cleavage on nick-flap substrates even in the absence of synthesis, as tested using the pol β synthesis-defective mutant (Fig. 2D) and in the absence of dNTPs (Fig. 2B). Although pol β stimulated FEN1 cleavage, we also observed the counter-stimulation from FEN1 to increase strand displacement synthesis (Fig. 2) (10). This mutual stimulation results in very efficient conversion of a THF-nick substrate to a nicked product for ligation. The efficiency of these mutual effects provides a reasonable explanation for the inability of 9-1-1 to augment FEN1 activity in the fully reconstituted system. This conclusion is consistent with results of measurements of flap cleavage with selected combinations of LP-BER components. When pol β and FEN1 were present together, and then 9-1-1 was added at the start of the reaction, cleavage was only slightly augmented compared with the activity without the addition of 9-1-1 (Fig. 2E). On incubating FEN1 and 9-1-1 prior to adding pol β, we also did not observe a significant effect of the polymerase (data not shown). This analysis confirms that the presence of either 9-1-1 or pol β achieved a nearly maximal stimulation of FEN1.
Substrates with progressively larger gap sizes were used to provide strong evidence that pol β is bound to the 3′ end of the upstream primer of a flap substrate when it stimulates FEN1. Human pol β is composed of two domains, an 8-kDa domain that has high affinity for single-stranded DNA and a 31-kDa domain that binds to double-stranded DNA regions and possesses the catalytic activity (38, 39). The enzyme binds its substrate in two different modes depending on the size of the gap between the upstream and downstream primer (40). When pol β was allowed to synthesize on flap substrates with varying gap lengths, ranging from 5 to 0, it stimulated FEN1 similarly (9-12-fold), irrespective of the size of the gaps in the flap substrate (Fig. 3A). This result was expected because synthesis converted each substrate to the same nick-flap, presumably the best substrate for stimulation.
When synthesis was not allowed, the ability of pol β to stimulate FEN1 decreased with gap size. In the absence of an upstream primer pol β could not stimulate FEN1 at all (Fig. 3B). Binding efficiencies of pol β on these substrates were similar, suggesting that the lack of interaction with the flap-bound FEN1 abrogated the stimulation and not an inability of pol β to bind to certain substrates (Table 2). Interestingly, the stimulatory capacity did not end abruptly at a specific gap size but rather fell off approximately linearly with distance. We hypothesize that the polymerase and nuclease could retain their stimulatory interaction by distorting the single-stranded region of the gap. As the gap became larger, this distortion required more energy. As the energy needed for template distortion approached the binding energy of the two proteins, the proteins experienced periods of transient dissociation. These periods became longer with the greater gap size. The stimulation then decreased proportionally to the fraction of time of the reaction that the two proteins were bound.
We demonstrated co-immunoprecipitation of pol β and FEN1, indicating a direct physical interaction of the proteins. Co-immunoprecipitation was also observed in situ (Fig. 4), suggesting biological relevance. These results are consistent with a previous observation that FEN1 and pol β co-purify from a rat hepatoma cell extract (41). However, Prasad et al. (37) reported that they were unable to observe an interaction using the purified proteins. Very likely, this is an indication that the direct protein-protein interaction is weak. It is consistent with the expectation that the interaction has evolved to occur whereas the proteins are bound to adjacent positions on the substrate, so that substrate binding and protein-protein binding energies both contribute.
Our flap-blocking experiments demonstrate that pol β stimulates FEN1 product formation by two mechanisms. FEN1 can only cleave if it tracks from the 5′ end of a flap to the base. Blocking the 5′ end with a biotin-streptavidin conjugate in the presence of FEN1 allows quantitative assessment of how much productive loading of the nuclease has occurred. The first mechanism of stimulation was revealed by results showing that after a defined number of FEN1 molecules were trapped on the flap, addition of pol β allowed for more cleavage (Fig. 5). Because the number of FEN1 molecules trapped on the flap was unchanged, the extra cleavage could only have occurred if pol β activated nonfunctional nucleases. A likely mechanism is that interaction with the pol β alters protein conformation of FEN1.
Previous results indicated that PCNA, encircling the double-stranded region at the 3′ end of the upstream primer, stabilizes FEN1 binding to the flap base (42). The second mode of stimulation by pol β appears to occur by the same mechanism. When the nuclease tracks to the flap base it can interact directly with pol β. FEN1 activity, tested after the flap was blocked, was distinctly greater if the FEN1 had been loaded in the presence versus absence of pol β. This strongly suggests that pol β stabilizes FEN1 binding to the flap base. 9-1-1 has a very similar structure to PCNA, and it is loaded onto double-stranded DNA by a similar mechanism. It is reasonable to assume that it also stimulates FEN1 by stabilizing nuclease binding to the base of the flap. Because pol β and 9-1-1 seem to employ at least one identical mechanism to stimulate FEN1, it appears that once FEN1 is stabilized by pol β, additional stabilization by 9-1-1 may not be possible. We also note that the first mechanism, activation of FEN1 catalytic activity, was not observed with PCNA (42) and so may be a stimulatory mechanism unique to pol β.
Using a nicked DNA substrate, we showed that various LP-BER proteins stimulate LigI activity. The greatest individual stimulations derived from APE1 were followed by the 9-1-1 complex (Fig. 6) (15, 27). We also measured a modest LigI stimulation by pol β. This is consistent with a previously reported direct association between pol β and LigI in purified bovine testis extracts (11). Significantly, LigI was maximally stimulated by APE1 and pol β to about 16-fold. Both APE1 and 9-1-1 also produced a 16-fold stimulation in the activity of Lig1 (Fig. 6). As with FEN1, these results clearly support the concept of an absolute stimulation maximum, achievable by more than one combination of proteins. The fact that it can be reached in the presence of APE1 and pol β explains the inability of 9-1-1 to additionally stimulate ligation in the reconstituted system.
FEN1 and pol β were reported to employ a “hit and run” mechanism in LP-BER (10). In this mechanism, the pol β binds to the APE1-cleaved substrate containing a 5′-dRP gap substrate. pol β fills in the 1-nt gap creating a dRP flap substrate. However, if the sugar residue is oxidized or reduced, β elimination is not possible and pol β quickly dissociates from this substrate. This is followed by the binding and cleavage of the nicked THF-flap by FEN1. A 1-nt gap is created after FEN1 cleavage, which again requires the binding of pol β to fill in the gap. Our results agree with the concept that these proteins mechanistically function in a defined sequential manner, an essential aspect of the hit and run mechanism. However, structurally they act as a part of a large multiprotein complex. If the proteins operated by fully independent sequential association with the substrate, it would be impossible for protein components acting early in the pathway to stimulate those acting later. Moreover, appropriate proteins could not act together to achieve maximum stimulation of downstream functions.
Taking into consideration all the different reported stimulations of LP-BER proteins, we present both a mechanistic (Fig. 8A) and structural (Fig. 8B) model for the LP-BER pathway. The proposed LP-BER pathway has many sub-pathways that use different sets of enzymes. Because our minimal reconstitution system involves the pathway containing the 9-1-1 complex and pol β, we have confined our model to this pathway. BER is initiated by the recognition of base damage by a specific DNA glycosylase that cleaves the N-glycosydic bond forming an abasic site (10). The 9-1-1 complex is then recruited to the site of base damage and can be loaded onto the substrate with the help of Rad17-RFC2-5 (43-45). In the cell, the Rad17-RFC2-5 might be important in the recognition of DNA lesions and the recruitment and loading of the 9-1-1 complex to that region (46-49). Our minimal reconstitution system does not need Rad17-RFC2-5, because the 9-1-1 complex can load onto the double strands of the substrate by sliding in from the ends. The 9-1-1 complex can physically interact with APE1 both in vivo and in vitro and stimulate cleavage (15). 9-1-1 then interacts with pol β and increases its affinity for the 3′-OH primer end (16). The strand displacement synthesis activity of pol β can be stimulated by the 9-1-1 complex (15, 16), APE1, LigI (data not shown), and FEN1 (10). This creates a flap for FEN1. FEN1 cleavage can also be stimulated by 9-1-1 (15, 18), APE1 (27), and pol β (current results). Cleavage of the flap creates a nick for LigI. LigI has also been shown to be stimulated by 9-1-1 (15), APE1 (27), and pol β (current results and see Ref. 11). Ligation of the 3′ upstream and 5′ downstream primer creates a fully repaired product. Even though these proteins interact with the substrate in sequence, they also interact with other proteins in the multienzyme complex modifying their activities. In this respect the LP-BER complex works like a clock-driven circuit board, with each protein firing (functioning) at the right time, yet part of a fixed structure in which components activate each other.
FIGURE 8.

A, mechanistic model for LP-BER. B, reported stimulations in the LP-BER repairosome. Arrows pointing in a single direction imply a protein (blunt end) stimulating another protein (arrow end). Arrows pointing in both directions imply reported stimulations and counter-stimulations by proteins in the repairosome.
Our results also suggest that the primary roles of 9-1-1 in LP-BER do not include stimulating FEN1 or LigI. Instead, its main activities must be in marking damage sites for recruitment of the BER repair complex, stimulating other BER proteins, and possibly in coordinating the sequential actions of the BER proteins (19, 43).
In conclusion, our results show that pol β structurally interacts with FEN1 and functionally stimulates its cleavage activity. FEN1 is maximally stimulated by pol β in the fully reconstituted LP-BER system accounting for the diminished responsiveness to the 9-1-1 complex. Similarly, LigI is also maximally stimulated by the combination of APE1 and pol β thereby showing no additive stimulation by 9-1-1. Taken together these results suggest that even though the LP-BER proteins stimulate various repair proteins in isolation, there is a remarkable synergy of coordinated functions in the multienzyme repair complex.
Additionally, although LP-BER has been proposed to be a significant repair pathway in cells, there is little concrete evidence of the extent of its utilization. The direct interaction of pol β with FEN1, resulting in stimulation of nuclease activity, provides strong evidence that the polymerase of short patch BER is also involved in a pathway that requires flap creation and cleavage. The previously reported stimulation of pol β by FEN1 is also consistent with this conclusion. However, we feel that the stimulation of FEN1 provides more compelling support for the involvement of pol β in a flap-creating repair pathway. This is because FEN1 might be anticipated to promote strand displacement synthesis, as it does with pol β. However, it is difficult to rationalize why pol β would stimulate FEN1 only when the polymerase is bound on an immediately adjacent upstream primer, unless the two proteins had evolved to work together in an LP-BER pathway. The functional cooperation between these two proteins is the central hallmark of efficient repair of a base damage via the LP-BER pathway.
Acknowledgments
We thank Drs. Samuel H. Wilson and Yuan Liu for the generous gift of the wild-type and mutant DNA polymerase β used in our studies. In addition, we thank members of the Bambara laboratory for helpful discussions.
This work was supported by National Institutes of Health Grant GM024441 (to R. A. B.), with additional support from Grant GM032833 (to A. S.).
Footnotes
The abbreviations used are: BER, base excision repair; LP-BER, long patch-BER; nt, nucleotide(s); pol β, DNA polymerase β; FEN1, flap endonuclease 1; AP, apurinic/apyrimidinic; APE1, AP endonuclease 1; 9-1-1, Rad 9-Rad1-Hus1; Lig1, DNA ligase 1; dRP, deoxyribose phosphate; DTT, dithiothreitol; BSA, bovine serum albumin; THF, tetrahydrofuran; EMSA, electrophoretic mobility shift assay; PCNA, proliferating cell nuclear antigen; IP, immunoprecipitation.
References
- 1.Scharer, O. D., and Jiricny, J. (2001) BioEssays 23 270-281 [DOI] [PubMed] [Google Scholar]
- 2.Matsumoto, Y., and Kim, K. (1995) Science 269 699-702 [DOI] [PubMed] [Google Scholar]
- 3.Piersen, C. E., Prasad, R., Wilson, S. H., and Lloyd, R. S. (1996) J. Biol. Chem. 271 17811-17815 [DOI] [PubMed] [Google Scholar]
- 4.Wilson, S. H. (1998) Mutat. Res. 407 203-215 [DOI] [PubMed] [Google Scholar]
- 5.Memisoglu, A., and Samson, L. (2000) Mutat. Res. 451 39-51 [DOI] [PubMed] [Google Scholar]
- 6.Sung, J. S., and Demple, B. (2006) FEBS J. 273 1620-1629 [DOI] [PubMed] [Google Scholar]
- 7.Wilson, D. M., 3rd, and Barsky, D. (2001) Mutat. Res. 485 283-307 [DOI] [PubMed] [Google Scholar]
- 8.Sobol, R. W., Horton, J. K., Kuhn, R., Gu, H., Singhal, R. K., Prasad, R., Rajewsky, K., and Wilson, S. H. (1996) Nature 379 183-186 [DOI] [PubMed] [Google Scholar]
- 9.Sattler, U., Frit, P., Salles, B., and Calsou, P. (2003) EMBO Rep. 4 363-367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu, Y., Beard, W. A., Shock, D. D., Prasad, R., Hou, E. W., and Wilson, S. H. (2005) J. Biol. Chem. 280 3665-3674 [DOI] [PubMed] [Google Scholar]
- 11.Prasad, R., Singhal, R. K., Srivastava, D. K., Molina, J. T., Tomkinson, A. E., and Wilson, S. H. (1996) J. Biol. Chem. 271 16000-16007 [DOI] [PubMed] [Google Scholar]
- 12.Chang, D. Y., and Lu, A. L. (2005) J. Biol. Chem. 280 408-417 [DOI] [PubMed] [Google Scholar]
- 13.Guan, X., Madabushi, A., Chang, D. Y., Fitzgerald, M. E., Shi, G., Drohat, A. C., and Lu, A. L. (2007) Nucleic Acids Res. 35 6207-6218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi, G., Chang, D. Y., Cheng, C. C., Guan, X., Venclovas, C., and Lu, A. L. (2006) Biochem. J. 400 53-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gembka, A., Toueille, M., Smirnova, E., Poltz, R., Ferrari, E., Villani, G., and Hubscher, U. (2007) Nucleic Acids Res. 35 2596-2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Toueille, M., El-Andaloussi, N., Frouin, I., Freire, R., Funk, D., Shevelev, I., Friedrich-Heineken, E., Villani, G., Hottiger, M. O., and Hubscher, U. (2004) Nucleic Acids Res. 32 3316-3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Friedrich-Heineken, E., Toueille, M., Tannler, B., Burki, C., Ferrari, E., Hottiger, M. O., and Hubscher, U. (2005) J. Mol. Biol. 353 980-989 [DOI] [PubMed] [Google Scholar]
- 18.Wang, W., Brandt, P., Rossi, M. L., Lindsey-Boltz, L., Podust, V., Fanning, E., Sancar, A., and Bambara, R. A. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 16762-16767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smirnova, E., Toueille, M., Markkanen, E., and Hubscher, U. (2005) Biochem. J. 389 13-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, W., Lindsey-Boltz, L. A., Sancar, A., and Bambara, R. A. (2006) J. Biol. Chem. 281 20865-20872 [DOI] [PubMed] [Google Scholar]
- 21.Jaiswal, A. S., Bloom, L. B., and Narayan, S. (2002) Oncogene 21 5912-5922 [DOI] [PubMed] [Google Scholar]
- 22.Bornarth, C. J., Ranalli, T. A., Henricksen, L. A., Wahl, A. F., and Bambara, R. A. (1999) Biochemistry 38 13347-13354 [DOI] [PubMed] [Google Scholar]
- 23.Lindsey-Boltz, L. A., Bermudez, V. P., Hurwitz, J., and Sancar, A. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 11236-11241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tom, S., Ranalli, T. A., Podust, V. N., and Bambara, R. A. (2001) J. Biol. Chem. 276 48781-48789 [DOI] [PubMed] [Google Scholar]
- 25.Beard, W. A., and Wilson, S. H. (1995) Methods Enzymol. 262 98-107 [DOI] [PubMed] [Google Scholar]
- 26.Menge, K. L., Hostomsky, Z., Nodes, B. R., Hudson, G. O., Rahmati, S., Moomaw, E. W., Almassy, R. J., and Hostomska, Z. (1995) Biochemistry 34 15934-15942 [DOI] [PubMed] [Google Scholar]
- 27.Ranalli, T. A., Tom, S., and Bambara, R. A. (2002) J. Biol. Chem. 277 41715-41724 [DOI] [PubMed] [Google Scholar]
- 28.Kao, H. I., Henricksen, L. A., Liu, Y., and Bambara, R. A. (2002) J. Biol. Chem. 277 14379-14389 [DOI] [PubMed] [Google Scholar]
- 29.Harrington, J. J., and Lieber, M. R. (1994) EMBO J. 13 1235-1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrington, J. J., and Lieber, M. R. (1995) J. Biol. Chem. 270 4503-4508 [DOI] [PubMed] [Google Scholar]
- 31.Murante, R. S., Huang, L., Turchi, J. J., and Bambara, R. A. (1994) J. Biol. Chem. 269 1191-1196 [PubMed] [Google Scholar]
- 32.Henricksen, L. A., Tom, S., Liu, Y., and Bambara, R. A. (2000) J. Biol. Chem. 275 16420-16427 [DOI] [PubMed] [Google Scholar]
- 33.Murante, R. S., Rust, L., and Bambara, R. A. (1995) J. Biol. Chem. 270 30377-30383 [DOI] [PubMed] [Google Scholar]
- 34.Wu, W., Blumberg, B. M., Fay, P. J., and Bambara, R. A. (1995) J. Biol. Chem. 270 325-332 [DOI] [PubMed] [Google Scholar]
- 35.Klungland, A., and Lindahl, T. (1997) EMBO J. 16 3341-3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petermann, E., Z. M., and Oei, S. L. (2003) DNA Repair 2 1101-1114 [DOI] [PubMed] [Google Scholar]
- 37.Prasad, R., Dianov, G. L., Bohr, V. A., and Wilson, S. H. (2000) J. Biol. Chem. 275 4460-4466 [DOI] [PubMed] [Google Scholar]
- 38.Casas-Finet, J. R., Kumar, A., Morris, G., Wilson, S. H., and Karpel, R. L. (1991) J. Biol. Chem. 266 19618-19625 [PubMed] [Google Scholar]
- 39.Kumar, A., Abbotts, J., Karawya, E. M., and Wilson, S. H. (1990) Biochemistry 29 7156-7159 [DOI] [PubMed] [Google Scholar]
- 40.Rajendran, S., Jezewska, M. J., and Bujalowski, W. (1998) J. Biol. Chem. 273 31021-31031 [DOI] [PubMed] [Google Scholar]
- 41.Rein, D. C., Recupero, A. J., Reed, M., and Meyer, R. R. (1990) in The Eukaryotic Nucleus (Strauss, P. R., and Wilson, S. H., eds) Vol. 1, pp. 95-124, Telford Press, Inc., Caldwell, NJ [Google Scholar]
- 42.Tom, S., Henricksen, L. A., and Bambara, R. A. (2000) J. Biol. Chem. 275 10498-10505 [DOI] [PubMed] [Google Scholar]
- 43.Bermudez, V. P., Lindsey-Boltz, L. A., Cesare, A. J., Maniwa, Y., Griffith, J. D., Hurwitz, J., and Sancar, A. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 1633-1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou, L., Liu, D., and Elledge, S. J. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 13827-13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellison, V., and Stillman, B. (2003) PLoS Biol. 1 E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burtelow, M. A., Kaufmann, S. H., and Karnitz, L. M. (2000) J. Biol. Chem. 275 26343-26348 [DOI] [PubMed] [Google Scholar]
- 47.Dahm, K., and Hubscher, U. (2002) Oncogene 21 7710-7719 [DOI] [PubMed] [Google Scholar]
- 48.Meister, P., Poidevin, M., Francesconi, S., Tratner, I., Zarzov, P., and Baldacci, G. (2003) Nucleic Acids Res. 31 5064-5073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou, L., Cortez, D., and Elledge, S. J. (2002) Genes Dev. 16 198-208 [DOI] [PMC free article] [PubMed] [Google Scholar]